

Abstract

Antibody–drug conjugates (ADCs) that incorporate the exatecan derivative DXd in their payload are showing promising clinical results in solid tumor indications. The payload has an F-ring that also contains a second chiral center, both of which complicate its synthesis and derivatization. Here we report on new camptothecin-ADCs that do not have an F-ring in their payloads yet behave similarly to DXd-bearing conjugates in vitro and in vivo. This simplification allows easier derivatization of camptothecin A and B rings for structure–activity relationship studies and payload optimization. ADCs having different degrees of bystander killing and the ability to release hydroxyl or thiol-bearing metabolites following peptide linker cleavage were investigated.
Keywords: Antibody−drug conjugates, camptothecin peptide
Antibody–drug conjugates (ADCs) are composed of a monoclonal antibody (mAb) attached via a linker to a cytotoxic payload and are typically used in cancer indications.1,2 ADCs bind to a target antigen on the surface of cells (Ag+ cells), but not to cells lacking the antigen (Ag– cells). Once bound, the conjugate is internalized and degraded in catabolic vesicles to release metabolite(s). Studies with radiolabeled monoclonal antibodies in patients have shown that only a small amount localizes at the tumor (∼0.003–0.08% of the injected dose/g tumor).3 Since the accumulation of an ADC is expected to be similar, or even lower, to achieve efficacy at a practical dose, ADC payloads are typically highly cytotoxic.1
The majority of ADCs currently in clinical evaluation use microtubule inhibitors or DNA damaging agents as the payload. Recently a few ADCs have incorporated topoisomerase I (TOPO 1) inhibitors.1 The enzyme TOPO 1 cleaves one strand of double stranded DNA, partially unwinds, and then reanneals the strand to relieve tension.4 Camptothecin (1) and its derivatives bind to the TOPO 1/DNA complex to prevent reannealing, which can cause cell death due to the accumulation of partially cleaved DNA.4 This class of compounds has at least five rings (A–E), and in aqueous solution, the E-ring is in equilibrium between the closed and open forms, Figure 1. The E-ring open form is reported to be far less active, in part because its charge inhibits diffusion into cells and >99% of it is bound by human serum albumin (HSA) in plasma.5 Structure–activity relationship (SAR) studies indicate that a fluorine substituent at the C11 position typically increases cytotoxicity several fold, and the carbon 20 center must be in the S configuration for the molecule to maintain activity.6 Exatecan (2) bears an additional F ring and was found to be less prone to hydrolysis in human plasma, with ∼30% remaining in the E-ring closed form at equilibrium.7 The improved lactone stability has been attributed to effects of the C11 fluorine substituent and the F-ring.8
Figure 1.

Ring opened and closed structures of camptothecin and exatecan. Letter designations of camptothecin rings and atom numbering are also shown.
A few ADCs that use camptothecin derivatives as the payload are currently in clinical evaluation, and promising data is emerging.9−12 One such ADC, DS-8201a (3a), depicted in Figure 2, binds to human epidermal growth factor receptor 2 (HER2) on targeted cells and is then internalized into catabolic vesicles. Proteases cleave the peptide linker, followed by concomitant amine immolation to release the highly cytotoxic metabolite DXd (4), Figure 2. This ADC has shown impressive results in clinical trials, including a 53% overall response rate (ORR) in HER2+ breast cancer patients who no longer respond to the ADC T-DM1,11 and a 58% ORR in HER2+ nonsmall cell lung cancer.10 We therefore wished to conduct SAR studies to provide new camptothecin-linker constructs and to determine if ADCs bearing these derivatives would have a similar or improved therapeutic index (TI) in preclinical models.
Figure 2.

Depiction of DXd-bearing ADCs and processing in cells. DAR stands for drug to antibody ratio.
The F-ring of DXd has a chiral center, which complicates synthetic efforts and SAR studies. Also, two ADCs bearing the less potent camptothecin SN-38, via a linkage that stabilizes the E-ring, or through a linkage that does not, were found to have similar in vivo efficacies.13 This calls into question if the F-ring of DXd, which potentially stabilizes the E-ring, is required. We therefore investigated if an F-ring was beneficial for camptothecin-ADCs. Another important aspect of ADC design is its ability to induce bystander killing, an effect where ADCs not only kill targeted antigen positive (Ag+) cells but also some of the nearby (bystander) cells in a tumor, which may be Ag+ or antigen negative (Ag−). There is mounting evidence that ADCs with this capability are more efficacious in tumor xenograft models.14−17 For bystander killing to occur, ADC metabolites must be membrane permeable in order to diffuse into proximal nontargeted cells. Compounds typically become more membrane permeable as their hydrophobicity increases.18 Consequently, if other attributes are not affected, an ADC’s bystander killing should be enhanced by increasing the hydrophobicity of its metabolite(s). We therefore synthesized payloads with different hydrophobicities to determine if ADCs using them would induce varying degrees of bystander killing. Lastly, although it is not dose limiting in the clinic, DXd bearing ADCs have significant gastrointestinal (GI) toxicity,10,11,19 and DXd itself is not metabolized by liver.20 Liver inactivation of camptothecin-ADC metabolites could potentially reduce GI toxicity. Compounds containing sulfide moieties are typically easily oxidized21 and can often be inactivated in liver.22,23 Also, thiol-bearing compounds can be S-methylated by cells, facilitating inactivation through oxidation to sulfoxides and sulfones in the liver.22,23 We therefore designed ADCs that could efficiently release camptothecin metabolites that contain a sulfide or thiol moiety. All synthetic and analytical procedures are detailed in the supplementary section and briefly described herein.
A set of camptothecin derivatives was prepared as depicted in Scheme 1. The aniline 5 was acylated with chloroacetonitrile (6a) or 5-bromopentylnitrile (6b) in the presence of Lewis acids, BCl3 and AlCl3, to give 7a or 7b, respectively. Each compound was then reacted with commercially available 8 in the presence of a catalytic amount of pyridinium p-toluene sulfonate (PPTS) to give 9a or 9b, respectively.24 Compound 9a was reacted with sodium azide, then reduced to give 10, which was coupled to glycolic acid giving 11. Reaction of 9b with 15% water in HMPA at 101 °C overnight gave 12. The chlorine atom of 9a was displaced with 2-mercaptoethanol or 1,2-dimercaptoethane in N,N-dimethylformamide (DMF) and N,N-diisopropylethylamine (DIPEA) to give 13 or 14a, respectively. Methylation of 14a with iodomethane gave 14b.
Scheme 1. Synthesis of Camptothecin Derivatives.

Reagents and conditions: (a) BCl3, AlCl3, ∼39%, (b) PPTS, ∼78% (c) NaN3; P(OEt)3; HCl/H2O 57%, (d) glycolic acid DMTMM 93%, (e) HMPA/H2O 101 °C, 51%, (f) 2-mercaptoethanol, TEA, 78%, (g) 1,2-dimercaptoethane, DIPEA, 47%, (h) CH3I, DIPEA 54%.
Camptothecin payloads, capable of being conjugated to a mAb, were synthesized as shown in Scheme 2. The protected peptide (15, Fmoc-Ala-Ala-Ala-Gly-OH) was prepared by solid phase synthesis using standard procedures.25 Oxidative decarboxylation of 15 using lead tetraacetate gave 16,26 which was then reacted with the benzyl ester of glycolic acid in a solution of 20% trifluoroacetic acid (TFA) in dichloromethane to give 17. Deprotection of 17 with morpholine, followed by hydrogenation with 10% palladium on carbon gave 18, which was then reacted with the heterobifunctional reagent 19 to give 20. Coupling of 20 with compound 10 using (4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride) (DMTMM) gave payload 21a. Displacement of the acetate moiety of 16 with 12 in DMF containing 4% HCl gave 22, which was then deprotected with morpholine and reacted with 19 to give 21b. Compound 19 has five methylene units, making it and the payloads that incorporate it fairly hydrophobic. ADCs that are highly hydrophobic can form aggregates and are potentially cleared from circulation in vivo faster than more hydrophilic conjugates.27 Adding a polar moiety to the linker was therefore desired, but preferably at a location that would not affect the hydrophobicity of an ADC’s released metabolite. Consequently, a more hydrophilic derivative, bearing a polyhydroxyl moiety and a maleimide, to enable conjugation was prepared. Z-l-Glutamic acid benzyl ester 23 was coupled to N-methyl-d-glucamine (24) using DMTMM to give 25, which was deprotected by hydrogenation in methanol/water to 26. The heterobifunctional linker 27 was then coupled to 26 to give 28. To prepare a sulfide-bearing payload, 16 was reacted with 14a in dilute acid to give 29. Deprotection of 29 with morpholine, followed by coupling to 28 using DMTMM gave 21c.
Scheme 2. Synthesis of Camptothecin Payloads.

Reagents and conditions: (a) Pb(OAc)4, AcOH, Cu(OAc)2 62%, (b) HOCH2COOBn, 20% TFA in CH2Cl2, 59%, (c) 15% morpholine in DMF; 10% Pd–C/H2 20 PSI, 69%, (d) DMF, DIPEA 63%, (e) 10, DMTMM, TEA, 36%, (f) 12, 4% HCl in DMF, 48%, (g) 15% morpholine in DMF; 19, DIPEA, 42%, (h) DMTMM 68%, (i) 10% Pd–C/H2 20 PSI, 94%, (j) DIPEA 55%, (k) 20% TFA in CH2Cl2, 56%, (l) 15% morpholine in DMF; 28, TEA, DMTMM, 24%.
Exatecan mesylate was purchased from Abzena and used to prepare the F-ring bearing camptothecins DXd (4) and the linkable payload deruxtecan (21d) as described by Agatsuma et al.28 The conjugates mAbE-21d, mAbF-21d, and mAbctrl-21d depicted in Figure 2 and ADCs bearing new payloads were prepared as shown schematically in Figure 3.28 The humanized anti-huEGFR and anti-huFRα antibodies, which bind to human epidermal growth factor receptor 1 (EGFR) or to human folate receptor-alpha (FRα), respectively, were used to prepare ADCs capable of targeting EGFR+ or FRα+ cells. The four interchain disulfide bonds of each mAb were reduced with tris(2-carboxyethyl)phosphine (TCEP). The eight free cysteine thiols of the mAb were then reacted with one of the payloads (21a–21d), and side products were removed by size exclusion chromatography (SEC). The anti-chKTI antibody, a chimeric antibody targeting Kunitz soybean trypsin inhibitor, was also conjugated to 21d to give a control ADC (ADCCtrl), which does not bind to mouse or human cells. The drug to antibody ratio (DAR) for each ADC is shown in Figure 2 and Scheme 2. Similar to the published conjugation procedure, full 8.0 DAR ADCs were not obtained presumably because some free thiols reformed interchain disulfide bonds.
Figure 3.

Preparation of mAb-21a–mAb-21c. Reagents and conditions (a) TCEP, one of 21a–21c, SEC. Conjugations with 21d were performed identically using an anti-huEGFR, anti-huFRα, or anti-chKTI mAb. The value n indicates the drug per antibody ratio (DAR).
The ADCs (mAbE-21a–mAbE-21d) were assayed for cytotoxicity and bystander killing, Table 1. As expected, they were highly potent against targeted Ag+ cells and much less potent against the cells in the presence of unconjugated antibodies that block binding of the conjugate, or toward nontargeted Ag– cells. In the bystander killing assay, several concentrations of a conjugate were incubated with a coculture of Ag+ cells and Ag–/luc cells, (Ag– cells transfected with luciferase gene) in U-bottom 96-well plates. The ADC is taken up by Ag+ cells, then metabolites are released inside the cell and kill it, and potentially diffuse into and kill the Ag– cells. Survival of Ag–/luc cells was measured with an assay that detects only luciferase-positive cells. ADCs mAbE-21b and mAbE-21c induced the highest level of bystander killing even though they were slightly less cytotoxic to Ag+ cells (Table 1). The metabolites from these conjugates have not yet been identified. However, if these new conjugates are metabolized by cells, similarly to ADC 3a, then mAbE-21a, mAbE-21b, mAbE-21c, and mAbE-21d should predominantly give metabolites 11, 12, 14a, and 4 (DXd), respectively. Also, 14a could potentially be S-methylated in cells to give 14b. Compounds 12, 14a, and 14b, lacking a polar amide in their C7 side chains, are expected to be more hydrophobic than 11 or DXd. We postulate that the metabolites from each ADC will have similar cytotoxicities when produced inside cells, but those that are more hydrophobic diffuse more efficiently into Ag– cells.
Table 1. In Vitro Cytotoxicity and Bystander Killing Activity of ADCs mAbE-21a–mAbE-21d.
| cell lines IC50 (M) |
bystander IC50 (M)c | ||
|---|---|---|---|
| ADC | NLa Ag– | Mbb Ag+ | Ag–:Ag+ |
| mAbE-21a | 1 × 10–7 | 4 × 10–10 | 4 × 10–9 |
| mAbE-21b | 5 × 10–7 | 7 × 10–10 | 5 × 10–10 |
| mAbE-21c | 6 × 10–7 | 1 × 10–9 | 6 × 10–10 |
| mAbE-21d | 1 × 10–7 | 7 × 10–10 | 2 × 10–9 |
Namalwa/luc cells (NL).
MDA-MB-468 cells (Mb).
Bystander killing indicates the IC50 for killing Ag– cells in the mixture of Ag– and Ag+ cells (Ag–:Ag+). The Ag– and Ag+ cells are NL and M,b respectively.
Thiol-bearing compounds, such as 14a, can react with cystine or other charged disulfide containing molecules in cell culture, to become poorly membrane permeable.23 Therefore, only the in vitro cytotoxicities of 11, 12, 13, 14b, and DXd were determined, Table 2. The more hydrophobic compounds 12, 13, and 14b had higher potencies than 11 and DXd against the two cell lines tested. Further studies will be needed to determine how efficiently metabolites are released from ADCs mAb-21a–mAb-21d, and if the postulated metabolites are indeed formed.
Table 2. In Vitro Cytotoxicities of Unconjugated Compounds against HSC-2 and Namalwa/luc (NL) Cells.
| cell line IC50, M |
||
|---|---|---|
| compound | HSC-2 | NLa |
| DXd (4) | 1 × 10–9 | 6 × 10–10 |
| 11 | 2 × 10–9 | 9 × 10–10 |
| 12 | 5 × 10–10 | 2 × 10–10 |
| 13 | 4 × 10–10 | 2 × 10–10 |
| 14b | 6 × 10–10 | 2 × 10–10 |
Namalwa/luc cells (NL).
The antitumor activity of mAbE-21a and mAbE-21d was evaluated in nude mice bearing human head and neck squamous cell carcinoma EGFR-positive HSC-2 xenografts, Figure 4 (top), which display a high level of antigen expression (H score of 270), as determined using immunohistochemical methods as previously described.29 The activities of mAbE-21a, mAbE-21b, and mAbE-21d were also evaluated in nude mice bearing nonsmall cell lung cancer (NSCLC) squamous cell H1703 xenografts, wherein the antigen expression was lower (H score of 125) Figure 4 (bottom). Groups of six mice per test article were dosed as described in the figures. Control groups were dosed with vehicle or the nontargeted conjugate ADCCtrl. Data from these studies was interpreted using standardized methods.30 Tables containing T/C, PRs, and CRs for each study are shown in the Supporting Information. In both models, little efficacy was seen for the nontargeted ADCCtrl group, demonstrating targeting specificity. mAbE-21a and mAbE-21d showed similar dose-dependent antitumor activity in both the HSC-2 and H1703 EGFR+ xenograft models. The T/C and number of PRs and CRs were not significantly different between the mAbE-21a and mAbE-21d groups in either model, see Supporting Information. mAbE-21b was only tested in the lower antigen expressing H1703 model and was found to be significantly more active than the other two ADCs tested. Thus, at a dose of 75 μg/kg it had comparable activity, including T/C, PRs, and CRs, to that obtained with a higher dose (250 μg/kg) of the mAbE-21a and mAbE-21d groups. Also, no significant body weight loss was observed for mice in any group, indicating that the conjugates were well tolerated (data not shown).
Figure 4.
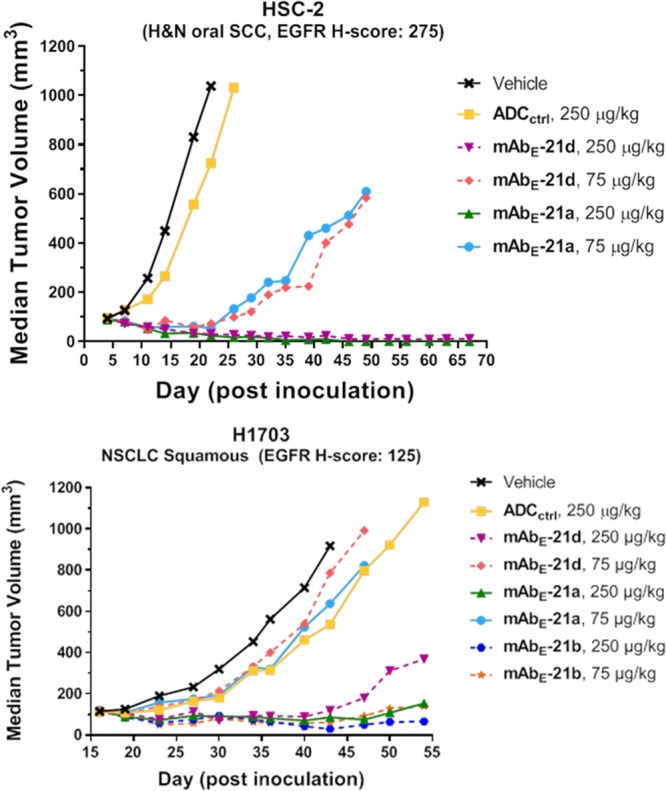
Antitumor activity of vehicle, ADCCtrl, mAbE-21a, and mAbE-21d at 75 and 250 μg/kg in HSC-2 (top); and vehicle, ADCCtrl, mAbE-21a, mAbE-21b, and mAbE-21c at 75 and 250 μg/kg in H1703 (bottom) mouse xenograft models. Dosing based on payload (75 and 250 μg/kg are ∼3 and ∼10 mg/kg based on antibody).
A pharmacokinetic study was conducted in mice to determine the stability and clearance rates of ADCs mAbE-21a and mAbE-21d. Groups of mice (3 mice/group) were dosed with the two ADCs at 10 mg/kg to determine the averaged clearance of their antibody components and retained bioactivity over time. Blood samples were taken at 2 min, 1 day, and 3 days post-inoculation and assayed for total anti-huEGFR mAb (μg/mL), which was determined by a sandwich enzyme-linked immunosorbent assay (ELISA), as previously described.31 The clearance of the antibody component was found to be similar for both conjugates (Figure 5). Retained bioactivity of the ADC in the plasma samples over time was determined in cytotoxicity assays with Ag+ or Ag– cells. The ADCs retained most of their activity against Ag+ cells at each of the time points while remaining over 200-fold less active against Ag– cells, indicating that the cytotoxicities were due to intact ADC, with little or no contribution from any released payload (Figures 5 and 6).
Figure 5.
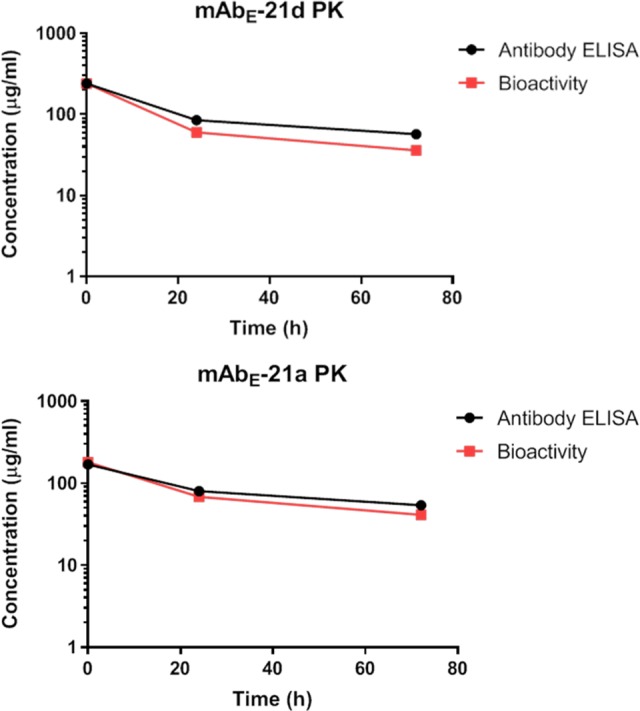
Plots of concentration (μg/mL) vs time of the mAb component (average) and retained bioactivity (pooled samples) of ADCs at 2 min, 1 day, and 3 day time points post administration in mice: mAbE-21d (top) and mAbE-21a (bottom).
Figure 6.
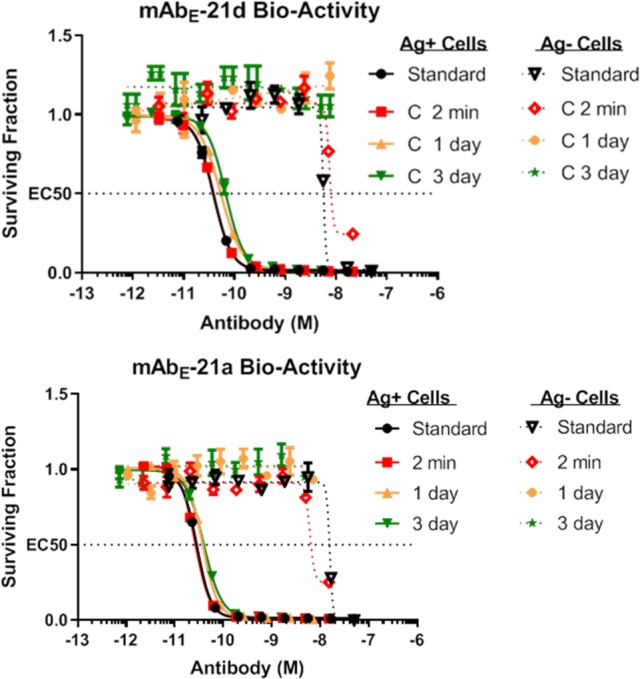
In vitro cytotoxicities of ADCs against Ag+ and Ag– cells. ADC standard in formulation (standard) or blood serum (pooled) containing ADC taken at 2 min, 1 da, y or 3 days post administration into mice for mAbE-21d (top) or mAbE-21a (bottom).
The tolerability of the ADCs was then assessed in mice. Groups of mice (3/group) were administered a single intravenous dose of vehicle control, mAbF-21a, or mAbF-21d at 5000 μg/kg based on payload (∼200 mg/kg based on Ab), Figure 7. Both conjugates were well tolerated. The maximum tolerated dose (MTD), as defined by body weight loss of 20% or more, was not reached for either ADC group 14 days post injection. However, the mice treated with mAbF-21d lost significantly more body weight than those treated with vehicle or mAbF-21a in these initial studies.
Figure 7.
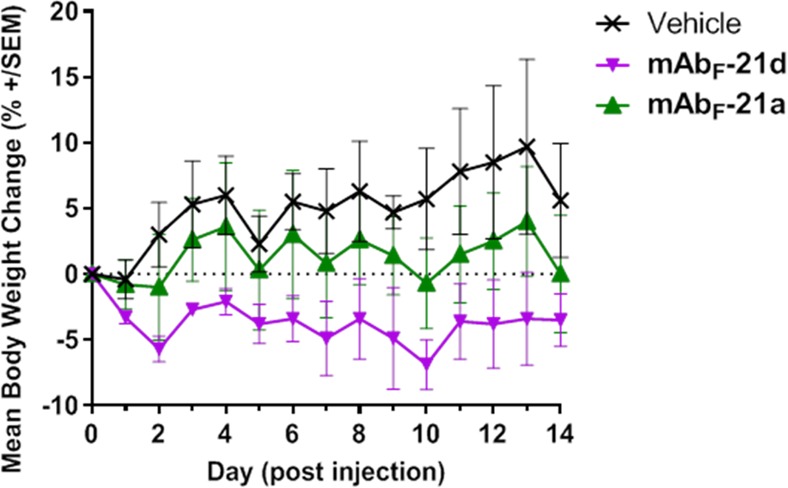
Mouse tolerability of noncross-reactive ADCs at 5000 μg/kg payload dose (∼200 mg/kg based on the Ab component).
In conclusion, camptothecins that do not have an F-ring are highly promising ADC payloads, which are easily derivatized on their A and B rings. This allows simplified preparation of camptothecin ADCs for SAR studies. The TI of conjugates that use payload 21a appears to be comparable to those containing the F-ring bearing payload 21d. An initial study indicated that an ADC with 21b was ∼3-fold more efficacious than ADCs bearing the 21a or 21d payloads. Additional studies will be needed to evaluate these ADCs, such as identification of the structures and formation efficiencies of their metabolites and comparisons of their pharmacokinetics and tolerabilities. We have previously used the concept of increasing metabolite hydrophobicity to increase the bystander killing of conjugates bearing maytansinoids and believe this strategy will be generally applicable to ADCs bearing other payloads.32,33
Acknowledgments
We would like to thank Richard Gregory and Joseph Kenny for their valuable suggestions.
Glossary
ABBREVIATIONS
- ADC
antibody–drug conjugate
- Bn
benzyl
- DAR
drug per antibody ratio
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DCM
dichloromethane
- DMSO
dimethyl sulfoxide
- DMTMM
(4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride)
- Fmoc
9-fluorenylmethoxycarbonyl
- HMPA
hexamethyl phosphoramide
- NHS
N-hydroxysuccinimide ester
- NMM
N-methyl morpholine
- PPTS
pyridinium p-toluene sulfonate
- TCEP
tris(2-carboxyethyl)phosphine
- TEA
triethylamine
- TFA
trifluoroacetic acid
- SEC
size-exclusion chromatography
- Z
benzyloxycarbonyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00301.
Detailed experimental procedures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This study was supported by ImmunoGen, Inc.
The authors declare no competing financial interest.
Supplementary Material
References
- Chari R. V.; Miller M. L.; Widdison W. C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–71. 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlacek H. H.; Steinstraesser A.; Kuhlmann L.; Schwarz A.; Seidel L.; Seemann G.; Kraemer H.-P.; Bosslet K.. Monoclonal antibodies in tumor therapy. Contributions in Oncology, Eckhardt S., Holzner J. H., Nagel G. A., Eds.; Karger: Munchen, 1988; Vol. 32, pp 81–100. [Google Scholar]
- Hsiang Y. H.; Hertzberg R.; Hecht S.; Liu L. F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–8. [PubMed] [Google Scholar]
- Burke T. G.; Mi Z. Preferential binding of the carboxylate form of camptothecin by human serum albumin. Anal. Biochem. 1993, 212, 285–7. 10.1006/abio.1993.1325. [DOI] [PubMed] [Google Scholar]
- Yaegashi T.; Sawada S.; Nagata H.; Furuta T.; Yokokura T.; Miyasaka T. Synthesis and antitumor activity of 20(S)-camptothecin derivatives. A-ring-substituted 7-ethylcamptothecins and their E-ring-modified water-soluble derivatives. Chem. Pharm. Bull. 1994, 42, 2518–25. 10.1248/cpb.42.2518. [DOI] [PubMed] [Google Scholar]
- Minami H.; Fujii H.; Igarashi T.; Itoh K.; Tamanoi K.; Oguma T.; Sasaki Y. Phase I and pharmacological study of a new camptothecin derivative, exatecan mesylate (DX-8951f), infused over 30 minutes every three weeks. Clin. Cancer Res. 2001, 7, 3056–64. [PubMed] [Google Scholar]
- Venditto V. J.; Simanek E. E. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol. Pharmaceutics 2010, 7, 307–49. 10.1021/mp900243b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardia A.; Mayer I. A.; Diamond J. R.; Moroose R. L.; Isakoff S. J.; Starodub A. N.; Shah N. C.; O’Shaughnessy J.; Kalinsky K.; Guarino M.; Abramson V.; Juric D.; Tolaney S. M.; Berlin J.; Messersmith W. A.; Ocean A. J.; Wegener W. A.; Maliakal P.; Sharkey R. M.; Govindan S. V.; Goldenberg D. M.; Vahdat L. T. Efficacy and safety of anti-Trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2017, 35, 2141–2148. 10.1200/JCO.2016.70.8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D.; Li B. T.; Murakami H.; Shiga R.; Lee C. C.; Wang K.; Jänne P. A. 183TiPA phase II study of [fam-] trastuzumab deruxtecan (DS-8201a) in HER2-overexpressing or mutated advanced non-small cell lung cancer. Ann. Oncol. 2019, 30, mdz063.081. 10.1093/annonc/mdz063.081. [DOI] [Google Scholar]
- Tamura K.; Modi S.; Tsurutani J.; Takahashi S.; Krop I.; Iwata H.; Wada R.; Yin O.; Garimella T.; Sugihara M.; Zhang L.; Lee C.; Yver A.; Baselga J. Abstract P6–17–10: Dose justification for DS-8201a, a HER2-targeted antibody-drug conjugate, for HER2-positive breast cancer: Observed clinical data and exposure-response analyses. Cancer Res. 2019, 79, P6-17-10. 10.1158/1538-7445.SABCS18-P6-17-10. [DOI] [Google Scholar]
- Doi T.; Shitara K.; Naito Y.; Shimomura A.; Fujiwara Y.; Yonemori K.; Shimizu C.; Shimoi T.; Kuboki Y.; Matsubara N.; Kitano A.; Jikoh T.; Lee C.; Fujisaki Y.; Ogitani Y.; Yver A.; Tamura K. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522. 10.1016/S1470-2045(17)30604-6. [DOI] [PubMed] [Google Scholar]
- Lau U. Y.; Benoit L. T.; Stevens N. S.; Emmerton K. K.; Zaval M.; Cochran J. H.; Senter P. D. Lactone stabilization is not a necessary feature for antibody conjugates of camptothecins. Mol. Pharmaceutics 2018, 15, 4063–4072. 10.1021/acs.molpharmaceut.8b00477. [DOI] [PubMed] [Google Scholar]
- Ogitani Y.; Hagihara K.; Oitate M.; Naito H.; Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–46. 10.1111/cas.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdison W. C.; Ponte J. F.; Coccia J. A.; Lanieri L.; Setiady Y.; Dong L.; Skaletskaya A.; Hong E. E.; Wu R.; Qiu Q.; Singh R.; Salomon P.; Fishkin N.; Harris L.; Maloney E. K.; Kovtun Y.; Veale K.; Wilhelm S. D.; Audette C. A.; Costoplus J. A.; Chari R. V. Development of anilino-maytansinoid ADCs that efficiently release cytotoxic metabolites in cancer cells and induce high levels of bystander killing. Bioconjugate Chem. 2015, 26, 2261–78. 10.1021/acs.bioconjchem.5b00430. [DOI] [PubMed] [Google Scholar]
- Vasalou C.; Helmlinger G.; Gomes B. A mechanistic tumor penetration model to guide antibody drug conjugate design. PLoS One 2015, 10, e0118977 10.1371/journal.pone.0118977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovtun Y. V.; Audette C. A.; Mayo M. F.; Jones G. E.; Doherty H.; Maloney E. K.; Erickson H. K.; Sun X.; Wilhelm S.; Ab O.; Lai K. C.; Widdison W. C.; Kellogg B.; Johnson H.; Pinkas J.; Lutz R. J.; Singh R.; Goldmacher V. S.; Chari R. V. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–37. 10.1158/0008-5472.CAN-09-3546. [DOI] [PubMed] [Google Scholar]
- Bennion B. J.; Be N. A.; McNerney M. W.; Lao V.; Carlson E. M.; Valdez C. A.; Malfatti M. A.; Enright H. A.; Nguyen T. H.; Lightstone F. C.; Carpenter T. S. Predicting a drug’s membrane permeability: A computational model validated with in vitro permeability assay data. J. Phys. Chem. B 2017, 121, 5228–5237. 10.1021/acs.jpcb.7b02914. [DOI] [PubMed] [Google Scholar]
- Nakada T.; Sugihara K.; Jikoh T.; Abe Y.; Agatsuma T. The latest research and development into the antibody-drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem. Pharm. Bull. 2019, 67, 173–185. 10.1248/cpb.c18-00744. [DOI] [PubMed] [Google Scholar]
- Nagai Y.; Oitate M.; Shiozawa H.; Ando O. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS-8201a), a HER2-targeting antibody-drug conjugate, in cynomolgus monkeys. Xenobiotica 2019, 49, 1086. 10.1080/00498254.2018.1531158. [DOI] [PubMed] [Google Scholar]
- Barnard D.; Bateman L.; Cunneen J. I.. Oxidation of organic sulfides. In Organic Sulfur Compounds, Kharasch N., Ed.; Pergamon Press: New York, 1961; Vol. 1. [Google Scholar]
- Sun X.; Widdison W.; Mayo M.; Wilhelm S.; Leece B.; Chari R.; Singh R.; Erickson H. Design of antibody-maytansinoid conjugates allows for efficient detoxification via liver metabolism. Bioconjugate Chem. 2011, 22, 728–35. 10.1021/bc100498q. [DOI] [PubMed] [Google Scholar]
- Widdison W. C.; Wilhelm S.; Veale K.; Costoplus J.; Jones G.; Audette C.; Leece B.; Bartle L.; Kovtun Y.; Chari R. J. Metabolites of antibody-maytansinoid conjugates: characteristics and in vitro potencies. Mol. Pharmaceutics 2015, 12, 1762–73. 10.1021/mp5007757. [DOI] [PubMed] [Google Scholar]
- Ejima A.; Terasawa H.; Sugimori M.; Tagawa H. Antitumor agents. 1. Asymmetric synthesis of (S)-camptothecin. Tetrahedron Lett. 1989, 30, 2639–2640. 10.1016/S0040-4039(00)99086-5. [DOI] [Google Scholar]
- Benoiton N. L.Chemistry of Peptide Synthesis; CRC Press, 2006. [Google Scholar]
- Needles H. L.; Ivanetich K. Decarboxylation of N-acetylamino-acids with lead tetra-acetate in N,N-dimethylformamide. Chem. Ind. 1967, 14, 581. [PubMed] [Google Scholar]
- Lyon R. P.; Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-LaFord H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–5. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Agatsuma T.; Takahashi S.; Hasegawa J.; Okajima D.; Hamada H.; Yamagushi M.. Anti-trop2 antibody-drug. US Patent 2016/0297890 A1, October 13, 2016.
- Detre S.; Saclani Jotti G.; Dowsett M. A ″quickscore″ method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J. Clin. Pathol. 1995, 48, 876–8. 10.1136/jcp.48.9.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissery M. C.; Guenard D.; Gueritte-Voegelein F.; Lavelle F. Experimental antitumor activity of taxotere (RP 56976, NSC 628503), a taxol analogue. Cancer Res. 1991, 51, 4845–52. [PubMed] [Google Scholar]
- Xie H.; Audette C.; Hoffee M.; Lambert J. M.; Blattler W. A. Pharmacokinetics and biodistribution of the antitumor immunoconjugate, cantuzumab mertansine (huC242-DM1), and its two components in mice. J. Pharmacol. Exp. Ther. 2004, 308, 1073–82. 10.1124/jpet.103.060533. [DOI] [PubMed] [Google Scholar]
- Widdison W. C.; Costoplus J. A.; Ponte J. F.; Lanieri L.; Setiady Y.; Dong L.; Skaletskaya A.; Wu R.; Qiu Q.; Kovtun Y.; Chari R. V. Peptide-cleavable maytansinoid (ADCs) induce high bystander killing leading to improved anti-tumor activity in vivo. Cancer Res. 2017, 77 (13 Suppl), 2186. 10.1158/1538-7445.AM2017-2186.28428271 [DOI] [Google Scholar]
- Manuscript submitted for publication.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.