

Abstract

Bruton’s tyrosine kinase (BTK) is a member of the TEC kinase family and is selectively expressed in a subset of immune cells. It is a key regulator of antigen receptor signaling in B cells and of Fc receptor signaling in mast cells and macrophages. A BTK inhibitor will likely have a positive impact on autoimmune diseases which are caused by autoreactive B cells and immune-complex driven inflammation. We report the design, optimization, and characterization of potent and selective covalent BTK inhibitors. Starting from the selective reversible inhibitor 3 binding to an inactive conformation of BTK, we designed covalent irreversible compounds by attaching an electrophilic warhead to reach Cys481. The first prototype 4 covalently modified BTK and showed an excellent kinase selectivity including several Cys-containing kinases, validating the design concept. In addition, this compound blocked FcγR-mediated hypersensitivity in vivo. Optimization of whole blood potency and metabolic stability resulted in compounds such as 8, which maintained the excellent kinase selectivity and showed improved BTK occupancy in vivo.
Keywords: BTK, covalent inhibitor, irreversible inhibitor, kinase inhibitor, kinase selectivity
Bruton’s tyrosine kinase (BTK) is a cytoplasmic tyrosine kinase and a member of the TEC kinase family consisting of BTK, tyrosine kinase expressed in hepatocellular carcinoma (TEC), bone marrow X-linked tyrosine kinase (BMX), interleukin-2-inducible T cell kinase (ITK), and tyrosine-protein kinase TXK (TXK).1 BTK is selectively expressed in a subset of immune cells, including macrophages, mast cells, basophils, platelets, and B cells, but not in plasma cells or T cells.2,3 BTK contains an N-terminal pleckstrin homology (PH) domain, a Src homology 3 (SH3) domain, a Src homology 2 domain, and a C-terminal kinase domain. Its function is regulated by membrane recruitment via the PH domain, phosphorylation of Tyr551 in the activation loop, and autophosphorylation of Tyr223 in the SH3 domain.4 BTK is a key regulator of antigen receptor signaling in B cells and of Fcγ and Fcε receptor signaling in macrophages and mast cells, respectively. Mutations in the BTK gene leading to BTK deficiency or nonfunctional BTK protein are the most frequent cause of X-linked agammaglobulinemia (XLA), a human primary immunodeficiency characterized by incomplete B cell development resulting in the absence of mature B cells and immunoglobulins.5 BTK deficient mice or mice carrying the hypomorphic xid mutation in the BTK PH domain exhibit defects in B cell development resulting in a phenotype similar to human XLA, but with reduced severity.6 Pharmacological intervention with BTK inhibitors has shown efficacy in animal models of rheumatoid arthritis and systemic lupus erythematosus.7,8 Based on this strong genetic and pharmacological validation, it is likely that a BTK inhibitor will have a positive impact on autoimmune diseases which are caused by autoreactive B cells or immune-complex driven inflammation. In addition, BTK inhibitors have been shown to be clinically efficacious in various B cell malignancies.9
Over the last few years, many pharmaceutical companies and academic groups embarked on the identification and development of BTK inhibitors, both irreversible and reversible.10 Ibrutinib (1) (Figure 1) is approved for several B cell malignancies including chronic lymphocytic leukemia and irreversibly inhibits BTK through covalent modification of the noncatalytic Cys481 residue in the ATP (adenosine triphosphate) binding site of the kinase domain.11 Due to its nonselective type I binding mode, 1 not only potently inhibits BTK but irreversibly inhibits all 10 kinases which carry a Cys at the same position as BTK (Supporting Information).11−13 In addition, 1 inhibits several kinases noncovalently at clinically relevant concentrations.12,14 Most importantly, 1 covalently inhibits epidermal growth factor receptor (EGFR) kinase, which is associated with clinical adverse effects of skin rash and diarrhea.15 The inhibition of TEC and proto-oncogene tyrosine-protein kinase Src (SRC) is being discussed in the context of clinical bleeding events with 1.14,16,17 Second generation clinical-stage covalent inhibitors such as spebrutinib and acalabrutinib keep a similar binding mode to BTK as 1 and therefore, despite offering an overall improved kinase selectivity profile, still inhibit several Cys-containing kinases.13,18
Figure 1.

Selected BTK inhibitors.
Reversible BTK inhibitors with high kinase selectivity have been described. CGI1746 (2) (Figure 1) was the first compound to be reported to bind to a specific inactive conformation of BTK which resulted in an outstanding kinase selectivity profile, superior to the clinical irreversible inhibitors.19
While reversible inhibitors require continuous exposure of the drug over the whole dosing interval in order to maintain a high degree of target inhibition, the pharmacodynamic (PD) effect of irreversible inhibitors can be decoupled from their pharmacokinetics (PK). A short systemic exposure could be sufficient for a sustained PD effect, since the duration of the PD effect depends on the turnover of the target–inhibitor adduct, rather than the duration of exposure of the inhibitor. In addition, irreversible inhibitors offer higher potency and therefore potentially lower human doses.20
Hence, during the course of our BTK inhibitor program the strategy was selected to combine the highly specific binding mode of CGI1746-like reversible inhibitors with the potency and PK/PD advantage of the irreversible mode of action. As starting point we used the selective reversible inhibitor 3 (Figure 2, see Supporting Information for selectivity data), a combination of an internal pyrrolopyrimidine screening hit with the tail fragment of 2.21
Figure 2.

Reversible starting point 3, covalent prototype 4, and its reversible analog 5.
The cocrystal structure of 3 with the human BTK kinase domain (Figure 3) revealed the stabilization of an inactive BTK conformation similar to 2 leading to a sequestration of Tyr551, which shields it from phosphorylation by the upstream kinases in the pathway. Based on this cocrystal structure, the pyrrolopyrimidine hinge binder and the phenyl ring occupying the hydrophobic channel of the protein are in close enough proximity to Cys481 to provide suitable exit vectors for linkers containing electrophilic warheads. Since we intended to keep our inhibitors as small as possible to allow for good physicochemical properties, we decided to attach the linker directly to the pyrrolopyrimidine while removing the hydrophobic channel substituent.
Figure 3.
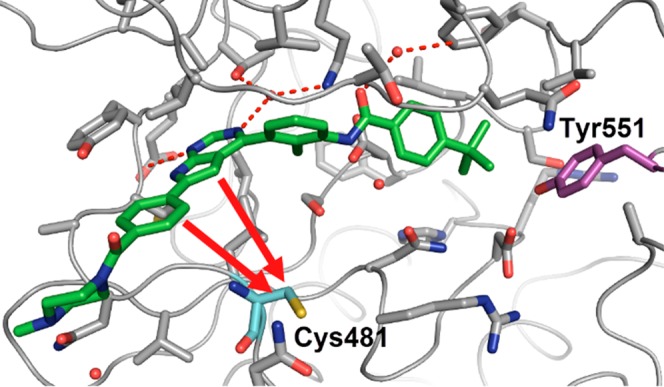
Co-crystal structure of the human BTK kinase domain in complex with 3 (PDB ID 6S90). Potential attachment points toward Cys481 (cyan) are shown with red arrows.
Based on this design concept we generated our first prototype (4) with an acrylamide warhead attached to the pyrrolopyrimidine via a methylene linker (Figure 2). In a BTK enzymatic assay 4 exhibited an IC50 of 16 nM compared to 13 nM of the reversible starting point 3. The propionamide derivative 5, which is devoid of an electrophilic warhead, inhibited BTK only very weakly with an IC50 of 3.9 μM, suggesting an important contribution of the acrylamide warhead to the potency of 4.
Additional evidence for a covalent mode of action was generated by incubation of 4 with the human BTK kinase domain protein, revealing a single peak mass corresponding to the 1:1 adduct of BTK and 4 in LC/MS analysis (Supporting Information). Furthermore, after preincubation of full-length human BTK and 4 at high concentrations, the enzymatic activity was not rescued by dilution (Supporting Information). This indicates an irreversible mode of action for 4, similar to rac-1 and in contrast to the reversible inhibitor 5.
We next assessed whether 4 maintains the binding mode of 3 to the inactive BTK conformation. In Ramos cells, 4 inhibited phosphorylation of Tyr551 over time in contrast to the results obtained for rac-1 (Figure 4). This is in line with the expected sequestration of Tyr551 as previously reported for 2.19 In addition, this suggests that the cellular turnover of Tyr551 phosphorylation is very rapid and allows for fast binding and inhibition of cellular BTK in the presence of pathway activation.
Figure 4.

Time course of Tyr551 phosphorylation in Ramos cells in the presence of 0.5 μM compound (in % of a maximally stimulated control).
As a consequence of its specific binding mode, 4 exhibits an excellent kinase selectivity profile. This is probably due to a higher dissimilarity of the kinases within the so-called “H3” pocket formed around Tyr551 compared to the ATP binding site as well as the possible inability of kinases other than BTK to adopt this specific conformation.19 In a panel of 61 non-Cys containing kinases including SRC, 4 did not show any IC50 < 10 μM (Supporting Information). Moreover, it also did not inhibit the Cys-containing kinases EGFR, receptor tyrosine-protein kinase erbB-2 (ERBB2), receptor tyrosine-protein kinase erbB-4 (ERBB4), and Janus kinase 3 (JAK3) and showed a 312-fold selectivity over BMX, the most related kinase to BTK (Table 1 and Supporting Information). The promising biochemical selectivity profile could be confirmed in cellular assays. While 4 potently blocked the BTK-dependent FcγR-induced IL8 release in THP1 cells with an IC50 of 0.08 μM, it did not inhibit EGFR phosphorylation in A431 cells up to the highest concentration of 8 μM. In contrast, 1 inhibited BMX, EGFR, and ERBB4 with similar potency to BTK in biochemical assays, as well as cellular pEGFR. Taken together, these data validate our design concept of transforming a selective reversible scaffold into a selective irreversible inhibitor.
Table 1. Potency and Selectivity Data for 1 and 4.
| IC50 [μM]a | 1 | 4 |
|---|---|---|
| BTKb | 0.005f | 0.014 (0.016) |
| BMXb | 0.001 (0) | 5.0 (0) |
| EGFRb | 0.016f | >10 |
| ERBB4b | 0.007f | >10f |
| Cellular FcγR/IL8c | <0.005 | 0.080 (0.0007) |
| Cellular pEGFR/A431d | 0.045 (0.015) | >8f |
| Human blood CD69e | 0.066 (0.022) | 0.934 (0.407) |
IC50 determined as a mean (n ≥ 2) (SD).
Enzyme assay.
FcγR-induced IL8 release in THP1 cells.
EGFR phosphorylation in A431 cells.
anti-IgM/IL-4 induced CD69 expression on B cells in human blood.
Single experiment.
Since our data demonstrated that 4 blocks the cellular FcγR-dependent response, we tested its effect on FcγR-mediated hypersensitivity in vivo. For that, we chose the mouse reverse passive Arthus reaction (RPA), where an acute hypersensitivity reaction is induced by intravenous injection of ovalbumin (OVA) antigen, followed by intradermal injection of a polyclonal Immunoglobulin G (IgG) anti-OVA antibody. For 1, efficacy in the mouse RPA has been described previously.22 We used rac-1 as a positive control, which exhibits very similar in vitro and in vivo properties compared to the (R)-enantiomer 1. Treatment with a single oral dose of 4, given 2 h prior to the induction of the RPA response, dose dependently reduced skin swelling measured 5 h after dosing the compound. Maximal inhibition of 65% was seen with the 60 mg/kg dose (Figure 5). In comparison, rac-1 reached 84% inhibition of swelling with a 30 mg/kg dose. Terminal blood concentrations of 4 were 4, 14, and 118 nM for the 6, 20, and 60 mg/kg doses, respectively (compared to 187 nM for rac-1).
Figure 5.

Dose dependent inhibition of the reverse passive Arthus reaction (RPA) in mice (FcγR-dependent skin swelling) 5 h after a single p.o. dose of 4 compared to rac-1 and vehicle control (error bars indicate SD). *p ≤ 0.05, ***p ≤ 0.001.
With our prototype 4 we had identified a selective covalent BTK inhibitor with in vivo efficacy after oral dosing. Nevertheless, several properties of 4 required further optimization. First, the cellular potency with an IC50 of 0.934 μM in human blood was significantly weaker than for 1 (IC50 of 0.066 μM) (Table 1). Second, the ADME properties of 4 were suboptimal, as the compound was highly unstable in mouse, rat, and human microsomes with intrinsic clearance values of 554, 693, and >700 μL min–1 mg–1, respectively. Although covalent irreversible inhibitors do not require continuous exposure, the fast elimination of 4 resulted in rather high doses for efficacy in the mouse RPA. The PK profile was even poorer in rat, where 4 showed an extremely high clearance of >300 mL min–1 kg–1 resulting in a very low oral bioavailability (BAV) of 2% (Supporting Information).
Although the in vivo clearance was too high to be solely driven by hepatic metabolism, we aimed to increase the stability in liver microsomes first. An in vitro metabolite identification study after incubation with rat liver microsomes (RLM) revealed the product of oxidation of the tert-butyl group as the most prominent metabolite. Therefore, we replaced it with a more stable cyclopropyl group in 6, which did not affect stability in RLM but interestingly slightly reduced human microsomal clearance (Table 2). The loss of potency of 6 could be compensated by the introduction of a fluorine at the central aromatic ring (7), which could be due to a multipolar interaction of the fluorine with the carbonyl group of Gly409 of the P-loop.23
Table 2. SAR for Covalent Pyrrolopyrimidines.


BTK enzyme inhibition IC50 mean (SD) (n ≥ 2).
anti-IgM/IL-4 induced CD69 expression in human blood B cells IC50 mean (SD) (n ≥ 2).
intrinsic clearance in rat and human liver microsomes [μL·min–1·mg–1 protein], n = 1.
Single experiment.
Mean of two experiments.
We then focused our attention on the linker between the pyrrolopyrimidine hinge binder and the acrylamide warhead. We hypothesized that by modification of the linker we could not only increase the metabolic stability but also increase the potency of the compounds by providing a more optimal orientation of the warhead toward Cys481, allowing for a faster formation of the covalent bond. We quickly realized that by optimizing the compounds we hit the limit of the biochemical BTK inhibition assay which was in the range of an IC50 of 1–2 nM (half of the enzyme concentration present in the assay). Therefore, our optimization of these potent compounds was mostly led by the more relevant and discriminating human blood B cell inhibition assay. Extension of the linker by one carbonyl group (8) provided a compound with highly improved biochemical and cellular potency, in addition to a good stability in liver microsomes. The hydroxy-azetidine 9 further improved microsomal stability but lost nearly 20-fold in human blood potency vs 8. Increasing the ring size to a 3-pyrrolidine (10) did not affect in vitro potency, while the 3-piperidine (11) clearly provided a less suitable positioning as indicated by a ∼300-fold loss in biochemical potency compared to 9 and 10. Both the 4-piperidine (12) and the 4-tetrahydropyridine (13) linker showed similar biochemical potency as 9 and 10, but these compounds were significantly more potent in the human blood assay with IC50 values of 19 and 32 nM, respectively. The high potency of compounds such as 13 allowed the introduction of substituted, less-reactive warheads. Both the aminomethyl-substituted acrylamide 14 and the but-2-enamide 15 showed a remarkably high potency in the human blood assay with IC50 values of 26 and 66 nM, respectively. Unfortunately, none of the compounds 10–15 showed good stability in rat or human liver microsomes.
As expected from the in vitro microsomal stability, 13, 14, and 15 showed fast elimination after intravenous dosing in rat PK studies with clearance values of >300, 176, and >300 mL min–1 kg–1, respectively. Unexpectedly, even 8 (300 mL min–1 kg–1) and 9 (183 mL min–1 kg–1) did not show improved in vivo clearance despite good stability in rat liver microsomes (Supporting Information). Since the clearance values were considerably above the hepatic blood flow of the rats (50–100 mL min–1 kg–1), it is likely that extrahepatic pathways contributed significantly to the fast elimination. Additional in vitro profiling did not provide indications for the extrahepatic clearance mechanism, since the compounds generally showed significant instability neither in plasma nor in liver S9 fractions.
Despite not being able to significantly improve the rat clearance compared to the prototype 4, the compounds were considerably more potent in the in vitro human blood CD69 assay. To assess a potential progress in in vivo efficacy of the compounds compared to 4, we measured BTK occupancy (Supporting Information), which has been shown to correlate with pathway inhibition in several BTK-dependent animal models.24 We assessed BTK occupancy in spleen homogenates of female rats 5 h after an oral dose of 10 mg kg–1 of the compounds (Figure 6). Compared to a mean splenic occupancy of 93% for rac-1, no significant BTK occupancy could be measured for 4. This was in line with the compound concentrations in blood we detected 0.5, 2, and 5 h after dosing. The blood levels for rac-1 were 67, 82, and 15 nM, whereas no exposure could be measured for 4. Among other factors such as suboptimal permeability and solubility, the exposure of 4 in rat might be limited by a significant first-pass metabolism. Despite very low blood levels (4.2, 2.2, and 1.5 nM), 8 showed occupancy of 44%. In a separate experiment, 14 showed no significant BTK occupancy, whereas 15 provided 70% (blood levels 14 and 15 at 5 h were 3 and 16 nM, respectively). Finally, 13 achieved the same BTK occupancy as rac-1 (96%), despite significantly lower blood levels at 5 h (6 nM for 13, 86 nM for rac-1).
Figure 6.

BTK occupancy in rat spleen homogenate 5 h after a 10 mg kg–1 p.o. dose of compounds compared to the respective vehicle control. Circles represent single animal values; lines indicate average ± SD.
Finally, we confirmed the kinase selectivity of our optimized inhibitors by testing 8 in a biochemical kinase panel containing 60 kinases, including Cys-containing kinases BMX, EGFR, ERBB2, and JAK3 (Supporting Information). 8 showed an excellent profile providing a 40-fold selectivity over BMX (BMX IC50 49 nM) and a >25,000-fold selectivity for all other kinases tested.
In summary, we converted pyrrolopyrimidine-based selective reversible inhibitors into a series of potent and selective irreversible BTK inhibitors with in vivo activity. Similar to what has in the meantime been published by others,25 the combination of binding to an inactive conformation of BTK with the covalent irreversible MoA resulted in compounds with a high kinase selectivity even over several closely related Cys-containing kinases. In addition, these compounds provided high BTK occupancy after oral dosing, similar to 1. Due to the very high clearance of these compounds, we decided to abandon this series in favor of an alternative scaffold. This resulted in the discovery of a highly potent and selective clinical candidate, which will be described in due course.
Acknowledgments
We thank E. Blum, O. Decoret, F. Gruber, J. Karrer, M.-L. Manisse, W. Miltz, B. Niepold, F. Ossola, M. Regenscheit, G. Rose, and S. Wildpreth for their synthetic support, P. Drueckes, D. Eichlisberger, S. Gaveriaux, and A.-S. Mangold for their efforts toward developing and performing biochemical and cellular assays, P. Ramseier and M. Vogelsanger for in vivo pharmacology, and G. Weckbecker for scientific guidance. We are grateful to the MX beamline team at the Swiss Light Source (PSI Villigen, Switzerland) for outstanding support at the beamline and Expose GmbH (Switzerland) for diffraction data collection.
Glossary
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion;
- ATP
adenosine triphosphate
- BAV
bioavailability
- BCR
B-cell receptor
- BMX
bone marrow X-linked tyrosine kinase
- BTK
Bruton’s tyrosine kinase
- CD69
cluster of differentiation 69
- EGFR
epidermal growth factor receptor
- ERBB2
receptor tyrosine-protein kinase erbB-2
- ERBB4
receptor tyrosine-protein kinase erbB-4
- ITK
interleukin-2-inducible T cell kinase
- IgG
immunoglobulin G
- JAK3
Janus kinase 3
- OVA
ovalbumin
- PAMPA
parallel artificial membrane permeability assay
- PH
pleckstrin homology
- PK/PD
pharmacokinetic/pharmacodynamic
- RLM
rat liver microsomes
- RPA
reverse passive Arthus reaction
- SH3
Src homology 3
- SRC
proto-oncogene tyrosine-protein kinase Src
- TEC
tyrosine-protein kinase Tec
- TXK
tyrosine-protein kinase TXK
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00317.
Full descriptions of all biological assays and in vivo studies. Kinase selectivity data. Crystallographic data collection and refinement statistics for crystal structures. Experimental procedures and characterization of compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Robinson D. R.; Wu Y.-M.; Lin S.-F. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene 2000, 19, 5548–5557. 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A. B. Independent and Opposing Roles For Btk and Lyn in B and Myeloid Signaling Pathways. J. Exp. Med. 1998, 188, 833–844. 10.1084/jem.188.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprulu A. D.; Ellmeier W. The Role of Tec Family Kinases in Mononuclear Phagocytes. Crit. Rev. Immunol. 2009, 29, 317–333. 10.1615/CritRevImmunol.v29.i4.30. [DOI] [PubMed] [Google Scholar]
- Mano H. Tec Family of Protein-Tyrosine Kinases: An Overview of Their Structure and Function. Cytokine Growth Factor Rev. 1999, 10, 267–280. 10.1016/S1359-6101(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Conley M. E.; Dobbs A. K.; Farmer D. M.; Kilic S.; Paris K.; Grigoriadou S.; Coustan-Smith E.; Howard V.; Campana D. Primary B Cell Immunodeficiencies: Comparisons and Contrasts. Annu. Rev. Immunol. 2009, 27, 199–227. 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- Mestas J.; Hughes C. C. W. Of Mice and Not Men: Differences between Mouse and Human Immunology. J. Immunol. 2004, 172, 2731–2738. 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- Xu D.; Kim Y.; Postelnek J.; Vu M. D.; Hu D.-Q.; Liao C.; Bradshaw M.; Hsu J.; Zhang J.; Pashine A.; Srinivasan D.; Woods J.; Levin A.; O’Mahony A.; Owens T. D.; Lou Y.; Hill R. J.; Narula S.; DeMartino J.; Fine J. S. RN486, a Selective Bruton’s Tyrosine Kinase Inhibitor, Abrogates Immune Hypersensitivity Responses and Arthritis in Rodents. J. Pharmacol. Exp. Ther. 2012, 341, 90–103. 10.1124/jpet.111.187740. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A. B. Bruton’s Tyrosine Kinase, a Component of B Cell Signaling Pathways, Has Multiple Roles in the Pathogenesis of Lupus. Front. Immunol. 2018, 8, 1–10. 10.3389/fimmu.2017.01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aw A.; Brown J. R. Current Status of Bruton’s Tyrosine Kinase Inhibitor Development and Use in B-Cell Malignancies. Drugs Aging 2017, 34, 509–527. 10.1007/s40266-017-0468-4. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Zhang D.; Liu Y.; Yang D.; Ran F.; Wang M. L.; Zhao G. Targeting Bruton’s Tyrosine Kinase for the Treatment of B Cell Associated Malignancies and Autoimmune Diseases: Preclinical and Clinical Developments of Small Molecule Inhibitors. Arch. Pharm. (Weinheim, Ger.) 2018, 351, 1700369. 10.1002/ardp.201700369. [DOI] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Médard G.; Pachl F.; Ruprecht B.; Klaeger S.; Heinzlmeir S.; Helm D.; Qiao H.; Ku X.; Wilhelm M.; Kuehne T.; Wu Z.; Dittmann A.; Hopf C.; Kramer K.; Kuster B. Optimized Chemical Proteomics Assay for Kinase Inhibitor Profiling. J. Proteome Res. 2015, 14, 1574–1586. 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- Nicolson P. L. R.; Hughes C. E.; Watson S.; Nock S. H.; Hardy A. T.; Watson C. N.; Montague S. J.; Clifford H.; Huissoon A. P.; Malcor J.-D.; Thomas M. R.; Pollitt A. Y.; Tomlinson M. G.; Pratt G.; Watson S. P. Inhibition of Btk by Btk-Specific Concentrations of Ibrutinib and Acalabrutinib Delays but Does Not Block Platelet Aggregation Mediated by Glycoprotein VI. Haematologica 2018, 103, 2097–2108. 10.3324/haematol.2018.193391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur V.; Swami A. Ibrutinib in CLL: A Focus on Adverse Events, Resistance, and Novel Approaches beyond Ibrutinib. Ann. Hematol. 2017, 96, 1175–1184. 10.1007/s00277-017-2973-2. [DOI] [PubMed] [Google Scholar]
- Shatzel J. J.; Olson S. R.; Tao D. L.; McCarty O. J. T.; Danilov A. V.; DeLoughery T. G. Ibrutinib-Associated Bleeding: Pathogenesis, Management, and Risk Reduction Strategies. J. Thromb. Haemostasis 2017, 38, 1–13. 10.1111/jth.13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson B. T.; Ellmeier W.; Watson S. P. Tec Regulates Platelet Activation by GPVI in the Absence of Btk. Blood 2003, 102, 3592–3599. 10.1182/blood-2003-04-1142. [DOI] [PubMed] [Google Scholar]
- Evans E. K.; Tester R.; Aslanian S.; Karp R.; Sheets M.; Labenski M. T.; Witowski S. R.; Lounsbury H.; Chaturvedi P.; Mazdiyasni H.; Zhu Z.; Nacht M.; Freed M. I.; Petter R. C.; Dubrovskiy A.; Singh J.; Westlin W. F. Inhibition of Btk with CC-292 Provides Early Pharmacodynamic Assessment of Activity in Mice and Humans. J. Pharmacol. Exp. Ther. 2013, 346, 219–228. 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A. D. D.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. Specific Btk Inhibition Suppresses B Cell– and Myeloid Cell–Mediated Arthritis. Nat. Chem. Biol. 2011, 7, 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Thakkar M.; Koul S.; Bhuniya D.; Singh U.. Preparation of Substituted Heterobicyclic Compounds, Compositions and Medicinal Applications Thereof. WO2013/157021 A1, 2013.
- Chang B. Y.; Huang M. M.; Francesco M.; Chen J.; Sokolove J.; Magadala P.; Robinson W. H.; Buggy J. J. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Ameliorates Autoimmune Arthritis by Inhibition of Multiple Effector Cells. Arthritis Res. Ther. 2011, 13, R115. 10.1186/ar3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulini R.; Müller K.; Diederich F. Orthogonal Multipolar Interactions in Structural Chemistry and Biology. Angew. Chem., Int. Ed. 2005, 44, 1788–1805. 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]
- Goess C.; Harris C. M.; Murdock S.; McCarthy R. W.; Sampson E.; Twomey R.; Mathieu S.; Mario R.; Perham M.; Goedken E. R.; Long A. J. ABBV-105, a Selective and Irreversible Inhibitor of Bruton’s Tyrosine Kinase, Is Efficacious in Multiple Preclinical Models of Inflammation. Mod. Rheumatol. 2019, 29, 510–522. 10.1080/14397595.2018.1484269. [DOI] [PubMed] [Google Scholar]
- Liang Q.; Chen Y.; Yu K.; Chen C.; Zhang S.; Wang A.; Wang W.; Wu H.; Liu X.; Wang B.; Wang L.; Hu Z.; Wang W.; Ren T.; Zhang S.; Liu Q.; Yun C.-H.; Liu J. Discovery of N-(3-(5-((3-Acrylamido-4-(morpholine-4-carbonyl)phenyl)amino)-1-methyl-6-oxo-1,6-dihydropyridin-3-yl)-2-methylphenyl)-4-(tert-butyl)benzamide (CHMFL-BTK-01) as a Highly Selective Irreversible Bruton’s Tyrosine Kinase (BTK) Inhibitor. Eur. J. Med. Chem. 2017, 131, 107–125. 10.1016/j.ejmech.2017.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.