

Abstract
In this study, a series of 49 five-membered heterocyclic compounds containing either a pyridine- or a pyrrole-type nitrogen were synthesized and tested against Mycobacterium tuberculosis. Among them, only the 1,3,5-trisubstituted pyrazoles 5–49 exhibited minimum inhibitory concentration values in the low micromolar range, and some also exhibited an improved physicochemical profile without cytotoxic effects. Three pyrazoles were subjected to an animal tuberculosis efficacy model, and compound 6 induced a statistically significant difference in lung bacterial counts compared with untreated mice. Moreover, to determine the target of this series, resistors were generated, and whole genome sequencing revealed mutations in the mmpL3 gene.
Keywords: Tuberculosis, pyrazoles, MmpL3, drug discovery, antimycobacterials
Tuberculosis (TB) is the leading cause of death worldwide from a single infectious agent and ranks above HIV/AIDS. An estimated 1.3 million people died of TB and ∼10 million fell ill with TB in 2017. Luckily, TB mortality has fallen 47% since 1990, with an estimated 53 million saved lives between 2000 and 2017.1 Despite that, the emergence and spread of multi- and extensively drug-resistant (MDR and XDR) strains have made the control of the disease more difficult. Therefore, there is a high priority on the development of new anti-TB drugs with bactericidal mechanisms different from those of the presently available agents.2
Our interest in developing new antitubercular agents reflects our work on developing a class of 1,5-diphenyl pyrroles. An initial phenotypic screening of an in-house chemical library identified BM212 (Figure 1) as a “soft hit”,3 and further medicinal chemistry efforts provided an improved hit (BM635, Chart 1) active against both replicating and nonreplicating bacilli and efficacious in a murine model of TB infection.4,5 These pyrroles act by inhibiting the Mycobacterium membrane protein Large 3 (MmpL3).6,7 MmpL3 belongs to the resistance, nodulation, and cell division protein superfamily and is likely involved in trehalose monomycolate flipping across the inner membrane of M. tuberculosis (Mtb).8 We have previously demonstrated that BM212 binds MmpL3 directly and inhibits its activity.8
Figure 1.
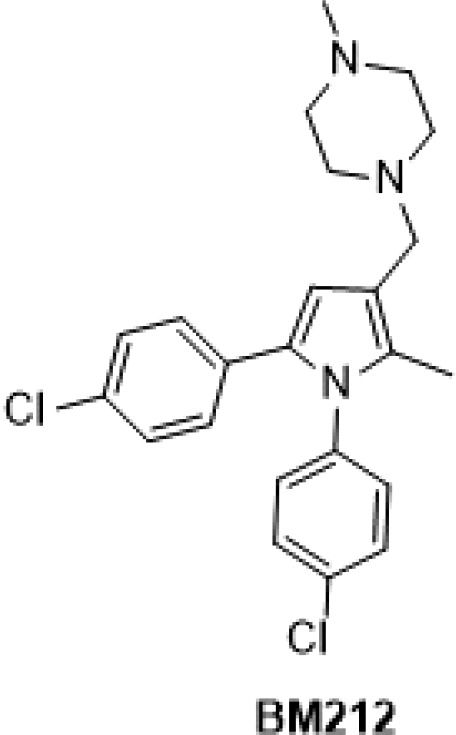
Chemical structure of BM212.
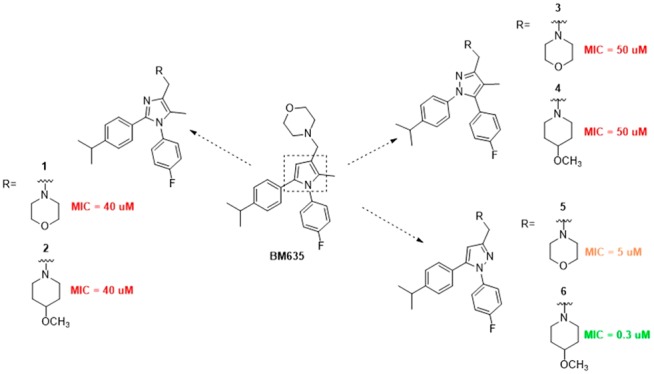
Chart 1. Chemical Structures of BM635 and Compounds 1–6.

After the discovery of BM635, we followed diverse strategies for improving BM635’s drug-like profile (low water solubility and high lipophilicity), such as preparing different pharmaceutical salts9 and making modifications around the central pyrrole core.10
To further develop this class of compounds, and with the aim of improving both the solubility and the polarity of the BM series, we have designed new compounds containing a second nitrogen atom on the heterocyclic moiety that could easily lead to the formation of hydrogen bonds. Therefore, the central core has been replaced with two five-membered heterocycles containing either a pyridine- or a pyrrole-type nitrogen:11−15 imidazole and pyrazole. Herein we report the synthesis and the full biological and pharmacological profiling of such novel five-membered heterocyclic MmpL3 inhibitors.
We first investigated which five-membered ring could replace the central pyrrole core of BM635 by preparing compounds 1–6 (Chart 1), but only the 1,3,5-trisubstituted pyrazoles 5 and 6 showed good activity against Mtb, with 6 providing a minimum inhibitory concentration (MIC) of 0.3 μM (Table 1).
Table 1. Activity against Mtb H37RV (MIC) and Cytotoxicity on HepG2 Cells (CC50) of Compounds 1–6, BM635, and Isoniazid.
| 1 | 2 | 3 | 4 | 5 | 6 | BM635 | isoniazid | |
|---|---|---|---|---|---|---|---|---|
| MIC (μM)a | 40 | 40 | 50 | 50 | 5 | 0.3 | 0.12 | 1.8 |
| CC50 (μM)b | 50.1 | ndc | >100 | ndc | 50.1 | 31.6 | 40 | |
| PI (CC50/MIC)d | 1.25 | >2 | 10 | 105 | 333 |
Minimum inhibitory concentration required to inhibit Mtb growth.
Concentration of compound resulting in 50% inhibition of HepG2 cell line.
Not determined.
Protection Index (CC50/MIC).
Thus we selected the 1,3,5-trisubstituted pyrazole as a suitable central core and prepared analogues 7–49 (Tables 2 and 3) decorated with the best substituents resulting from the previous works. We explored which amine could replace the morpholine by introducing different six-membered cyclic amines at position 3. To date, compounds 13, 23, 40, and 47 incorporate a silicon amine, which was shown, in a previous work by Ramesh and coworkers, to be responsible for a remarkable increase in activity.16 Then, we examined positions 1 and 5 by introducing substituents that in the pyrrole series produced the best activities (compounds 14–49, Tables 2 and 3).4,10,17
Table 2. Activity against Mtb H37RV (MIC) and Cytotoxicity on HepG2 Cells (CC50) of Compounds 7–29.

Minimum inhibitory concentration required to inhibit Mtb growth.
Concentration of compound resulting in 50% inhibition of HepG2 cells.
Not determined.
Protection Index (CC50/MIC).
Table 3. Activity against Mtb H37RV (MIC) and Cytotoxicity on HepG2 Cells (CC50) of Compounds 30–49.

Minimum inhibitory concentration required to inhibit Mtb growth.
Concentration of compound resulting in 50% inhibition of HepG2 cells.
Protection Index (CC50/MIC).
Compounds were evaluated in vitro to determine the minimum concentration required to inhibit Mtb growth in the culture as well as the compound cytotoxicity by measuring the concentration of the compound resulting in 50% inhibition (CC50) of HepG2 cells. Moreover, compounds with MICs < 1 μM were evaluated to determine the drug metabolism and pharmacokinetics (DMPK) profile (Chrom log DpH7.4 values, artificial membrane permeability, percentage of binding to human serum albumin (HSA), kinetic solubility, solubility in the biorelevant medium to simulate fasted states in vivo (FaSSIF), and human ether-a-go-go-related gene (hERG)) binding, and those providing the best profile were selected to determine their efficacy in vivo. Finally, having modified the main chemical motif of the parent compounds, we verified if the new pyrazoles act by inhibiting MmpL3 by generating spontaneous resistant mutants and whole genome sequencing.
Structure–activity relationship (SAR) studies were performed on a series of 1,3,5-trisubstituted pyrazoles. Compounds 1–49 were prepared as reported in the Supporting Information. Antituberculosis assays revealed that within the different substitutions at position five, only the 4-isopropylphenyl (compounds 6, 7, 11–17, 20, 22–24, and 26–29) and the trifluoromethylphenyl (compounds 36, 37, 39–47, and 49) ones were well tolerated, confirming a trend already seen for the pyrrole series (Tables 2 and 3).10 Compounds 6, 12–14, 16, 22, 23, 27, 36, 40, and 47 showed remarkable activities against Mtb (0.00925 to 0.6 μM, Tables 1–3) with 6, 12, 14, 22, 27, and 36 providing low cytotoxicities (3.6–64 μM, Tables 1–3), whereas, a different trend was observed for the substituent at position one (compounds 24–26, 48, and 49): The isopropyl chain, which provided one of the best pyrrole compounds, did not work well for the pyrazoles (MIC = 1.75, 50, 1, 23, and 2.5 μM, respectively). In general, the activity of this series of pyrazoles seemed to be more influenced by the cyclic amine at position three. All of the compounds incorporating a silicon amine (13, 23, 40, and 47) showed a remarkable activity increase (0.00925, 0.04, 0.02, and 0.07 μM, respectively), but unfortunately they exhibited high cytotoxicity together with high levels of hERG activity, low water solubility, as well as low membrane permeability (Table 4). Both the high toxicity and the DMPK may be explained by the high lipophilicity displayed by silicon compounds. In view of the limited safety and poor DMPK, silicon-incorporating compounds were not further progressed. Instead, compounds bearing either the 4-methoxypiperidine or the pyridinepiperazine at position three (6, 12, 16, 22, and 36) showed the best results. In general, these compounds showed good activities and low cytotoxicities combined with good DMPKs and low hERG activities (Table 4). Surprisingly, when moved to the pyrazoles, the morpholine ring that did work well for the previous pyrrole compounds4,10 did provide only two active compounds: 14 and 27.
Table 4. DMPK and Safety Profile of Compounds 6, 12, 13, 14, 22, 23, 27, 36, 40, 47 and BM635.
| comp | Chrom log DpH7.4a | membrane permeability cm/sb | %HSAc | KS (μM)d | FaSSIF (μg/mL)e | hERG IC50 (μM)f |
|---|---|---|---|---|---|---|
| 6 | 7.31 | 4.4 × 10–5 | 96.42 | 152 | ndg | 6.31 |
| 12 | 8.76 | <0.3 × 10–5 | 98.45 | 6 | ndg | 25.1 |
| 13 | 8.82 | 4-Far | 97.49 | 41 | ndg | 2 |
| 14 | 8.29 | 3.5 × 10–5 | 97.11 | 78.5 | 100 | >50 |
| 22 | 9.83 | <0.03 × 10–5 | 98.41 | 15 | ndg | >50 |
| 23 | ndg | 4-Far | 97.35 | 46 | ndg | 8 |
| 27 | 7.37 | 6.1 × 10–5 | 96.72 | 305 | 139 | 32 |
| 36 | 6.67 | 1.7 × 10–5 | 96.47 | 258 | ndg | 3.2 |
| 40 | 8.13 | 4-Far | 96.65 | 53 | ndg | 1.26 |
| 47 | 8.49 | 4-Far | 96.32 | 69 | ndg | 2.5 |
| BM635 | 8.1 | 2.4 × 10–5 | 98.37 | <1 | 5 | 10 |
Lipophilicity expressed as Chrom log DpH7.4.
Artificial membrane permeability.
Percentage of binding to human serum albumin (HSA).
Kinetic solubility.
Solubility in the biorelevant medium to simulate the fasted states in vivo (FaSSIF).
Human ether-a-go-go-related gene (hERG) binding.
Not determined.
We sought to establish physicochemical, DMPK, and hERG profiles for selected compounds. Compounds showing MICs <1 μM were profiled for physicochemical properties, in vitro DMPK properties, and hERG activities, as shown in Table 4. Overall, the introduction of a free pyrrole-type nitrogen led to both increased water solubility and reduced binding to HSA, as for compounds 6, 14, 27, and 36 (Table 4), compared with the previous pyrroles.10 The ability of compounds to cross biological membranes was assessed by an artificial membrane permeability assay, which is based on permeation through planar polycarbonate-filter-supported lipid bilayers. Most of the tested compounds proved to be highly permeable, with values in the 10–5 range, except for silicon-containing compounds 13, 23, 40, and 47 (Table 4). Finally, whereas compounds 6, 13, 23, 36, 40, and 47 were found to be low micromolar inhibitors of the potassium channel, 12, 14, 22, and 27 had a clean profile in the hERG activity (Table 4).
Because the pyrazoles and pyrroles have different SARs, they could possibly have different targets. To determine this, we isolated resistant mutants to two of the compounds, 6 (at 3× MIC) and 22 (at 3× and 5× MIC), with a resistance frequency of 10–7 and performed whole genome sequencing on DNA from isolated colonies (Table 5). We found no mutations in a colony grown on the higher concentration of 22. At the lower compound concentrations, we found multiple mutations; however, both had mutations that altered the coding sequence of MmpL3. For 6, we found a substitution of alanine for valine at position 240 (along with a missense mutation in ppsB). For 22, we found a substitution at the adjacent position, 239, of histidine for proline (along with a missense mutation in lprM and an insertion mutation in fadD26). These mutations both lie in the fourth transmembrane helix, where other resistance mutations to other MmpL3-targeting compounds are known to exist.19
Table 5. MICs and Amino Acid Substitutions Found in Mtb Mutants Isolated as Resistant to Compounds 6 and 22.
| comp | MIC (μM) | gene | mutation |
|---|---|---|---|
| 6 | 12.5 | mmpL3 | V240A |
| 6 | 12.5 | ppsB | L1476S |
| 22 | 20 | mmpL3 | P239H |
| 22 | 25 | lprM | V86V |
| 22 | 20 | fadD26 | +g in aa 336 |
Finally, to evaluate the potential of these compounds, we selected 6, 14, and 27 to be tested in a fast standardized 9 day acute assay intended to measure the efficacy of drugs against M. tuberculosis growing in the lungs of immunocompetent mice.18 Compounds 6, 14, and 27 where chosen because of their low cytotoxicities (31.60, 63.10, and 40 μM) but especially because of the high water solubility (152, 78.50, and 305.50 μM), which is expected to play a crucial role in the in vivo efficacy. Compounds were administered at a dose of 200 mg/kg per day for 4 days starting on day 5 after infection. Surprisingly, only 6 induced a statistically significant difference in lung bacterial counts compared with untreated mice (Table 6).
Table 6. Therapeutic Efficacy of 6, 14, and 27 and Moxifloxacina.
| comp | dose (mg/kg) | administration | difference in log CFU/lungs | P2 |
|---|---|---|---|---|
| 6 | 200 | once per day | 1.5 | p < 0.05 |
| 14 | 200 | once per day | 0.4 | p < 0.05 |
| 27 | 200 | once per day | 0.3 | p < 0.05 |
| moxifloxacin | 30 | once per day | 3.5 | p < 0.05 |
Differences in the lung microorganism burden (log 10 CFUs/lungs) with respect to untreated controls (day 9 after infection).
Despite the good permeability and good water solubility, 14 and 27 proved to be inactive. Because poor metabolic stability may account for the lack of in vivo efficacy, we tested 14 and 27 on mouse and human liver microsomes to determine the intrinsic clearance (Clint) and the metabolic stability (%LBF). On the basis of clearance in mouse microsomes (7.98 and 7.04 mL/min/g) and applying a well-stirred model, an in vivo blood clearance of ∼75% for both compounds is predicted (Table 7). This would explain the lack of efficacy of those compounds related to an expected high first-pass liver effect and a quick elimination process.
Table 7. Intrinsic Clearance (Clint) in Mouse and Human Microsomes of Compounds 14 and 27.
| Clint (mL/min/g) |
%LBFa |
|||
|---|---|---|---|---|
| comp | mouse | human | mouse | human |
| 14 | 7.98 | 3.4 | 76.3 | 82.2 |
| 27 | 7.04 | 2.8 | 74 | 79.2 |
Liver blood flow, predicted in vivo blood clearance.
In summary, following up on our medicinal chemistry campaign based on the implementation of a class of potent MmpL3 inhibitors, we sought to replace the central pyrrole core and prepared 49 five-membered heterocyclic compounds. The introduction of a free pyrrole-type nitrogen gave several compounds that proved to be active against Mtb with a low level of cytotoxicity but, above all, with improved water solubility and reduced hERG activity—two of the major concerns in TB drug development. Moreover, one of the tested pyrazoles showed efficacy in a murine model of acute TB infection. This, combined with the results of whole genome sequencing highlighting MmpL3 as the possible target, confirms the high potential of the MmpL3 inhibitor class for development in TB drug discovery.
Acknowledgments
This work was supported by the Tres Cantos Open Lab Foundation (grant number TC028). This work has been supported by the Italian Ministry of Education, Universities and Research - Dipartimenti di Eccellenza - L. 232/2016
Glossary
Abbreviations
- CFUs
colony forming units
- DMPK
drug metabolism and pharmacokinetics
- FaSSIF
fasted simulated fluid
- hERG
human ether-a-go-go-related gene
- HSA
human serum albumin
- LBF
liver blood flow
- MDR
multi-drug-resistant
- MIC
minimum inhibitory concentration
- MmpL3
Mycobacterium membrane protein large 3
- Mtb
M. tuberculosis
- SAR
structure–activity relationship
- TB
tuberculosis
- WHO
World Health Organization
- XDR
extensively drug resistant.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00204.
Detailed synthetic procedure, spectroscopic data, and full biological characterizations of all new compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland; 2018.
- Koch A.; Cox H.; Mizrahi V. Drug-Resistant Tuberculosis: Challenges and Opportunities for Diagnosis and Treatment. Curr. Opin. Pharmacol. 2018, 42, 7–15. 10.1016/j.coph.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda D.; Lampis G.; Fioravanti R.; Biava M.; Porretta G. C.; Zanetti S.; Pompei R. Bactericidal Activities of the Pyrrole Derivative BM212 against Multidrug-Resistant and Intramacrophagic Mycobacterium tuberculosis Strains. Antimicrob. Agents Chemother. 1998, 42, 3035–3037. 10.1128/AAC.42.11.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G.; Bates R. H.; Alfonso S.; Cocozza M.; Porretta G. C.; Ballell L.; Rullas J.; Ortega F.; De Logu A.; Agus E.; La Rosa V.; Pasca M. R.; De Rossi E.; Wae B.; Franzblau S. G.; Manetti F.; Botta M.; Biava M. Improved BM212 MmpL3 Inhibitor Analogue Shows Efficacy in Acute Murine Model of Tuberculosis Infection. PLoS One 2013, 8, e56980. 10.1371/journal.pone.0056980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaro G.; Poce G.; Biava M.; Giannoni F.; Fattorini L. Activity of Lipophilic and Hydrophilic Drugs against Dormant and Replicating Mycobacterium tuberculosis. J. Antibiot. 2015, 68, 711–714. 10.1038/ja.2015.52. [DOI] [PubMed] [Google Scholar]
- La Rosa V.; Poce G.; Canseco J. O.; Buroni S.; Pasca M. R.; Biava M.; Raju R. M.; Porretta G. C.; Alfonso S.; Battilocchio C.; Javid B.; Sorrentino F.; Ioerger T. R.; Sacchettini J. C.; Manetti F.; Botta M.; De Logu A.; Rubin E. J.; De Rossi E. MmpL3 Is the Cellular Target of the Antitubercular Pyrrole Derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G.; Consalvi S.; Biava M. MmpL3 Inhibitors: Diverse Chemical Scaffolds Inhibit the Same Target. Mini-Rev. Med. Chem. 2016, 16, 1274–1283. 10.2174/1389557516666160118105319. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Meshcheryakov V. A.; Poce G.; Chng S.-S. MmpL3 Is the Flippase for Mycolic Acids in Mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 7993–7998. 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G.; Consalvi S.; Cocozza M.; Fernandez-Menendez R.; Bates R. H.; Ortega Muro F.; Barros Aguirre D.; Ballell L.; Biava M. Pharmaceutical Salt of BM635 with Improved Bioavailability. Eur. J. Pharm. Sci. 2017, 99, 17–23. 10.1016/j.ejps.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Poce G.; Cocozza M.; Alfonso S.; Consalvi S.; Venditti G.; Fernandez-Menendez R.; Bates R. H.; Barros Aguirre D.; Ballell L.; De Logu A.; Vistoli G.; Biava M. In Vivo Potent BM635 Analogue with Improved Drug-like Properties. Eur. J. Med. Chem. 2018, 145, 539–550. 10.1016/j.ejmech.2017.12.075. [DOI] [PubMed] [Google Scholar]
- Castagnolo D.; De Logu A.; Radi M.; Bechi B.; Manetti F.; Magnani M.; Supino S.; Meleddu R.; Chisu L.; Botta M. Synthesis, Biological Evaluation and SAR Study of Novel Pyrazole Analogues as Inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2008, 16, 8587–8591. 10.1016/j.bmc.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Castagnolo D.; Manetti F.; Radi M.; Bechi B.; Pagano M.; De Logu A.; Meleddu R.; Saddi M.; Botta M. Synthesis, Biological Evaluation, and SAR Study of Novel Pyrazole Analogues as Inhibitors of Mycobacterium tuberculosis: Part 2. Synthesis of Rigid Pyrazolones. Bioorg. Med. Chem. 2009, 17, 5716–5721. 10.1016/j.bmc.2009.05.058. [DOI] [PubMed] [Google Scholar]
- Castagnolo D.; Radi M.; Dessì F.; Manetti F.; Saddi M.; Meleddu R.; De Logu A.; Botta M. Synthesis and Biological Evaluation of New Enantiomerically Pure Azole Derivatives as Inhibitors of Mycobacterium Tuberculosis. Bioorg. Med. Chem. Lett. 2009, 19, 2203–2205. 10.1016/j.bmcl.2009.02.101. [DOI] [PubMed] [Google Scholar]
- De Vita D.; Pandolfi F.; Cirilli R.; Scipione L.; Di Santo R.; Friggeri L.; Mori M.; Fiorucci D.; Maccari G.; Arul Christopher R. S.; Zamperini C.; Pau V.; De Logu A.; Tortorella S.; Botta M. Discovery of in Vitro Antitubercular Agents through in Silico Ligand-Based Approaches. Eur. J. Med. Chem. 2016, 121, 169–180. 10.1016/j.ejmech.2016.05.032. [DOI] [PubMed] [Google Scholar]
- Manetti F.; Magnani M.; Castagnolo D.; Passalacqua L.; Botta M.; Corelli F.; Saddi M.; Deidda D.; De Logu A. Ligand-Based Virtual Screening, Parallel Solution-Phase and Microwave-Assisted Synthesis as Tools to Identify and Synthesize New Inhibitors of Mycobacterium tuberculosis. ChemMedChem 2006, 1, 973–989. 10.1002/cmdc.200600026. [DOI] [PubMed] [Google Scholar]
- Ramesh R.; Shingare R. D.; Kumar V.; Anand A.; B S.; Veeraraghavan S.; Viswanadha S.; Ummanni R.; Gokhale R.; Srinivasa Reddy D. Repurposing of a Drug Scaffold: Identification of Novel Sila Analogues of Rimonabant as Potent Antitubercular Agents. Eur. J. Med. Chem. 2016, 122, 723–730. 10.1016/j.ejmech.2016.07.009. [DOI] [PubMed] [Google Scholar]
- Biava M.; Porretta G. C.; Poce G.; Battilocchio C.; Alfonso S.; de Logu A.; Manetti F.; Botta M. Developing Pyrrole-Derived Antimycobacterial Agents: A Rational Lead Optimization Approach. ChemMedChem 2011, 6, 593–599. 10.1002/cmdc.201000526. [DOI] [PubMed] [Google Scholar]
- Rullas J.; García J. I.; Beltrán M.; Cardona P.-J.; Cáceres N.; García-Bustos J. F.; Angulo-Barturen I. Fast Standardized Therapeutic-Efficacy Assay for Drug Discovery against Tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 2262–2264. 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Li J.; Yang X.; Wu L.; Zhang J.; Yang Y.; Zhao Y.; Zhang L.; Yang X.; Yang X.; Cheng X.; Liu Z.; Jiang B.; Jiang H.; Guddat L.; Yang H.; Rao Z. Crystal Structures of Membrane Transporter MmpL3, an Anti-TB Drug Target. Cell 2019, 176, 636–648.e13. 10.1016/j.cell.2019.01.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.