

Abstract

Recent reports have highlighted the dual bromodomains of TAF1 (TAF1(1,2)) as synergistic with BET inhibition in cellular cancer models, engendering interest in TAF/BET polypharmacology. Here, we examine structure activity relationships within the BI-2536 PLK1 kinase inhibitor scaffold, previously reported to bind BRD4. We examine binding by this ligand to TAF1(2) and apply structure guided design strategies to discriminate binding to both the PLK1 kinase and BRD4(1) bromodomain while retaining activity on TAF1(2). Through this effort we discover potent dual inhibitors of TAF1(2)/BRD4(1), as well as biased derivatives showing marked TAF1 selectivity. We resolve X-ray crystallographic data sets to examine the mechanisms of the observed TAF1 selectivity and to provide a resource for further development of this scaffold.
Keywords: Bromodomain, kinase inhibitor, epigenetics, BI-2536
Featured in 46 human proteins, the bromodomain is the largest class of binding module, or “reader domain”, for acetylated lysines (KAc), a pervasive modification in epigenetic control.1 The development of JQ1 and related probes for the bromodomain and extra terminal domain (BET) subfamily first demonstrated the bromodomain’s chemical tractability, competively occupying the hydrophobic KAc pocket, which is formed by a rim of loops between domain’s common four-α-helical bundle structure. These probes have provided widely used tools for the study of BET function, in particular BRD4’s role as a master transcriptional coactivator, as well as broad therapeutic investiations of BET inhibition in disease.2−4 Subsequent clinical translations are currently underway.5−7 Broadly ensuing efforts to extend bromodomain probe development have now succeeded in providing selective tools for nearly all existing subfamilies, enabling direct study of domain specific contributions to protein function.8
Several recent studies have reported small molecule ligands for the dual bromodomain containing TAF1.9−13 As the largest of approximately 13 Tata-binding protein (TBP) associated factors “TAFs” that assemble to comprise the general transcription factor IID (TFIID),14,15 TAF1 scaffolds the pioneer factor for general eukaryotic transcription.16−18 In-vitro transcription and temperature sensitive mutant experiments have implied that TAF1 may be dispensable for basal transcription19 but directly mediates transcription downstream of RB,20 B-Myb,21 and C-Jun22 and is also critical to transcription downstream of key cell cycle drivers.23,24 Despite these roles, no direct phenotypes of TAF1 bromodomain inhibition have been reported, suggesting that as recently observed for other non-BET bromodomain targets, existing probes may not dramatically impact protein function.25−27 However, recent studies have suggested that TAF1 may collaborate with BRD4 to control cancer cell proliferation in a bromodomain dependent manner, potentially through cooperating transicriptional effects.10 Indeed, two structurally dissimilar TAF1 inhibitors have now been independently demonstrated to potentiate the activity of BET inhibition in cultured cancer models.10,13 These findings have engendered interest in compounds that may offer dual BRD4/TAF1 inhibition. Toward this objective, we herein report structure activity relationships (SAR) toward dual TAF1/BRD4 inhibitors, as well as TAF1-biased derivatives, from a single kinase inhibitor derived scaffold.
In the course of our own studies on bromodomain ligand development, we reported the first small molecule ligand for TAF1, a midnanomolar binder accessed through biased multicomponent library synthesis.9 However, further exploration of the compound’s dimethylisoxazole warhead revealed a challenging BRD4 bias. Among other chemotypes previously described were several kinase inhibitor scaffolds,28,29 examples of which have now seen in-depth study and development as polypharmacological agents.30,31 In particular, previous work suggested that the PLK1 kinase inhibitor BI-2536 harbored activity on TAF1 through broad phage display (Ember, et al) and melt temperature (TM) shifts panels (Ciceri, et al) .28,29,32
To assess the ability of BI-2536 to bind TAF1(2), we first profiled the compound in dose via competitive phage display (DiscoverX), confirming activity on the second bromodomain of TAF1 (TAF1(2)) (170 nM) in addition to BRD4(1) (24 nM) (Table S1). To facilitate further exploration of SAR within TAF1(2), we implemented an AlphaScreen assay leveraging a biotin functionalized derivative of BI-2536 (SI, Synthetic Procedures A). Guided by previously published BRD4 cocrystal structures, we sought to determine the influence of substituents buried within the acetyl-lysine recognition pocket, as well as those positioned within the “ZA channel”, a known hotspot for bromodomain selectivity (Figure 1).1,8 Within both TAF1(2) and BRD4(1), the imbedded methyl amide acetyl lysine mimetic showed high sensitivity to modification (Table 1; 2,3). Modifications of the ZA alkoxy substituent (4–7) were better accommodated. Truncation of the buried asymmetric ethyl group to a methyl substituent (8,9) improved potency, while substitution of the solvent projected 4-amino-methylpipiridine solubilizing group was also well tolerated (Table S2).
Figure 1.
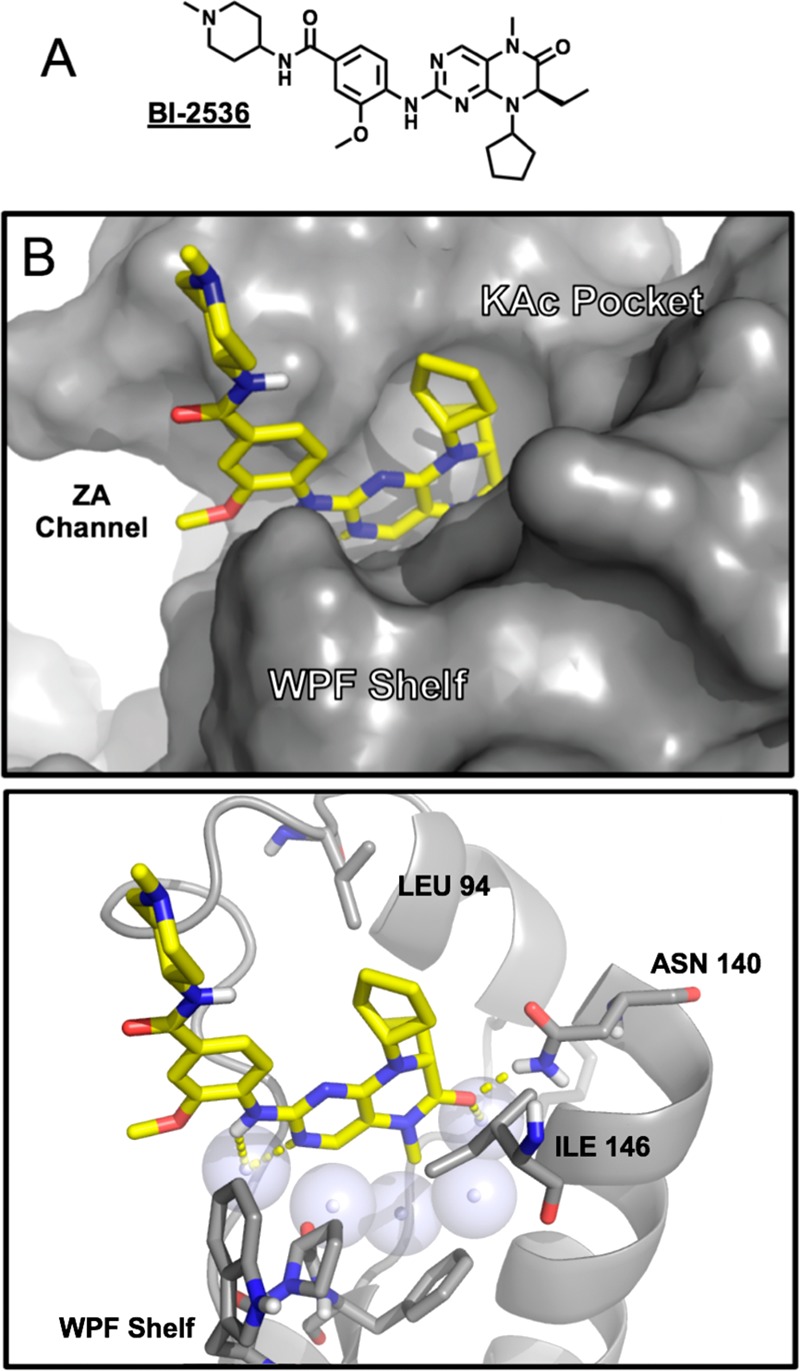
(A) Chemical structure of BI-2536. (B) Surface representation of BI-2536-BRD4(1) cocrystal structure (4O74).29 (C) Cartoon cutaway of view as in (B) showing conserved waters (spheres), key residues (sticks), and H-bonding (dashed).
Table 1. Substitutions to the BI-2536 KAc Mimetic (R1), ZA-Channel Projecting Alkoxy Substituent (R2), and Asymmetric Ethyl (R3).
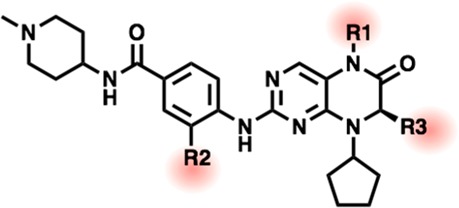
| Cmpd | R1 | R2 | R3 | TAF1(2)a (μM) | BRD4(1)a (μM) |
|---|---|---|---|---|---|
| 1 BI-2536 | Me | Ome | Et | 0.16 | 0.15 |
| 2 | Et | Ome | Et | 0.41 | 0.69 |
| 3 | H | Ome | Et | >10 | >10 |
| 4 | Me | H | Et | 0.15 | 0.28 |
| 5 | Me | O-iPr | Et | 0.21 | 0.063 |
| 6 | Me | O-cycloBu | Et | 0.11 | 0.062 |
| 7 | Me | O-cycloPent | Et | 0.10 | 0.033 |
| 8 | Me | H | Me | 0.087 | 0.068 |
| 9 | Me | O-iPr | Me | 0.32 | 0.22 |
AlphaScreen IC50s from quadruplicate measurement.
We next considered that the potent cytostatic activity arising from the PLK1 kinase activity of this scaffold could obscure biological studies of bromodomain inhibition. We therefore sought to alter the H-bonding capabilities of the imbedded aminopyrimidine hinge-binding motif, a well-known approach for disrupting kinase binding (Table 2).33
Table 2. Kinase Inactivation by Aminopyrimidine Hinge-Binding Motif Replacementa.
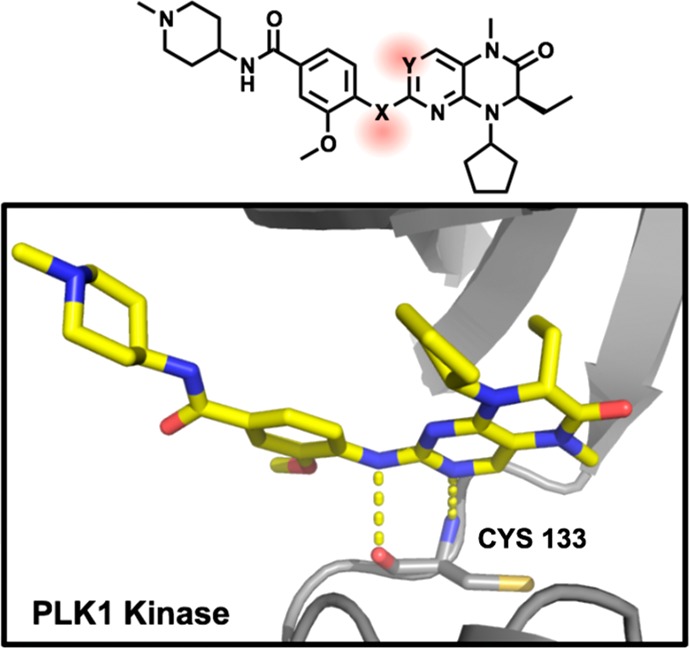
| Cmpd | X | Y | TAF1(2)b (μM) | BRD4(1)b (μM) | PLK1c (μM) |
|---|---|---|---|---|---|
| BI2536 | NH | N | 0.16 | 0.15 | 0.005 |
| 10 | NMe | N | >10 uM | 0.10 | 0.73 |
| 11 | O | N | >10 uM | 0.62 | >10 uM |
| 12 | NH | CH | 0.45 | 0.15 | >10 uM |
Inset image: cartoon cutaway of BI-2536 bound to PLK1 (2RKU) showing hinge binding H-bonding (dashed) to key Cys residue backbone (sticks).
AlphaScreen IC50s from quadruplicate measurement.
Z-Lyte assays (Invitrogen) with [ATP] of KM for each kinase, duplicate measurement.
Replacement of the key hydrogen bond donating NH with NMe as we previously reported,34 or with O,35 largely abolished kinase activity but resulted in both cases in dramatic and preferential loss of TAF1 bromodomain activity (versus BRD4) (10,11). As this NH engages in water-mediated H-bonding within BRD4, we considered that a similar interaction may underlie an imporant contribution to TAF1 affinity (Table 2, Figure 1C). We therefore applied an alternative strategy of removing hinge H-bond accepting to the pyrimidine core through N to CH substitution36 and were pleased to find that this approach successfully diminished activity against the PLK1 kinase while better retaining TAF1 affinity (Table 2; 12).
Advancing this bromodomain selective scaffold, we sought to assess further avenues toward potency on TAF1 as well as to identify strategies to bias for TAF1 activity relative to BRD4. Introducing the favored asymmetric ethyl to methyl substitution into the kinase-selective pyridopiperazone scaffold resulted in a potent dual inhibitor of TAF1(2) (16 nM IC50) and BRD4(1) (37 nM IC50) (Table 3; 13). Unexpectedly however, previously examined substitutions of the methoxy ZA substituent, particularly H replacement, were no longer favored, potentially reflecting rearrangement of the water mediated interaction network involving this substituent (14).29 To better understand the binding mode of this series, we obtained an X-ray cocrystal structure of 15 bound to TAF1(2). Interestingly, the resolved structure contains two protein molecules per unit cell, with 15 showing a conserved core binding mode but exhibiting two dissimilar poses of the substituted 4-aminobenzamide tail (Figure 2, 6P38).
Table 3. Alkoxy Substituent SAR for the Kinase Selective Dihydropyridopyrazinone Scaffold.
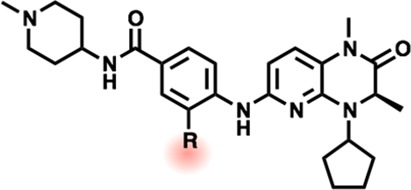
| Cmpd | R | TAF1(2)a (μM) | BRD4(1)a (μM) |
|---|---|---|---|
| 13 | OMe | 0.016 | 0.037 |
| 14 | H | 0.34 | 0.19 |
| 15 | O-iPr | 0.089 | 0.13 |
| 16 | O-cycloPent | 0.031 | 0.048 |
AlphaScreen IC50s from quadruplicate measurement.
Figure 2.
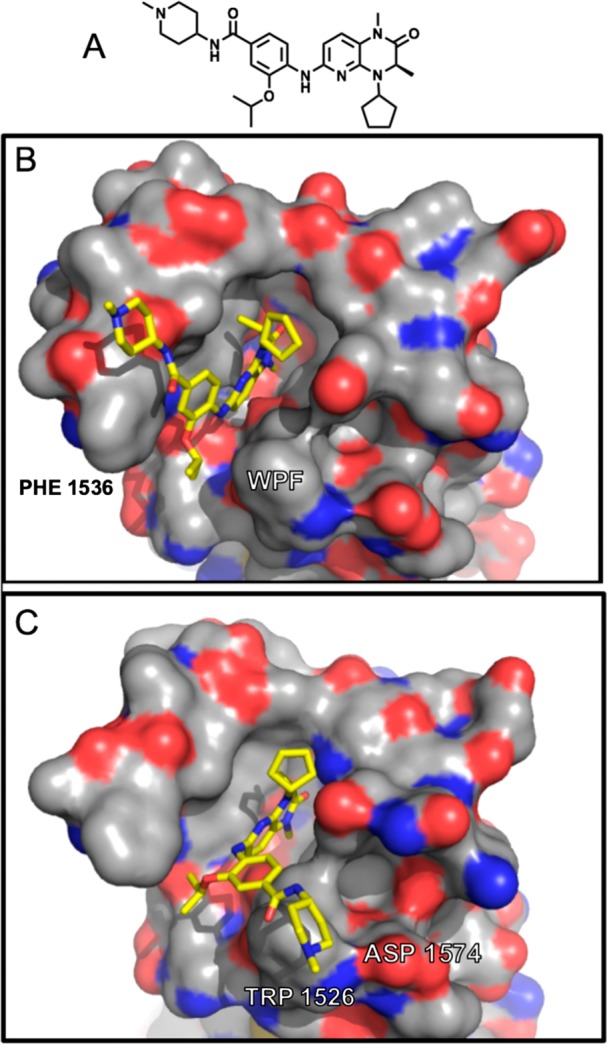
(A) Chemical structure of 15. (B) Cocrystal structure of 15 and TAF1(2) showing observed binding mode (I). (C) Observed binding mode (II) (6P38).
To further probe the ZA SAR in light of the apparent crystallographic flexibility, we undertook to study the role of tail group positioning in TAF1(2) binding. We thus examined substituents that would preclude ligand planarity, alternative tail compositions including addition of sulfonamide or methylene spacers which might allow improved “WPF shelf” access as in related efforts37 or which might engage Asp1524, as well as direct aryl tail appendages (Table S3). We observed broadly diminished affinity across these analogs, suggesting that while the parent methoxy-N-(1-methylpiperidin-4-yl)benzamide tail group likely samples a variety of conformations, its poses are nonetheless well positioned to contribute considerably to affinity. Key interactions likely include offset pi-stacking and backbone H-bonding to Phe1536 in Pose (I), and T-stacking with Trp1526 in Pose (II). We note, however, the influence of crystal packing contacts within the subcell featuring Pose (II) (Figure S2).
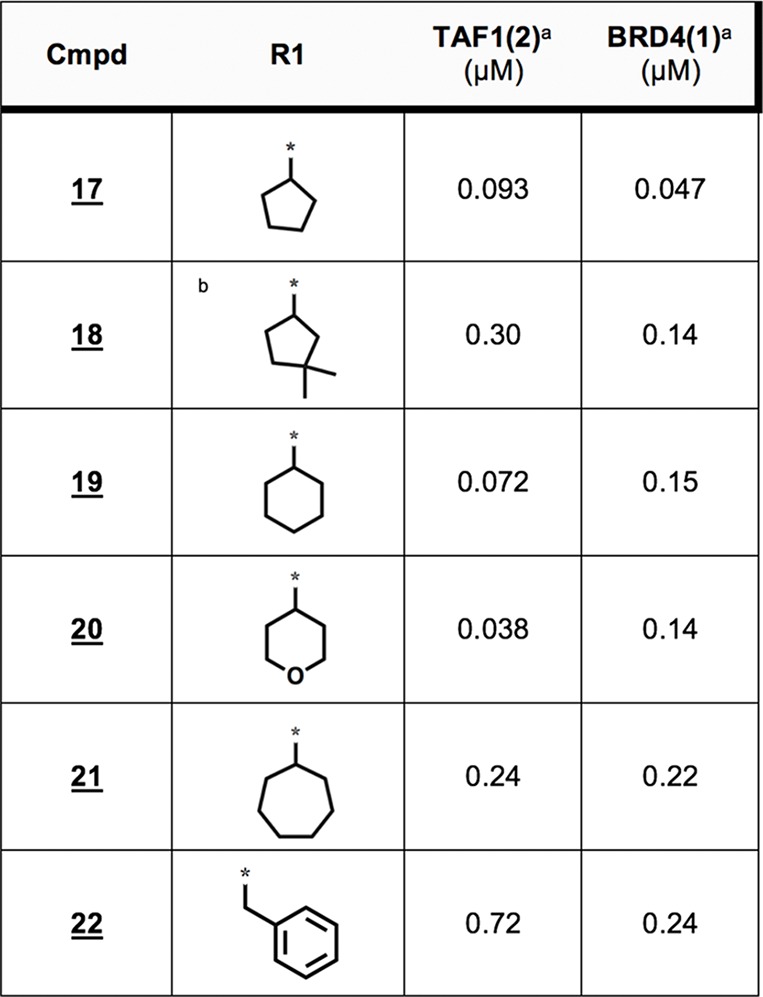
We next turned our attention to examine SAR within the dihydropyridopyrazone warhead, with a particular interest in TAF1-biasing modifications. Here, a neutral solubilizing tail was implemented to facilitate purifications, although we noted a cost in affinity against TAF1 (93 nM for 17 vs 16 nM for 13). We anticipated that TAF1’s Val1547 might better accommodate bulky replacements to the proximal cyclopentane moiety (Table 4; 17-22) relative to BRD4’s equivalently placed Leu94. Of this series, expansion to a six membered ring best improved TAF1 bias, with tetrahydropyran (THP) substitution further enhancing this effect (Table 4; 19,20). Further considering that Leu94 of BRD4 packs between the cyclopentyl and asymmetric ethyl substituents of BI-2536, we envisioned that more severe steric discrimination of BRD4 might be achieved by linking these positions through this space via the 4-carbon bridgehead (Figure 3). Gratifyingly the resulting tricyclic warhead-containing compound 23 gave a 66-fold improvement (vs parent 17) in measured selectivity (BRD4 IC50/TAF IC50).
Table 4. Cycloalkyl Moiety Expansions.
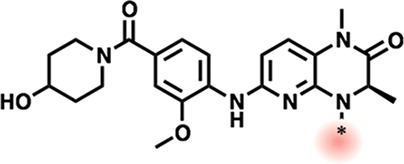
AlphaScreen IC50s from quadruplicate measurement.
Figure 3.
(A) TAF1(2)-15 cocrystal structure (6P38) showing proximal Val1547 (gray spheres). (B) Structural alignment (Pymol) of BRD4 (4O74) showing proximal Leu94 (red spheres). (C) Schematic of tricyclization approach. Inset table: aAlphaScreen IC50s from quadruplicate measurement. bIC50 Ratio: (BRD4(1)/TAF1(2)).
Alongside cycloalkyl modifications, we concurrently investigated substitutions to the warhead’s pyridine ring (24–30), as well as replacement of the embedded methyl amide with extended acyl mimetics following the approach of Crawford et al. (29,30) (Table 5).11 Although we found removal of the pyridyl nitrogen to be disfavored, we noted that 7-methoxy analog 27 showed largely abrogated BRD4 activity alongside only minor losses in TAF1 affinity relative to its parent analog (24). This SAR is consistent with the observations of Bennett et al.,37 who found that methoxy derivatization at an analogous position of a related bicyclic TRIM24 inhibitor also diminished BRD4 binding. Alternatively, we found that the extended 1-butenyl analog 30 also promoted TAF1 bias, as reported within an alternative pyrrolopyridinone scaffold.11 As discussed previously, this permissiveness likely relates to the ability of TAF1 to favorably rearrange the conserved water network, in line with its ability to recognize natural lysine butyrylation and crotonylation.11,38
Table 5. Bicyclic Core Exploration.
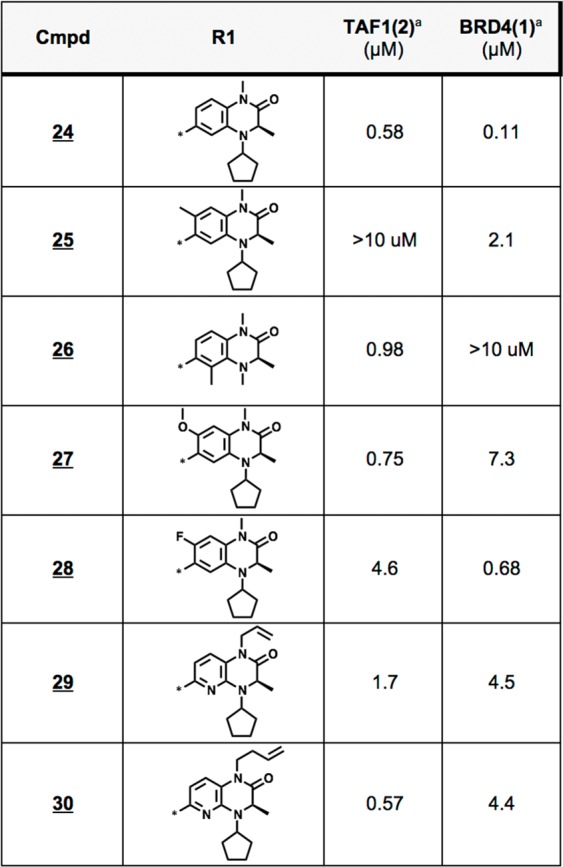
AlphaScreen IC50s from quadruplicate measurement.
Encouraged by their performance, we selected the tricyclic core and N4-THP modifications for further examination. Combining these features with the preferred BI-2536 tail group confirmed the respective contributions of the THP group to improved potency (31) and tricyclization to TAF1 selectivity (32) (Table 6). As THP featuring compound 31 offers attractive dual potency against both TAF1 and BRD4, it was prioritized for assessment of family wide selectivity. Profiling via the commercial phage-display BROMOscan (DiscoveRx) assay confirmed potent displacement to be largely restricted to TAF1(2) and the BET family bromodomains, with additional activity observed against the family IV bromodomain BRPF1 (Table S4).
Table 6. Lead Compounds.
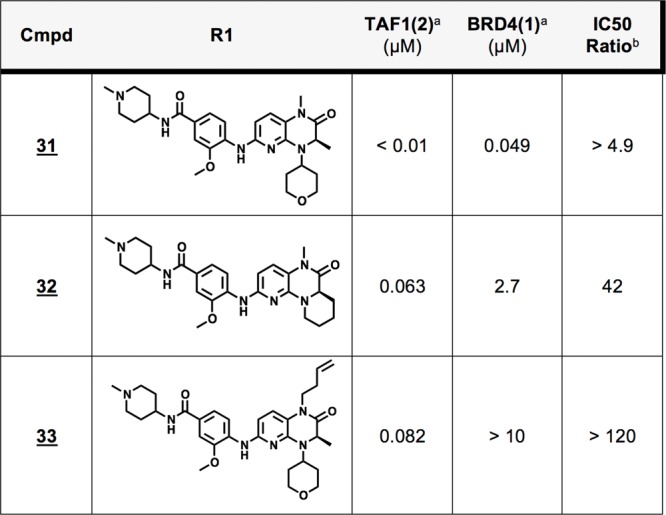
AlphaScreen IC50s from quadruplicate measurement.
Fold selectivity toward TAF1 given by IC50 ratio: BRD4(1)/TAF1(2).
To investigate whether structural features identified in this study might be favorably combined, we prepared analog 33 featuring both 1-butenyl and THP functionality. We were pleased to observe that this analog showed marked TAF1 bias, while still retaining considerable potency, as well as PLK1 inactivity (Table 6, Table S5). To pursue further structural insight into the selectivity promoted by analogs 32 and 33, we resolved X-ray cocrystal structures of binding modes within TAF1(2) (Figure 4, 6P39, 6P3A). The obtained 32 cocrystal reveals the cycloalkyl bridge to be asymmetrically projected as anticipated, affecting the intended steric contact with Val1574 (Figure 4 A,B). Indeed, an accommodating rearrangement of this side chain orientation is observed relative to the acyclic analog shown in Figure 2, supporting that increased burden of equivalent Leu94 underpins diminished binding within BRD4(1). Contrastingly, within the 33 cocrystal, we observe a distinct mode of bromodomain discrimination by projection of the 1-butenyl modification into the floor of the KAc binding pocket (Figure 4C,D). Here, structural comparisons reveal remarkable coherence to the 1-butenyl pose adopted in previous work (Figure S3).11 In addition to a nearly identical alkyl chain conformation, we note also close coherence of proximal residues, including shelf substituent Phe1528, backstop residue Ile1575, and pocket residues Tyr1589(“gatekeeper”)/Tyr1541, while in contrast distal ZA residues (Trp1526, Phe1536) show significant discrepancy (Figure S3). Although the current structural resolution does not permit assignment of water positions, these data strongly imply an equivalent rearrangement within the conserved water network as observed previously.11 Together, these structural data sets provide a resource for structure-based approaches toward kinase inactive dual (BRD4(1)/TAF1(2)) and TAF1 selective compound development. Here, we highlight 31 as a potent dual lead compound for TAF1/BRD4 inhibition (Table 6). Toward TAF1 selectivity, we report three independent approaches: 3- to 4-position cyclization (23,32), 7-position ZA channel methoxy projection (27), and methyl to 1-butenyl amide substitution as an extended acyl-lysine mimetic strategy (30, 33). Conversely, we find the NMe analog 10 to be over 100-fold selective for BRD4 over TAF1 by IC50 ratio, offering a potential starting point toward improving the selectivity of BET inhibitors based on this scaffold.
Figure 4.
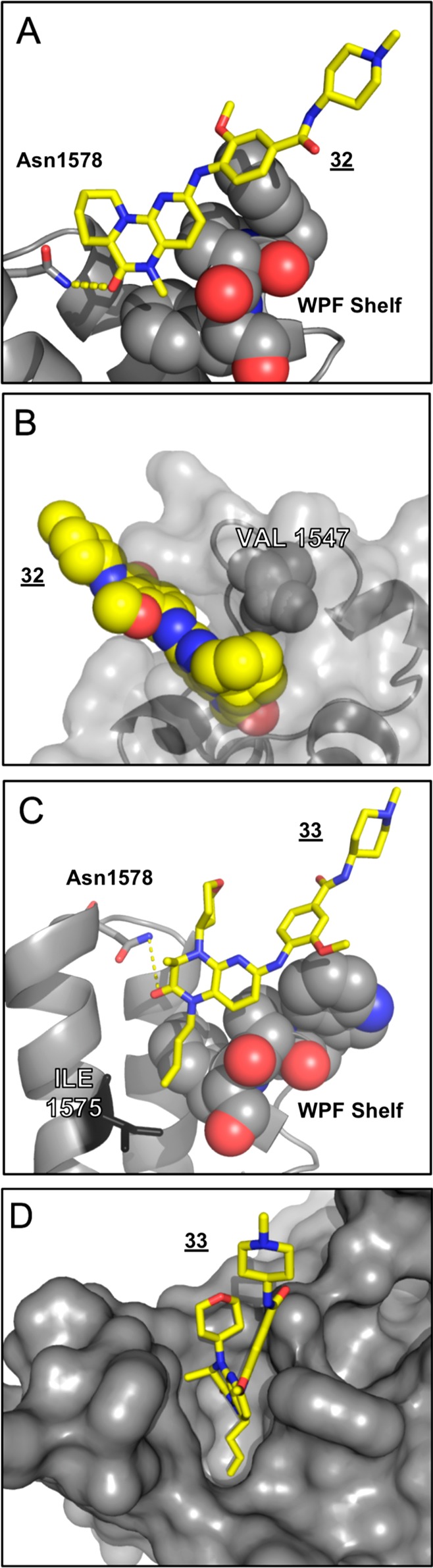
Cocrystal structures of 32 (A,B), and 33 (C,D) (6P39, 6P3A) showing binding modes to TAF1(2). (A) Cutaway of 32 binding within the KAc pocket, showing H-bonding (dashes) to conserved Asn (sticks), and packing against the WPF shelf (spheres). (B) Transparent surface view of 32 (spheres) packing against Val1547 (spheres). (C) Cutaway of 33 binding within the KAc pocket, showing H-bonding (dashes) to conserved Asn (sticks), packing against WPF shelf (spheres), and position of “backstop” residue Ile1575 (sticks). (D) Surface representation of 33 showing extended conformation of the 1-butenyl group into the floor of the KAc pocket.
Because of its role in transcription initiation, marked effects on cell cycle, and potentially selective effects on amplified gene expression, TAF1 is an attractive target for both basic and translational research. Further, the multiple purported enzymatic activities make TAF1 an excellent candidate for targeted degradation approaches.39−43 To this end, the compounds disclosed in this study offer new and advanced starting points for TAF1 probe development. In addition, previous studies of TAF1-BRD4 crosstalk have also outlined functional synergy of BRD4 and TAF1.10 Accordingly, the potent dual BRD4/TAF1 inhibitors reported herein may offer single agent efficacy in these contexts and merit further biological investigation in this regard.
Acknowledgments
The authors would like to thank Minoru Tanaka for technical discussions.
Glossary
Abbreviations
- KAc
acetyl-lysine
- BRD
bromodomain
- BET
bromodomain and extra-terminal domain
- TBP
tata-binding protein
- RB
retinoblastoma protein
- SAR
structure–activity relationships
- TM
thermal-melt
- THP
tetrahydropyran.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00243.
Supplemental data, experimental procedures, and synthetic procedures (PDF)
Author Present Address
∥ Scripps Research, La Jolla, CA.
Author Present Address
⊥ Novartis Institute for Biomedical Research, Cambridge, MA.
D.L.B. contributed to this work as a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2196-14). This work was supported by the Claudia Adams Barr Program for Innovative Cancer Research (to D.L.B.).
The authors declare the following competing financial interest(s): J.E.B. is now an executive and shareholder in Novartis AG.
Supplementary Material
References
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. a.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Vakoc C. R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54 (5), 72–736. 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S.; Polyak K. BET bromodomain proteins as cancer therapeutic targets. Cold Spring Harbor Symp. Quant. Biol. 2016, 81, 123–129. 10.1101/sqb.2016.81.030908. [DOI] [PubMed] [Google Scholar]
- Theodoulou N. H.; Tomkinson N. C.; Prinjha R. K.; Humphreys P. G. Clinical progress and pharmacology of small molecule bromodomain inhibitors. Curr. Opin. Chem. Biol. 2016, 33, 58–66. 10.1016/j.cbpa.2016.05.028. [DOI] [PubMed] [Google Scholar]
- Herait P. E.; Berthon C.; Thieblemont C.; Raffoux E.; Magarotto V.; Stathis A.; Thomas X.; Leleu X.; Gomez-Roca C.; Odore E.; Roumier C.; Bourrdel F.; Quesnel B.; Zucca E.; Michallet M.; Christian R.; Cvitkovic E.; Rezai K.; Preudhomme C.; Facon T.; Palumbo A.; Dombret H.. Abstract CT231: BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: interim results of an ongoing phase I trial in hematologic malignancies. AACR: 2014,CT231. 10.1158/1538-7445.AM2014-CT231 [DOI] [Google Scholar]
- Stathis A.; Zucca E.; Bekradda M.; Gomez-Roca C.; Delord J.-P.; Rouge T. d. L. M.; Uro-Coste E.; de Braud F.; Pelosi G.; French C. A. Clinical response of carcinomas harboring the BRD4–NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discovery 2016, 6 (5), 492–500. 10.1158/2159-8290.CD-15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Progress in the Development of non-BET Bromodomain Chemical Probes. ChemMedChem 2016, 11 (5), 477–487. 10.1002/cmdc.201500540. [DOI] [PubMed] [Google Scholar]
- McKeown M. R.; Shaw D. L.; Fu H.; Liu S.; Xu X.; Marineau J. J.; Huang Y.; Zhang X.; Buckley D. L.; Kadam A.; Zhang Z.; Blacklow S. C.; Qi J.; Zhang W.; Bradner J. E. Biased Multicomponent Reactions to Develop Novel Bromodomain Inhibitors. J. Med. Chem. 2014, 57 (21), 9019–9027. 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdelci S.; Lardeau C.-H.; Tallant C.; Klepsch F.; Klaiber B. o. r.; Bennett J.; Rathert P.; Schuster M.; Penz T.; Fedorov O.; Superti-Furga G.; Bock C.; Zuber J.; Huber K. V. M.; Knapp S.; ller S. M. u.; Kubicek S. Mapping the chemical chromatin reactivation landscape identifies BRD4-TAF1 cross-talk. Nat. Chem. Biol. 2016, 12 (7), 504–510. 10.1038/nchembio.2080. [DOI] [PubMed] [Google Scholar]
- Crawford T. D.; Tsui V.; Flynn E. M.; Wang S.; Taylor A. M.; Côté A.; Audia J. E.; Beresini M. H.; Burdick D. J.; Cummings R.; Dakin L. A.; Duplessis M.; Good A. C.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Kiefer J. R.; Jiang Y.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. Diving into the Water: Inducible Binding Conformations for BRD4, TAF1(2), BRD9, and CECR2 Bromodomains. J. Med. Chem. 2016, 59 (11), 5391–5402. 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- Bouché L.; Christ C. D.; Siegel S.; Fernández-Montalván A. E.; Holton S. J.; Fedorov O.; ter Laak A.; Sugawara T.; Stöckigt D.; Tallant C.; Bennett J. M.; Monteiro O. P.; Díaz-Sáez L.; siejka p.; Meier J.; Puetter V.; Weiske J.; Müller S.; Huber K. V. M.; Hartung I. V.; Haendler B. Benzo-isoquinoline-diones As Potent and Selective Inhibitors of BRPF2 and TAF1/TAF1L Bromodomains. J. Med. Chem. 2017, 60 (9), 4002–4022. 10.1021/acs.jmedchem.7b00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Tsui V.; Crawford T. D.; Audia J. E.; Burdick D. J.; Beresini M. H.; Côté A.; Cummings R.; Duplessis M.; Flynn E. M.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Jiang Y.; Joshi S.; Murray J.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Taylor A. M.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S.; Cochran A. G. GNE-371, a Potent and Selective Chemical Probe for the Second Bromodomains of Human Transcription-Initiation-Factor TFIID Subunit 1 and Transcription-Initiation-Factor TFIID Subunit 1-like. J. Med. Chem. 2018, 61 (20), 9301–9315. 10.1021/acs.jmedchem.8b01225. [DOI] [PubMed] [Google Scholar]
- Dynlacht B. D.; Hoey T.; Tjian R. Isolation of coactivators associated with the TATA-binding protein that mediate transcriptional activation. Cell 1991, 66 (3), 563–576. 10.1016/0092-8674(81)90019-2. [DOI] [PubMed] [Google Scholar]
- Struhl K.; Moqtaderi Z. The TAFS in the HAT. Cell 1998, 94, 1–4. 10.1016/S0092-8674(00)81213-1. [DOI] [PubMed] [Google Scholar]
- Segall J.; Matsui T.; Roeder R. Multiple factors are required for the accurate transcription of purified genes by RNA polymerase III. J. Biol. Chem. 1980, 255 (24), 11986–11991. [PubMed] [Google Scholar]
- Van Dyke M. W.; Roeder R. G.; Sawadogo M. Physical analysis of transcription preinitiation complex assembly on a class II gene promoter. Science 1988, 241 (4871), 1335–1338. 10.1126/science.3413495. [DOI] [PubMed] [Google Scholar]
- Buratowski S.; Hahn S.; Guarente L.; Sharp P. A. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 1989, 56 (4), 549–561. 10.1016/0092-8674(89)90578-3. [DOI] [PubMed] [Google Scholar]
- Wang E. H.; Tjian R. Promoter-selective transcriptional defect in cell cycle mutant ts13 rescued by hTAFII250. Science 1994, 263 (5148), 811–814. 10.1126/science.8303298. [DOI] [PubMed] [Google Scholar]
- Shao Z.; Ruppert S.; Robbins P. D. The retinoblastoma-susceptibility gene product binds directly to the human TATA-binding protein-associated factor TAFII250. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (April), 3115–3119. 10.1073/pnas.92.8.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartusel T.; Klempnauer K.-H. Transactivation mediated by B-Myb is dependent on TAF(II)250. Oncogene 2003, 22, 2932–2941. 10.1038/sj.onc.1206494. [DOI] [PubMed] [Google Scholar]
- Lively T. N.; Ferguson H. a.; Galasinski S. K.; Seto A. G.; Goodrich J. a. c-Jun Binds the N Terminus of Human TAFII250 to Derepress RNA Polymerase II Transcription in Vitro. J. Biol. Chem. 2001, 276 (27), 25582–25588. 10.1074/jbc.M100278200. [DOI] [PubMed] [Google Scholar]
- Sekiguchi T.; Yoshida M. C.; Sekiguchi M.; Nishimoto T. Isolation of a human X chromosome-linked gene essential for progression from G1 to S phase of the cell cycle. Exp. Cell Res. 1987, 169 (2), 395–407. 10.1016/0014-4827(87)90200-X. [DOI] [PubMed] [Google Scholar]
- Sekiguchi T.; Noguchi E.; Hayashida T.; Nakashima T.; Toyoshima H.; Nishimoto T.; Hunter T. D-type cyclin expression is decreased and p21 and p27 CDK inhibitor expression is increased when tsBN462 CCG1/TAFII250 mutant cells arrest in G1 at the restrictive temperature. Genes Cells 1996, 1 (7), 687–705. 10.1046/j.1365-2443.1996.00259.x. [DOI] [PubMed] [Google Scholar]
- Vangamudi B.; Paul T. A.; Shah P. K.; Kost-Alimova M.; Nottebaum L.; Shi X.; Zhan Y.; Leo E.; Mahadeshwar H. S.; Protopopov A.; Futreal A.; Tieu T. N.; Peoples M.; Heffernan T. P.; Marszalek J. R.; Toniatti C.; Petrocchi A.; Verhelle D.; Owen D. R.; Draetta G.; Jones P.; Palmer W. S.; Sharma S.; Andersen J. N. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75 (18), 3865–3878. 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard D.; Buckley D. L.; Paulk J.; Brien G. L.; Sonnett M.; Seo H. S.; Dastjerdi S.; Wühr M.; Dhe-Paganon S.; Armstrong S. A.; Bradner J. E. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew. Chem., Int. Ed. 2017, 56 (21), 5738–5743. 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gechijian L. N.; Buckley D. L.; Lawlor M. A.; Reyes J. M.; Paulk J.; Ott C. J.; Winter G. E.; Erb M. A.; Scott T. G.; Xu M.; Hyuk-Soo S.; Dhe-Paganon S.; Kwiatkowski N. P.; Perry J. A.; Qi J.; Gray N. S.; Bradner J. E. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14 (4), 405. 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciceri P.; Müller S.; O’Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. a.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10 (april), 305–12. 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W. J.; Zhu J.-y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schonbrunn E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9 (5), 1160–1171. 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W.; Lambert Q. T.; Berndt N.; Gunawan S.; Ayaz M.; Tauro M.; Zhu J.-Y.; Cranfill P. J.; Greninger P.; Lynch C. C.; Benes C. H.; Lawrence H. R.; Reuther G. W.; Lawrence N. J.; Schonbrunn E. Potent dual BET bromodomain-kinase inhibitors as value-added multitargeted chemical probes and cancer therapeutics. Mol. Cancer Ther. 2017, 16 (6), 1054–1067. 10.1158/1535-7163.MCT-16-0568-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaran A.; Talluri S. K.; Ayoub A. M.; Mishra N. K.; Cui H.; Widen J. C.; Berndt N.; Zhu J.-Y.; Carlson A. S.; Topczewski J. J.; Ernst S. K.; Harki D. A.; Pomerantz W. C. Molecular Basis for the N-Terminal Bromodomain-and-Extra-Terminal-Family Selectivity of a Dual Kinase–Bromodomain Inhibitor. J. Med. Chem. 2018, 61 (20), 9316–9334. 10.1021/acs.jmedchem.8b01248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffmann M.; Baum A.; Lénárt P.; Petronczki M.; Krššák M.; Gürtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J. M.; Rettig W. J. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr. Biol. 2007, 17, 316–322. 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Gray N. S.; Wodicka L.; Thunnissen A.-M. W.; Norman T. C.; Kwon S.; Espinoza F. H.; Morgan D. O.; Barnes G.; LeClerc S.; Meijer L.; Kim S.-H.; Lockhart D. J.; Schultz P. G. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science 1998, 281 (5376), 533–538. 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- Koblan L. W.; Buckley D. L.; Ott C. J.; Fitzgerald M. E.; Ember S. W. J.; Zhu J.-y.; Liu S.; Roberts J. M.; Remillard D.; Vittori S.; Zhang W.; Schonbrunn E.; Bradner J. E. Assessment of Bromodomain Target Engagement by a Series of BI2536 Analogues with Miniaturized BET-BRET. ChemMedChem 2016, 11 (23), 2575–2581. 10.1002/cmdc.201600502. [DOI] [PubMed] [Google Scholar]
- Chen L.; Yap J. L.; Yoshioka M.; Lanning M. E.; Fountain R. N.; Raje M.; Scheenstra J. A.; Strovel J. W.; Fletcher S. BRD4 Structure–Activity Relationships of Dual PLK1 Kinase/BRD4 Bromodomain Inhibitor BI-2536. ACS Med. Chem. Lett. 2015, 6 (7), 764–769. 10.1021/acsmedchemlett.5b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Wang Y.; Li Y.; Xu L.; Cao D.; Song S.; Damaneh M. S.; Wang X.; Meng T.; Chen Y.-L.; Shen J.; Miao Z.; Xiong B. Discovery of a series of dihydroquinoxalin-2(1H)-ones as selective BET inhibitors from a dual PLK1-BRD4 inhibitor. Eur. J. Med. Chem. 2017, 137, 176–195. 10.1016/j.ejmech.2017.05.049. [DOI] [PubMed] [Google Scholar]
- Bennett J.; Fedorov O.; Tallant C.; Monteiro O.; Meier J.; Gamble V.; Savitsky P.; Nunez-Alonso G. a.; Haendler B.; Rogers C.; Brennan P. E.; Müller S.; Knapp S. Discovery of a Chemical Tool Inhibitor Targeting the Bromodomains of TRIM24 and BRPF. J. Med. Chem. 2016, 59 (4), 1642–1647. 10.1021/acs.jmedchem.5b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn E. M.; Huang O. W.; Poy F.; Oppikofer M.; Bellon S. F.; Tang Y.; Cochran A. G. A Subset of Human Bromodomains Recognizes Butyryllysine and Crotonyllysine Histone Peptide Modifications. Structure 2015, 23 (10), 1801–1814. 10.1016/j.str.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Dikstein R.; Ruppert S.; Tjian R. TAF II 250 Is a Bipartite Protein Kinase That Phosphorylates the Basal Transcription Factor RAP74. Cell 1996, 84, 781–790. 10.1016/S0092-8674(00)81055-7. [DOI] [PubMed] [Google Scholar]
- Mizzen C. A.; Yang X.-j.; Kokubo T.; Brownell J. E.; Bannister A. J.; Owen-hughes T.; Workman J.; Wang L.; Berger S. L.; Kouzarides T.; Nakatani Y.; Allis C. D. The TAF II 250 Subunit of TFIID Has Histone Acetyltransferase Activity. Cell 1996, 87 (7), 1261–1270. 10.1016/S0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- Pham A.-D.; Sauer F. Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science 2000, 289 (5488), 2357–2360. 10.1126/science.289.5488.2357. [DOI] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien G. L.; Remillard D.; Shi J.; Hemming M. L.; Chabon J.; Wynne K.; Dillon E. T.; Cagney G.; Van Mierlo G.; Baltissen M. P.; Vermeulen M.; Qi J.; Frohling S.; Gray N. S.; Bradner J. E.; Vakoc C. R.; Armstrong S. A. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife 2018, 7, e41305 10.7554/eLife.41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.