

Abstract

The phenethylamine backbone is a privileged substructure found in a wide variety of G protein-coupled receptor (GPCR) ligands. This includes both endogenous neurotransmitters and active pharmaceutical agents. More than 20 structurally unique heterocyclic phenethylamine derivatives were broadly evaluated for GPCR affinity. Selective ligands for the 5-HT2B, 5-HT7, and σ1 receptors were identified, each with low nanomolar binding affinities. The σ1 receptor affinity was supported in a cellular assay that provided evidence for increased cell survival under oxidative stress.
Keywords: 5-HT7, serotonin receptors, σ receptor, σ1 receptor, G protein-coupled receptors, neuroprotection
G protein-coupled receptors (GPCRs) are a prominent pharmacological target. More than 30% of FDA approved drugs target at least one GPCR.1,2 Worldwide, more than 25% of drug sales come from GPCR modulating compounds.3 GPCR modulators are used to treat a wide variety of diseases and disorders including allergies,4 schizophrenia,5 depression,6 pain management,7 and asthma.8 Common GPCR targeting pharmaceuticals9 include the antihistamine loratadine,4 antidepressants fluoxetine and sertraline,6 antipsychotics aripiprazole, clozapine, and haloperidol,5 the opioids morphine, hydrocodone, and fentanyl,7 and bronchodilators salbutamol and tiotropium bromide.10 The phenethylamine core is a privileged substructure in GPCR ligands (Figure 1). This is partially because several endogenous neurotransmitters or neuromodulators are phenethylamines including dopamine11 and epinephrine12 (Figure 1a). The phenethylamine backbone is also found in a wide variety of GPCR targeting active pharmaceutical ingredients including morphine, salbutamol, and pseudoephedrine (Figure 1b).
Figure 1.

Phenethylamine containing GPCR ligands.
We recently developed a tandem Winstein rearrangement Friedel–Crafts alkylation that enabled the synthesis of differentially functionalized heterocyclic tertiary azides from an equilibrating mixture of allylic azides (Scheme 1a).13 This cascade was synthetically attractive because it constructed tetralins, chromanes, and tetrahydroquinolines featuring a tetra-substituted stereocenter while maintaining a diversifiable vinyl group. The heterocyclic products could be readily converted into substituted phenethylamine derivatives containing either a primary amine or pyrrolidine motif (Scheme 1b). It seemed prudent to investigate the potential GPCR affinity of these molecules because of their rigid structure, small molecular weight, and phenethylamine backbone. This report describes the results of an initial screen, which identified several individual molecules each with low nM binding affinity for 5-HT2B, 5-HT7, or σ1 receptor. Furthermore, the small family of compounds screened demonstrates divergent selectivity across the GPCR family. A new lead compound was identified for the 5-HT7 receptor, and a second lead compound was identified for the σ1 receptor. The σ1 receptor affinity was further supported by cellular assays.
Scheme 1. Source of Novel Phenethylamines.

The synthesis of these compounds began with the addition of vinyl Grignard to ketone 5, forming tertiary allylic alcohol 6 (Scheme 2). Subsequent cross metathesis with cis-1,4-but-2-enediol diacetate and Hoveyda–Grubbs second generation catalyst afforded allylic acetate 7. Azidation occurred upon exposure to TMSN3 and catalytic Zn(OTf)2 to generate allylic azide 8 as a mixture of equilibrating isomers. Methanolysis cleaved the acetate, and resulting alcohol 9 was activated as the trichloroacetimidate 1. The dynamic cyclization was performed with catalytic AgSbF6, yielding tertiary azide 2, typically in >25:1 dr. Global reduction with H2 and palladium on carbon afforded primary amine 3. Alternatively, exposure to HBCy2 resulted in pyrrolidine 4. The pyrrolidine could be subjected to N-methylation (10) or acetylation (11).
Scheme 2. Synthesis of (±)-GPCR Ligands.

Reagents and conditions: (a) vinyl MgCl, THF, 0 °C, 30 min, 83–91%; (b) cis-1,4-diacetoxy-2-butene, 1–2 mol % Hoveyda–Grubbs second generation catalyst, 40 °C, 18 h, 68–86%; (c) TMSN3, 10 mol % Zn(OTf)2, rt, 90 min, 29–87%; (d) K2CO3, MeOH, rt, 30 min, 97%–quant.; (e) NCCCl3, 20 mol % DBU, rt, 90 min, 73–97%; (f) 10 mol % AgSbF6, CHCl3, 40–60 °C, 24 h, 39–94%; (g) H2, 10% w/w Pd/C, MeOH, 18 h, 52%-quant; (h) HBCy2, DCM, 0 °C to rt, 18 h, 35–84%; (i) aq. CH2O, NaBH3CN, HOAc, NCCH3, 0 °C to rt, 30 min, 70–89%; (j) Ac2O, TEA, DMAP, 0 °C to rt, 18 h 73–96%.
The GPCR binding affinity of these compounds was assessed through the Psychoactive Drug Screening Program.14 Initially, three primary amines were screened (Table 1, see the Supporting Information for selected binding curves). Compounds 3a–3c represent one example each of the three synthetically accessible cores: tetralin (3a), chromane (3b), and tetrahydroquinoline (3c). These molecules were screened across a wide range of human GPCR including serotonin (5-HT), α- and β-adrenergic (Alpha and Beta), dopamine (D), histamine (H), muscarinic (M), opioid (OR), sigma (σ), and others (Table 1). Of the three compounds assayed, all exhibited at least moderate binding (Ki ≤ 200 nM) against one or more receptors. Tetralin 3a was the only compound to exhibit any affinity against the δ-, μ-, or κ-opioid receptor. This is consistent with the structures of other phenethyl opioid ligands that contain a cyclohexyl backbone (e.g., morphine, Figure 1b).7,15 Compound 3a exhibited moderate affinity toward the 5-HT2B and σ1 receptors. Chromane 3b exhibited moderate affinity against the 5-HT2B receptor but lacked any κ-opioid binding. Tetrahydroquinoline 3c exhibited the strongest initial hit, with a Ki of 74 nM against the 5-HT1A receptor. Based on these initial results and lack of opioid affinity, a subsequent structure activity relationship (SAR) study was conducted on the chromane and tetrahydroquinoline scaffolds, screening against the 5-HT1A, 5-HT2B, 5-HT7, σ1, and σ2 receptors.
Table 1. Initial GPCR Screena.

| pKia |
|||
|---|---|---|---|
| receptor | compound 3a | compound 3b | compound 3c |
| 5-HT1A | 6.2 ± 0.1b | 5.7 ± 0.1b | 7.13 ± 0.06b |
| 5-HT1B | <5 | <5 | <5 |
| 5-HT1D | <5 | <5 | <5 |
| 5-HT1E | <5 | <5 | <5 |
| 5-HT2A | <5 | <5 | <5 |
| 5-HT2B | 6.84 ± 0.06b | 6.75 ± 0.06b | 6.1 ± 0.1b |
| 5-HT2C | 5.6 ± 1.0b | 5.6 ± 3.5b | 5.4 ± 1.3b |
| 5-HT3 | <5 | <5 | <5 |
| 5-HT5A | 5.7 ± 1.5b | <5 | <5 |
| 5-HT6 | <5 | <5 | <5 |
| 5-HT7 | <5 | <5 | 6.0 ± 0.1b |
| Alpha1A | <5 | <5 | 5.8 ± 0.1b |
| Alpha1B | <5 | <5 | <5 |
| Alpha1D | <5 | <5 | <5 |
| Alpha2A | 5.2 ± 0.2b | <5 | 6.4 ± 0.1b |
| Alpha2B | 5.3 ± 0.1b | <5 | 6.4 ± 0.1b |
| Alpha2C | <5 | <5 | <5 |
| Beta1 | <5 | <5 | <5 |
| Beta2 | 5.3 ± 0.1b | <5 | <5 |
| Beta3 | <5 | <5 | 6.0 ± 0.1b |
| BZP | 5.8 ± 0.1b | <5 | 5.1 ± 0.1b |
| D1 | <5 | <5 | <5 |
| D2 | <5 | <5 | <5 |
| D3 | <5 | <5 | <5 |
| D4 | <5 | <5 | 5.7 ± 0.1b |
| D5 | <5 | <5 | <5 |
| DAT | 5.3 ± 0.2b | <5 | 5.5 ± 0.2b |
| GABAA | <5 | <5 | <5 |
| H1 | 5.5 ± 0.5b | <5 | <5 |
| H2 | <5 | <5 | <5 |
| H3 | <5 | <5 | 5.4 ± 0.2b |
| H4 | <5 | <5 | <5 |
| M1 | <5 | <5 | <5 |
| M2 | <5 | <5 | <5 |
| M3 | <5 | <5 | <5 |
| M4 | <5 | <5 | <5 |
| M5 | <5 | <5 | <5 |
| NET | 5.6 ± 0.1b | 6.5 ± 0.1b | 6.4 ± 0.1c |
| δ-OR | <5 | <5 | <5 |
| κ-OR | 6.5 ± 0.1b | <5 | <5 |
| μ-OR | 5.7 ± 0.1b | <5 | <5 |
| PBR | <5 | <5 | <5 |
| σ1 | 6.9 ± 0.1b | 6.0 ± 0.1b | 6.5 ± 0.1b |
| σ2 | <5 | <5 | 6.2 ± 0.1b |
For pKi values reported as <5, the compound did not demonstrate ≥50% binding in an initial assay at 10 μM concentration.
These data reflect triplicate measurements of a single experiment. The error reflects the error in the sigmoidal curve fit.
These data reflect the average of triplicate experiments run in triplicate. The error reflects the standard deviation of the three calculated pKi values. Values in italic type are noted for the reader’s convenience.
Assay results for five additional primary amine analogues are displayed (Table 2). Amines 3d and 3e did not demonstrate a Ki below 100 nM for any of the GPCRs assayed. Amine 3f bound the σ1 receptor with a Ki of 16 nM and demonstrated reasonable selectivity relative to the 5-HT2B receptor (10-fold), which was the second most sensitive receptor assayed. Amines 3g and 3h have higher affinities toward the 5-HT2B receptor (Ki = 3.5 and 20 nM, respectively). Compound 3g demonstrated ∼150-fold selectivity relative to the σ2 receptor, which was the second most sensitive GPCR assayed. The 5-HT2B receptor is a member of the 5-HT family of receptors that is known to be an essential receptor during development.16 Long-term consumption of 5-HT2B agonists can induce potentially fatal myofibroblast proliferation and valvular heart disease.17,18 Thus, the 5-HT2B receptor is considered an antitarget. Therefore, further optimization of this amine class was not pursued, opting instead to evaluate the σ1 affinity demonstrated by amine 3f on more rigid pyrrolidine analogues.
Table 2. Ki Data for Primary Amino-chromanesa.

For pKi values reported as <5, the compound did not demonstrate ≥50% binding in an initial assay at 10 μM concentration.
These data reflect triplicate measurements of a single experiment. The error reflects the error in the sigmoidal curve fit.
These data reflect the average of triplicate experiments run in triplicate. The error reflects the standard deviation of the three calculated pKi values. Values highlighted in red are noted for the reader’s convenience. Values not determined are noted as n.d.
The σ receptors were initially thought to be members of the opioid receptor family.19 Since, it has been shown that σ1 acts as a chaperone protein localized in the endoplasmic reticulum,20 and it affects a wide variety of cellular functions including regulation of opioid receptors,20 kinases,21 TRPV1,22 dopamine receptors,23 apoptosis,24 as well as cellular calcium and potassium levels.25 Modulating intracellular calcium levels implicated σ1 as a target for treating colon and breast cancer.26,27 Modulating the σ1 receptor also effects alcohol abuse,28 pain management,29 opioid analgesia,30,31 and neuroprotection in models of retinal neural degradation.32,33
Pyrrolidine containing chromanes were assessed for σ1 binding (Table 3). A direct analogue of amine 3f was essentially equipotent against σ1 (4f), while replacing the phenyl group with a methoxy group was disadvantageous (4e). Exploring substitution around the arene provided mixed results. Benzyloxy compound 4i was a high-affinity σ1 ligand. Removing the benzyl group was detrimental to affinity (4d). Some affinity could be restored through N-methylation (10d). Unsurprisingly, masking the basic amine as an amide removed affinity in both cases examined (11d and 11l). Compound 4j could be a lead compound. While compound 4j demonstrated reduced affinity relative to amine 3f as a σ1 ligand, it shows >200-fold selectivity versus the σ2 receptor. Changing the methoxy groups for methyl groups enhanced 5-HT2B binding (4k), and the other compounds assayed provided both reduced σ1 affinity and selectivity (4l and 4m).
Table 3. Ki Data for Pyrrolidine Containing Chromanesa.
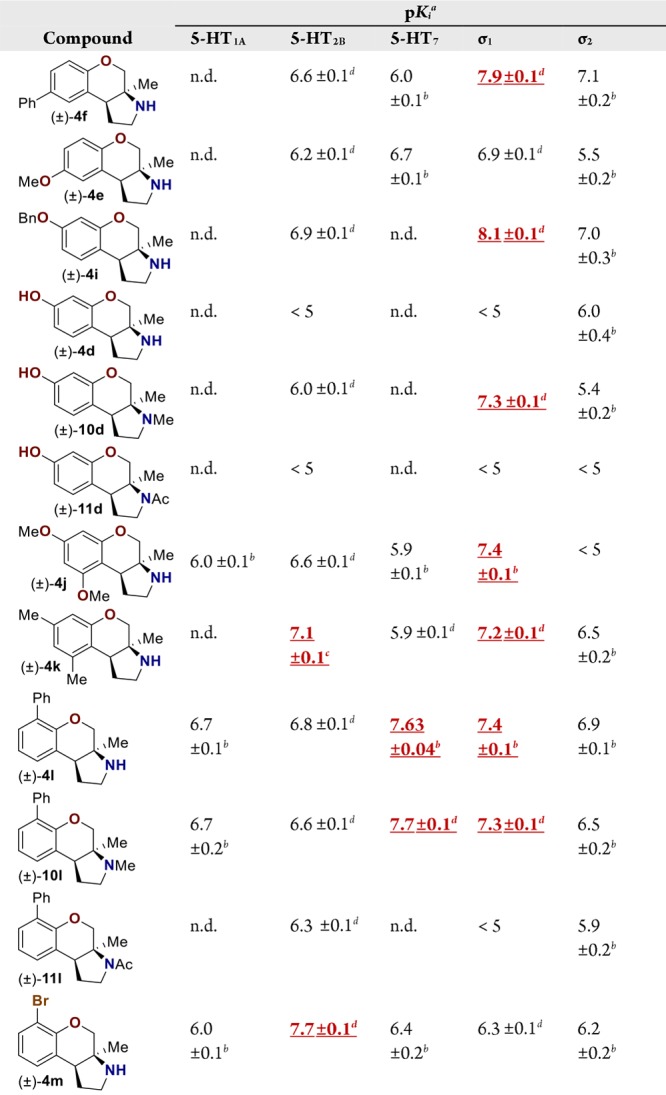
For pKi values reported as <5, the compound did not demonstrate ≥50% binding in an initial assay at 10 μM concentration.
These data reflect triplicate measurements of a single experiment. The error reflects the error in the sigmoidal curve fit.
These data reflect the average duplicate experiments run in triplicate. The error reflects one-half the difference between the two calculated pKi values.
These data reflect the average of triplicate experiments run in triplicate. The error reflects the standard deviation of the three calculated pKi values. Values highlighted in red are noted for the readers convenience. Values not determined are noted as n.d.
With an attempt to evaluate the 5-HT1A affinity demonstrated by amine 3c (Table 1), other tetrahydroquinolines were investigated (Table 4). As a direct comparison to amine 3c (Table 1), pyrrolidine 4c (Table 4) was assayed. While primary amine 3c was ∼15-fold selective for 5-HT1A over 5-HT7, pyrrolidine 4c was not a suitable ligand for 5-HT1A (Ki > 10 000 nM) and was instead a high-affinity 5-HT7 ligand (Ki = 6.3 nM). Therefore, the relatively small structural change resulted in more than a 20 000-fold relative difference in the 5-HT1A versus 5-HT7 selectivity. The 5-HT7 receptor has been implicated in the regulation of multiple biological functions including sleep, circadian rhythm, and mood.16 Various 5-HT7 antagonists have been investigated for depression treatment along with other disorders.34 The 5-HT7 affinity could be further enhanced though N-methylation (10c), while acylation removed affinity (11c). Other substituted arenes displayed reduced 5-HT7 affinity (4n and 4o). Even though compound 4c has a weaker affinity than compound 10c, compound 4c was selected for further assay because it exhibited enhanced selectivity, having a ∼75-fold lower affinity for the next most sensitive receptor, σ1.
Table 4. Ki Data for Tetrahydroquinolinesa.

For pKi values reported as <5, the compound did not demonstrate ≥50% binding in an initial assay at 10 μM concentration.
These data reflect triplicate measurements of a single experiment. The error reflects the error in the sigmoidal curve fit.
These data reflect the average of triplicate experiments run in triplicate. The error reflects the standard deviation of the three calculated pKi values. Values highlighted in red are noted for the reader’s convenience. Values not determined are noted as n.d.
Based on the results outlined in Tables 1–4, compounds 4c and 4j were screened more broadly against a wider array of GPCRs (Table 5). Gratifyingly, only minimal affinity was observed across the additional GPCRs that were investigated. This indicates that compound 4c could be considered a new lead targeting 5-HT7 due to the low nM binding affinity (Ki = 6.3 nM) and selectivity (∼75-fold versus next most sensitive receptor). Compound 4j could be considered a new lead targeting σ1 due to its respectable affinity (Ki = 44 nM), good σ1 versus σ2 selectivity (>200-fold), and good selectivity versus other GPCRs (>5-fold versus 5-HT2B, and >20-fold versus 38 other GPCRs).
Table 5. GPCR Screen for Selectivitya.

| pKia |
||
|---|---|---|
| receptor | compound 4c | compound 4j |
| 5-HT1A | <5 | 6.0 ± 0.1 |
| 5-HT1B | <5 | <5 |
| 5-HT1D | <5 | <5 |
| 5-HT1E | <5 | <5 |
| 5-HT2B | 6.3 ± 0.1 | 6.6 ± 0.1 |
| 5-HT2C | <5 | 5.8 ± 0.1 |
| 5-HT3 | <5 | <5 |
| 5-HT6 | <5 | <5 |
| 5-HT7 | 8.2 ± 0.1 | 5.9 ± 0.1 |
| Alpha1A | <5 | <5 |
| Alpha1B | <5 | <5 |
| Alpha1D | 6.0 ± 0.1 | <5 |
| Alpha2B | <5 | <5 |
| Alpha2C | 6.0 ± 0.1 | <5 |
| Beta1 | 5.5 ± 0.1 | 6.0 ± 0.1 |
| Beta2 | <5 | 6.24 ± 0.03 |
| Beta3 | <5 | <5 |
| BZP | <5 | 5.2 ± 0.1 |
| D1 | <5 | <5 |
| D2 | <5 | <5 |
| D3 | <5 | <5 |
| D4 | <5 | <5 |
| D5 | <5 | <5 |
| DAT | <5 | <5 |
| GABAA | <5 | <5 |
| H1 | <5 | <5 |
| H2 | 5.7 ± 0.1 | <5 |
| H4 | <5 | <5 |
| M1 | <5 | <5 |
| M2 | <5 | <5 |
| M3 | <5 | <5 |
| M4 | <5 | <5 |
| M5 | <5 | <5 |
| NET | 5.8 ± 0.1 | <5 |
| δ-OR | <5 | <5 |
| κ-OR | <5 | <5 |
| μ-OR | <5 | <5 |
| PBR | <5 | <5 |
| σ1 | 6.3 ± 0.1 | 7.4 ± 0.1 |
| σ2 | 6.0 ± 0.1 | <5 |
For pKi values reported as <5, the compound did not demonstrate ≥50% binding in an initial assay at 10 μM concentration. These data reflect triplicate measurements of a single experiment. The error reflects the error in the sigmoidal curve fit. Values in italic type are noted for the reader’s convenience.
The σ1 receptor is a target for protecting retinal cells from neural degradation.32,33,35 To date, the most studied agent is (+)-pentazocine (PTZ), which has shown promise in mice.36,37 Unfortunately, PTZ is a potent opioid and is probably unsuitable for clinical use as a treatment for retinopathy.7 The protective effects of compound 4j were demonstrated in the well-characterized cone photoreceptor cell line 661W.38 Treatment of 661W cells with up to 200 μM of compound 4j for 24 h did not alter cell viability as measured using the MTT assay (Figure 2). This indicated that compound 4j was not cytotoxic to 661W cells at the concentrations assayed.
Figure 2.
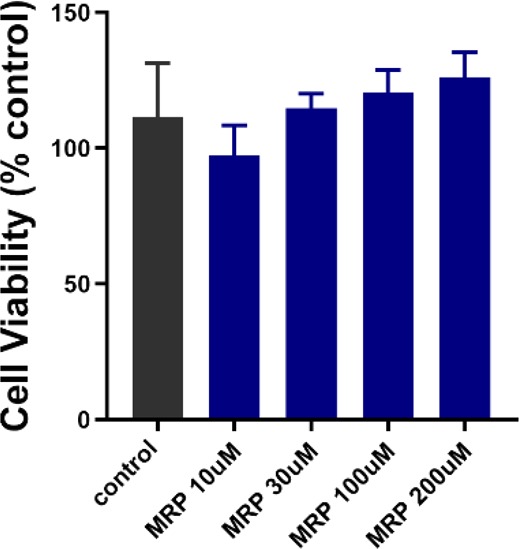
Cytotoxicity assay of compound 4j in 661W cells. 661W cells were treated with compound 4j (10, 30, 100, and 200 μM) for 24 h. Cell viability was assessed using the MTT assay. No significant difference in viability was observed at the 4j concentrations tested compared with nontreated cells. MRP = compound 4j.
Exposing 661W cells to tert-butyl hydroperoxide (tBHP, 55 μM) for 24 h induced oxidative stress, which decreased cell viability by more than 50% (Figure 3). Cotreatment with compound 4j resulted in a dose-dependent improvement in cell viability (see the Supporting Information for additional concentrations). At a 4j concentration of 30 μM, cell viability was significantly greater than tBHP-exposed cells and was >80% of nontreated cells. The beneficial effects of compound 4j (at 30 μM) were similar to those observed in cells treated with PTZ (50–100 μM, Figure 3). At the highest concentration tested (200 μM), the beneficial effects of compound 4j were no longer observed. The beneficial effects of compound 4j (30 μM) could be counteracted by the addition of BD1063 (10 μM), which is a known σ1 receptor antagonist (see the Supporting Information).
Figure 3.
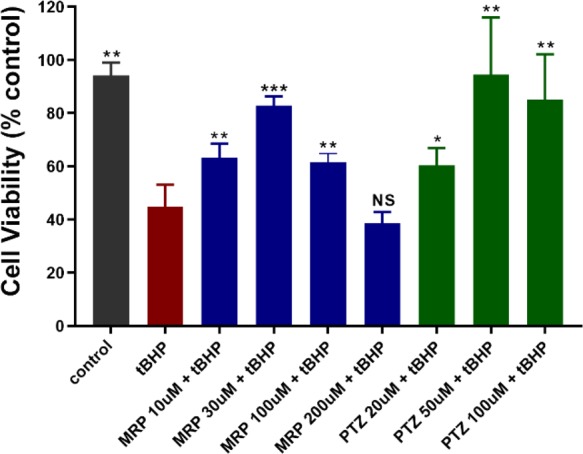
Enhanced 661W cell viability with compound 4j upon tBHP exposure. 661W cells were treated with tBHP in the presence/absence of increasing concentrations of compound 4j (10–200 μM) or the σ1 receptor ligand PTZ (20–100 μM) for 24 h before cell viability assessment. Cell viability was assessed using the MTT assay. Data are presented as mean ± standard deviation (SD) of triplicate measurements; *p < 0.05; ** p < 0.01; *** p < 0.001; ns = not significant as compared to the tBHP treatment. (tBHP = tert-butyl hydroperoxide; MRP = compound 4j; PTZ = (+)-pentazocine.)
Earlier studies demonstrated the beneficial effects of σ1 receptor modulation by PTZ in attenuating oxidative stress in 661W cells.39 Compound 4j was evaluated for similar effects. Reactive oxygen species (ROS) were detected using the CellROX green reagent in an immunocytochemical assay upon exposure of 661W cells to tBHP (55 μM). CellROX is a cell-permeant dye that is weakly fluorescent in a reduced state but exhibits bright green photostable fluorescence upon oxidation by ROS. Cells that received no tBHP showed minimal fluorescence, while cells exposed to tBHP showed a marked increase in green fluorescence (Figure 4A). Treatment with compound 4j resulted in decreased green fluorescence, especially at the 30 μM concentration. These data were similar to the fluorescence suppression observed with PTZ treatment (Figure 4A). Quantification of fluorescence intensity indicated a marked increase in ROS in the tBHP-exposed cells, whereas the level of ROS was significantly reduced in tBHP-exposed cells treated with compound 4j or PTZ (Figure 4B).
Figure 4.
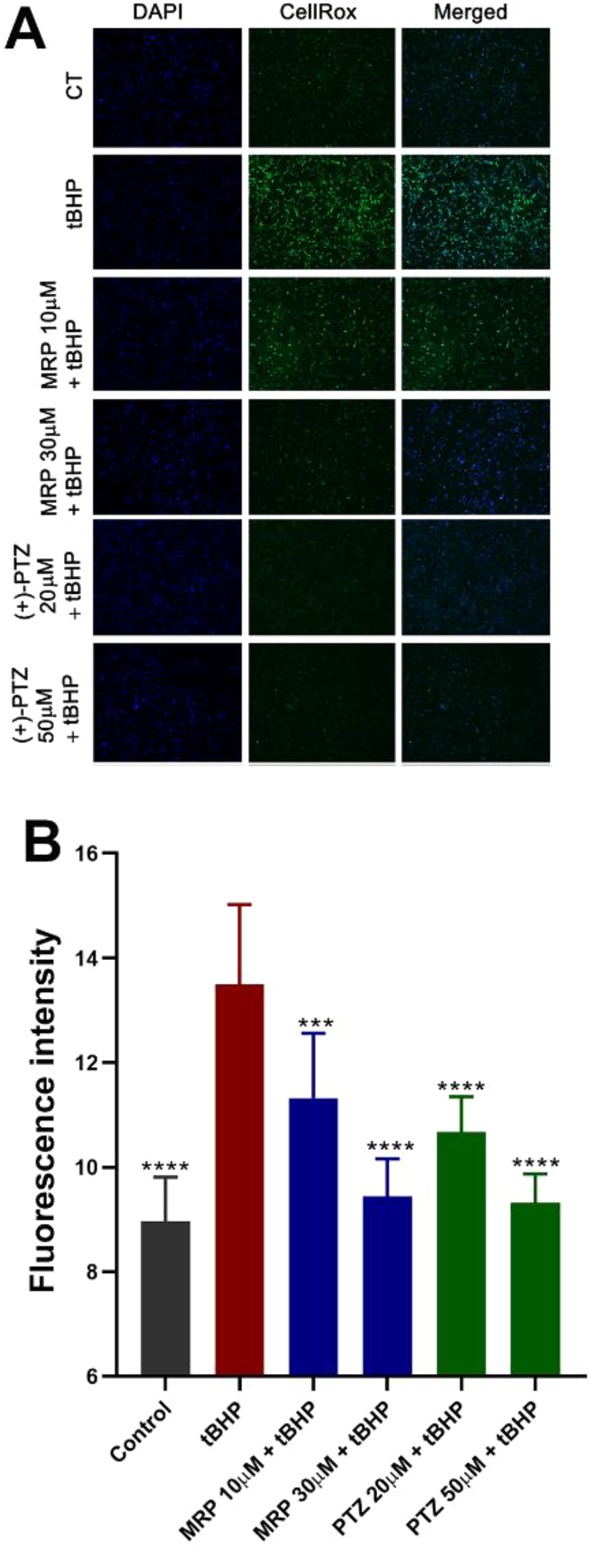
Attenuated oxidative stress induced by tBHP in 661W cells upon treatment with compound 4j or PTZ. 661W cells were seeded on coverslips for 18 h. Cells either were or were not (control) exposed for 2 h to tBHP (55 μM) in the presence/absence of compound 4j or PTZ. (A) Representative immunofluorescent images of cells incubated with CellROX green reagent to detect ROS; green fluorescent signals indicated ROS as visualized by epifluorescence. DAPI was used to label nuclei (blue). (B) Quantification of fluorescent intensity reflecting ROS levels of data shown in panel A. Data are presented as mean ± SD. Data represent three independent experiments performed in duplicate. Significant differences are indicated: *** p < 0.001; ****p < 0.0001 as compared to the tBHP treatment. (CT = control, tBHP = tert-butyl hydroperoxide; MRP = compound 4j; PTZ = (+)-pentazocine.)
In conclusion, the GPCR affinity was investigated for a variety of new phenethylamine-based primary amines and pyrrolidines. Several compounds demonstrated significant and selective binding against the 5-HT2B, 5-HT7, or σ1 receptor. Compound 4j protected against oxidative stress when assayed in 661W cells. The potency of compound 4j was comparable to PTZ in these assays.
Acknowledgments
Thanks is given to the National Institute of Mental Health’s Psychoactive Drug Screening Program (NIMH-PDSP) for the determination of the Ki values used in this paper. The PDSP is directed by Bryan L. Roth, MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda, MD. For more details on the PDSP, as well as experimental details on binding assays, see the PDSP website at https://pdspdb.unc.edu/pdspWeb/.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00225.
Experimental procedures, characterization data, and additional information on assays (PDF)
Author Contributions
MP performed the synthesis of new compounds. HX and JW performed assays with 661W cells. The project was directed by JT and SS. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM124718. This work was supported by the National Institutes of Health under award number R01EY028103 and the Foundation Fighting Blindness award number TA-NMT-0617–021-AUG.
The authors declare the following competing financial interest(s): A provisional patent has been filed by the University of Minnesota on the substructure disclosed in this report; M.R.P. and J.J.T. filed US patent No. 16/428,343, submitted 5/31/2019, on the compounds in this article.
Supplementary Material
References
- Santos R.; Ursu O.; Gaulton A.; Bento A. P.; Donadi R. S.; Bologa C. G.; Karlsson A.; Al-Lazikani B.; Hersey A.; Oprea T. I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discovery 2017, 16, 19–34. 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnadóttir T. K.; Gloriam D. E.; Hellstrand S. H.; Kristiansson H.; Fredriksson R.; Schiöth H. B. Comprehensive Repertoire and Phylogenetic Analysis of the G Protein-Coupled Receptors in Human and Mouse. Genomics 2006, 88, 263–273. 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Hauser A. S.; Chavali S.; Masuho I.; Jahn L. J.; Martemyanov K. A.; Gloriam D. E.; Babu M. M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. 10.1016/j.cell.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons F. E. R.; Simons K. J. Histamine and H1-Antihistamines: Celebrating a Century of Progress. J. Allergy Clin. Immunol. 2011, 128, 1139–1150. 10.1016/j.jaci.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Miyamoto S.; Duncan G. E.; Marx C. E.; Lieberman J. A. Treatments for Schizophrenia: A Critical Review of Pharmacology and Mechanisms of Action of Antipsychotic Drugs. Mol. Psychiatry 2005, 10, 79–104. 10.1038/sj.mp.4001556. [DOI] [PubMed] [Google Scholar]
- Turner E. H.; Matthews A. M.; Linardatos E.; Tell R. A.; Rosenthal R. Selective Publication of Antidepressant Trials and Its Influence on Apparent Efficacy. N. Engl. J. Med. 2008, 358, 252–260. 10.1056/NEJMsa065779. [DOI] [PubMed] [Google Scholar]
- Stein C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. 10.1146/annurev-med-062613-093100. [DOI] [PubMed] [Google Scholar]
- McCracken J. L.; Veeranki S. P.; Ameredes B. T.; Calhoun W. J. Diagnosis and Management of Asthma in Adults. J. Am. Med. Assoc. 2017, 318, 279–290. 10.1001/jama.2017.8372. [DOI] [PubMed] [Google Scholar]
- Fuentes A.; Pineda M.; Venkata K. Comprehension of Top 200 Prescribed Drugs in the US as a Resource for Pharmacy Teaching, Training and Practice. Pharmacy 2018, 6, 43. 10.3390/pharmacy6020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P. J. Drugs for Asthma. Br. J. Pharmacol. 2006, 147, S297–S303. 10.1038/sj.bjp.0706437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu J.-M.; Gainetdinov R. R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Small K. M.; McGraw D. W.; Liggett S. B. Pharmacology And Physiology of Human Adrenergic Receptor Polymorphisms. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 381–411. 10.1146/annurev.pharmtox.43.100901.135823. [DOI] [PubMed] [Google Scholar]
- Porter M. R.; Shaker R. M.; Calcanas C.; Topczewski J. J. Stereoselective Dynamic Cyclization of Allylic Azides: Synthesis of Tetralins, Chromanes, and Tetrahydroquinolines. J. Am. Chem. Soc. 2018, 140, 1211–1214. 10.1021/jacs.7b11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; et al. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492, 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak G. W.; Pan Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols D. E.; Nichols C. D. Serotonin Receptors. Chem. Rev. 2008, 108, 1614–1641. 10.1021/cr078224o. [DOI] [PubMed] [Google Scholar]
- Connolly H. M.; Crary J. L.; McGoon M. D.; Hensrud D. D.; Edwards B. S.; Edwards W. D.; Schaff H. V. Valvular Heart Disease Associated with Fenfluramine and Phentermine. N. Engl. J. Med. 1997, 337, 581–588. 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- Roth B. L. Drugs and Valvular Heart Disease. N. Engl. J. Med. 2007, 356, 6–9. 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- Gilbert P. E.; Martin W. R. The Effects of Morphine and Nalorphine-like Drugs in the Nondependent, Morphine-Dependent and Cyclazocine-Dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 1976, 198, 66–82. [PubMed] [Google Scholar]
- Kim F. J.; Kovalyshyn I.; Burgman M.; Neilan C.; Chien C.-C.; Pasternak G. W. 1 Receptor Modulation of G-Protein-Coupled Receptor Signaling: Potentiation of Opioid Transduction Independent from Receptor Binding. Mol. Pharmacol. 2010, 77, 695–703. 10.1124/mol.109.057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. W.; Kwon Y. B.; Roh D. H.; Yoon S. Y.; Han H. J.; Kim K. W.; Beitz A. J.; Lee J. H. Intrathecal Treatment with Σ1 Receptor Antagonists Reduces Formalin-Induced Phosphorylation of NMDA Receptor Subunit 1 and the Second Phase of Formalin Test in Mice. Br. J. Pharmacol. 2006, 148, 490–498. 10.1038/sj.bjp.0706764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortíz-Rentería M.; Juárez-Contreras R.; González-Ramírez R.; Islas L. D.; Sierra-Ramírez F.; Llorente I.; Simon S. A.; Hiriart M.; Rosenbaum T.; Morales-Lázaro S. L. TRPV1 Channels and the Progesterone Receptor Sig-1R Interact to Regulate Pain. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 1657–1666. 10.1073/pnas.1715972115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G.; Moreno E.; Aymerich M.; Marcellino D.; McCormick P. J.; Mallol J.; Cortes A.; Casado V.; Canela E. I.; Ortiz J.; et al. Direct Involvement of Σ1 Receptors in the Dopamine D1 Receptor-Mediated Effects of Cocaine. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18676–18681. 10.1073/pnas.1008911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher C. M.; Thomas J. D.; Haas D. A.; Longen C. G.; Oyer H. M.; Tong J. Y.; Kim F. J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255. 10.1158/1541-7786.MCR-17-0166. [DOI] [PubMed] [Google Scholar]
- Maurice T.; Su T. P. The Pharmacology of Sigma-1 Receptors. Pharmacol. Ther. 2009, 124, 195–206. 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock J. M.; Spino C. M.; Longen C. G.; Stabler S. M.; Marino J. C.; Pasternak G. W.; Kim F. J. Sequential Cytoprotective Responses to Sigma1 Ligand-Induced Endoplasmic Reticulum Stress. Mol. Pharmacol. 2013, 84, 751–762. 10.1124/mol.113.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguinou M.; Crottès D.; Chantôme A.; Rapetti-Mauss R.; Potier-Cartereau M.; Clarysse L.; Girault A.; Fourbon Y.; Jézéquel P.; Guérin-Charbonnel C.; et al. The SigmaR1 Chaperone Drives Breast and Colorectal Cancer Cell Migration by Tuning SK3-Dependent Ca2+ Homeostasis. Oncogene 2017, 36, 3640–3647. 10.1038/onc.2016.501. [DOI] [PubMed] [Google Scholar]
- Scott L. L.; Sahn J. J.; Ferragud A.; Yen R. C.; Satarasinghe P. N.; Wood M. D.; Hodges T. R.; Shi T.; Prakash B. A.; Friese K. M.; et al. Small Molecule Modulators of Σ2R/Tmem97 Reduce Alcohol Withdrawal-Induced Behaviors. Neuropsychopharmacology 2018, 43, 1867–1875. 10.1038/s41386-018-0067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahn J. J.; Mejia G. L.; Ray P. R.; Martin S. F.; Price T. J. Sigma 2 Receptor/Tmem97 Agonists Produce Long Lasting Antineuropathic Pain Effects in Mice. ACS Chem. Neurosci. 2017, 8, 1801–1811. 10.1021/acschemneuro.7b00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei J. Σ1 Receptor Modulation of Opioid Analgesia in the Mouse. J. Pharmacol. Exp. Ther. 2002, 300, 1070–1074. 10.1124/jpet.300.3.1070. [DOI] [PubMed] [Google Scholar]
- Chien C. C.; Pasternak G. W. Selective Antagonism of Opioid Analgesia by a Sigma System. J. Pharmacol Exp Ther 1994, 271, 1583–1590. [PubMed] [Google Scholar]
- Smith S. B.; Duplantier J.; Dun Y.; Mysona B.; Roon P.; Martin P. M.; Ganapathy V. Vivo Protection against Retinal Neurodegeneration by Sigma Receptor 1 Ligand (+)-Pentazocine. Invest. Ophthalmol. Visual Sci. 2008, 49, 4154–4161. 10.1167/iovs.08-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. B.; Wang J.; Cui X.; Mysona B. A.; Zhao J.; Bollinger K. E. Sigma 1 Receptor: A Novel Therapeutic Target in Retinal Disease. Prog. Retinal Eye Res. 2018, 67, 130–149. 10.1016/j.preteyeres.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modica M. N.; Lacivita E.; Intagliata S.; Salerno L.; Romeo G.; Pittalà V.; Leopoldo M. Structure-Activity Relationships and Therapeutic Potentials of 5-HT7 Receptor Ligands: An Update. J. Med. Chem. 2018, 61, 8475–8503. 10.1021/acs.jmedchem.7b01898. [DOI] [PubMed] [Google Scholar]
- Maurice T.; Su T.-P. The Pharmacology of Sigma-1 Receptors. Pharmacol. Ther. 2009, 124 (2), 195–206. 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Saul A.; Roon P.; Smith S. B. Activation of the Molecular Chaperone, Sigma 1 Receptor, Preserves Cone Function in a Murine Model of Inherited Retinal Degeneration. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (26), E3764–E3772. 10.1073/pnas.1521749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Zhao J.; Cui X.; Mysona B. A.; Navneet S.; Saul A.; Ahuja M.; Lambert N.; Gazaryan I. G.; Thomas B.; et al. The Molecular Chaperone Sigma 1 Receptor Mediates Rescue of Retinal Cone Photoreceptor Cells via Modulation of NRF2. Free Radical Biol. Med. 2019, 134, 604–616. 10.1016/j.freeradbiomed.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E.; Ding X.-Q.; Saadi A.; Agarwal N.; Naash M. I.; Al-Ubaidi M. R. Expression of Cone-Photoreceptor–Specific Antigens in a Cell Line Derived from Retinal Tumors in Transgenic Mice. Invest. Ophthalmol. Visual Sci. 2004, 45 (3), 764. 10.1167/iovs.03-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Ye X.; Liu R.; Chen H.-L.; Bai H.; Liang X.; Zhang X.-D.; Wang Z.; Li W.; Hai C.-X. Antioxidant Activities of Oleanolic Acid in Vitro: Possible Role of Nrf2 and MAP Kinases. Chem.-Biol. Interact. 2010, 184 (3), 328–337. 10.1016/j.cbi.2010.01.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.