

Abstract
Compound 3a, DV2-103, is a kinase inactive analogue of a potent Abl1/Src kinase inhibitor, PD173955, 2. Both compounds, 2 and 3a, are known to reduce production of beta amyloid (Aβ) peptide in cells and animal models. We have now prepared and evaluated a series of PD-173955 analogues, several of which reduced Aβ production potently. This occurs in cells expressing human full-length amyloid precursor protein (APP) and not in cells expressing APP β-C terminal fragment (APP-C99), suggesting that the kinase inactive analogues strongly affect β-secretase (BACE1) cleavage of APP, similarly to Gleevec. A combination of the kinase inactive analogues of PD173955 with a BACE1 inhibitor (BACEi), namely, BACE IV, strongly reduced Aβ levels in cells, as noted previously with Gleevec and analogues. Several potent compounds also penetrated and accumulated in mouse brain in high nanomolar to low micromolar concentration.
Keywords: Alzheimer’s disease, Aβ peptide, β-secretase, DV2-103, PD173955
Two potent Abl and Abl/Src kinase inhibitors, imatinib (Gleevec), 1,1 and PD-173955, 2,2 respectively, and a kinase inactive analogue of 2, i.e., 3a (DV2-103),3 were previously shown to potently reduce production of secreted β-amyloid (Aβ) peptide in cells.4,5 Compounds 1, 2, and 3a also reduced Aβ levels in guinea pigs4 or in mice5 that express three Alzheimer’s disease (AD)-related mutations.6 Aβ40 and Aβ42 peptides are two major isoforms of Aβ peptides found in AD brain.7 These peptides accumulate in AD brain in the form of amyloid plaques when levels of both isoforms as well as the Aβ42/Aβ40 ratio rise.8 Production of Aβ peptides from the amyloid precursor protein (APP), which is an integral membrane protein expressed abundantly in neurons,9 is mediated by β-secretase (BACE1) and γ-secretase (GS) proteases.10,11 However, a general inhibition of these proteases using their inhibitors, BACEi or GSi,12,13 have failed in clinical trials for the treatment of Alzheimer’s disease (AD).14−16 These inhibitors cause deleterious side-effects by inhibiting cleavage of other protein substrates, such as Neuregulin and Notch1.17,18 In contrast, compounds 1 and 3a(5) indirectly inhibited BACE1 cleavage of APP19 and did not affect processing of other BACE1 substrates tested.5 Additionally, a combination of compound 1 with a BACEi strongly reduced Aβ production in cells with a potency that is greater than the additive effects of each inhibitor.5,20 Now, we are developing 1 and 3a analogues for use as single agents, and in combination with a BACEi. When combined with a BACEi, the 1 and 3a analogues could reduce production of both Aβ40 and Aβ42 peptides, and enhance the efficacy of a BACEi while minimizing its associated side effects.21 Described in this communication are syntheses and evaluations of 3a analogues, including 3m, 5b, 5c, and 5f (Figure 1), showing that these compounds reduce production of both Aβ40 and Aβ42 at low micromolar concentrations. Moreover, compound 3a analogues strongly enhance the Aβ-lowering effect of BACE IV, a potent BACEi, in a nonadditive manner (analogue + BACEi). This finding is consistent with our prior reports where 1 and BACE IV combinations were used,5,20 and provides further rationale behind our ongoing efforts to develop and identify potent, selective and effective combination.20
Figure 1.

Structure of Abl or Abl/Src kinase inhibitors, Gleevec (Imatinib), and PD-173955, and the representative kinase inactive compound 3a (DV2-103) and PD-173955 analogues.
Chemistry
We designed and prepared a series of 3a analogues, including compounds 3b–p, 4a–m, and 5a–n (Figures 1 and 2). Unlike compound 2 and the previously described analogues of compound 2, which were shown to inhibit γ-secretase cleavage of APP without affecting Notch cleavage,22 all 3a analogues lack NH ‘H’ attached to pyrimidine (shown by an asterisk (*) in compound 2 in Figure 1). Because the crystal structure of the kinase domain of c-Abl kinase complexed with compound 2 has shown that the latter makes an important contact through “NH” attached to the pyrimidine ring with Met318 of the kinase,23 the 3a analogues were expected to bind c-Abl kinase less efficiently. Thus, we anticipated that the effects mediated by these compounds on Aβ production could be considered independent of Abl kinase inhibition as was shown previously for 3a.5 We prepared compounds 3b–p, 4a–m, and 5a–h using the readily available intermediates 6a–c and 8a (Figure 2). Compounds 6a(24) and 8a(25) were obtained as described,22,24,25 and converted to the sulfone derivatives 7a and 8b by reacting with m-CPBA. Compounds 6b and 6c were prepared similarly22 by modifying the synthesis of 6a and converted to 7b and 7c by reacting with m-CPBA (see Supporting Information Scheme S-1). Subsequently, compounds 7a–c were reacted with various piperazine derivatives or with amines to afford 3b–p, 4a–m, and 5a–f; and 8b was reacted with N-methylpiperazine to give 9. The latter underwent Suzuki–Miyaura coupling26 with aryllboronic acids affording compounds 5g–n.
Figure 2.

Synthesis of PD-173955 analogues: (A) 3b–p, 4a–m, and 5a–f, and (B) 5g–n. aCompounds 3l, 4e, 4k, and 5e are HCl salts, prepared by treating the Boc-protected coupled products with 4 M HCl in dioxane and EtOAc.
Evaluations
Initially, we screened all analogues to select potent compounds by determining their effects on Aβ production in N2a695 cells. The latter express sufficient Aβ40 peptide in 5–6 h. For this, we incubated compounds at 10 μM with N2a695 cells for 6 h at 37 °C, as described previously.5 Gleevec (10 μM) was used as a positive control in all experiments. Cell supernatants were collected and Aβ40 levels were measured using commercially available human Aβ40 ELISA kits. We found nine compounds, including 3c, 3l, 3m, 4m, 5b–d, 5f, and 5l, were equally or more potent than 1 (Figure S-1). To confirm that the reduction in Aβ40 production by all nine compounds was not due to the toxicity, we determined the toxicity of these compounds at 10 μM concentration by incubating with N2a cells for 5 and 24 h and determining the number of viable cells using the MTT assay. We also used 3a (10 μM) and 2 (1 μM) for a comparison. We found all compounds, except 3c and 5c, were nontoxic (Figure S-2). Compounds 3c and 5c showed some toxicity in 24 h, but not in 5 h. These results suggested that the toxicity of compounds 3c and 5c did not contribute to their effects on Aβ production in the 5 h assay.
For the sake of a preliminary structure–activity relationship (SAR) among the nine active analogues of 3a, i.e., 3c, 3l, 3m, 4m, 5b–d, 5f, and 5l, we pooled these compounds into group 1 and 2 depending upon whether the compounds lack or contain an additional (NHBoc or NHCOCMe3) substitution in ring D. Based on the screening data and further evaluation (see latter), we found that addition of NHBoc or NHCOCMe3 substitution in ring D generally increased the activity compared to the analogous compounds lacking one of these functions. However, this also increased molecular weight (MW) of the resulting products, as in 5b–d and 5f, and violated Lipinski’s rules.27 We further calculated the ADME properties, including gastrointestinal (GI) absorption, blood-brain barrier (BBB) permeation, and P-gp interaction, and synthetic accessibility of compounds 3c, 3l, 3m, 4m, 5b–d, 5f, and 5l, as compared to 2 and 3a using SwissADME (http://www.swissadme.ch/index.php) web tool.28 The calculated values, as shown in Table S-2, suggested that all these compounds could possess high GI absorption and likely to be distributed in the body readily. Second, group 1 compounds, including 3a, are likely to be BBB permeant, whereas compound 2 and the group 2 compounds are not. Third, all compounds, except 2 and the analogues 3a, 3m, 5d and 5l, were predicted as the P-gp substrates. Fourth, all compounds possess synthetic accessibility score less than 5 (on 1 to 10 scale for easy to difficult synthesis).29 Thus, we found compounds 3m and 5l suitable for further profiling based on both the activity and physicochemical properties of these compounds. We focused on compound 3m and compared its activity with 5b, 5c and 5f. Compound 3m possesses identical substitution in the piperazine “A” ring as in 5c and 5f, but lacks the NHBoc or NHCOCMe3 substitution in the D ring, whereas both 5b and 5c possess NHBoc substitution in the D ring and two different substitutions in the piperazine “A” ring.
We confirmed the activity of compounds 3m, 5b, 5c, and 5f by determining their effects on production of both Aβ40 and Aβ42. Compound 3a was used at 20 μM, and compounds 3m, 5b, 5c, and 5f were tested at 10 μM concentrations in these experiments. As shown in Figure 3A, all four compounds 3m, 5b, 5c, and 5f (10 μM) reduced Aβ40 and Aβ42 production more potently than 3a (20 μM) did. Separately, to confirm that 3a analogues reduce Aβ production through a pathway not requiring the inhibition of Abl or Src kinase activities, we examined the effects of these compounds on Abl and Src kinase activity. The kinase assay was performed (by an outsourcing company) with compounds set at a concentration of 10 μM, as described.30 The results shown in Figure 3B clearly reveal that compounds 3m, 5b, 5c, and 5f do not inhibit Abl or Src kinases potently, while these compounds reduce Aβ production by >50% in cellular assays. These data indicate that 3a analogues 5b, 5c and 5f reduce Aβ production independent of Abl and Src kinase and in that regard are similar to 1 and 3a.5
Figure 3.

PD-173955 analogues reduce Aβ production potently by affecting BACE1 cleavage of APP and do not inhibit Abl and Src kinases. (A) Compound 3a, 3m, 5b, 5c, and 5f reduced production of Aβ40 and Aβ42 peptides from N2a695 cells. Aβ40 and Aβ42 levels in cell supernatants post treatment with compounds were measured using human Aβ ELISA kits. (B) Compounds 3m, 5b, 5c and 5f did not inhibit or weakly inhibited Abl and Src kinase activity at 10 μM compound concentration (kinase activity assays were performed by Luceome Biotechnologies, Tucson, Arizona). Compound 3a is known to not inhibit Abl and Src kinase (ref (5)). (C, D) Effects of 3a analogues on Aβ production in N2a cells transfected with human (C) APP-FL or (D) APP-C99, measured by ELISA. DAPT is used as a positive control, and MK8931 is used as positive control in (C) and a negative control in (D). All experiments were performed in duplicate, and results shown here are the representative of two independent experiments. Statistical significance: *P < 0.05%; **P < 0.01%.
The amyloidogenic BACE1 cleavage of APP yielding soluble APPβ (sAPPβ) and the membrane bound β-C terminal fragment (βCTF) compete with a parallel nonamyloidogenic, α-secretase cleavage process that produces soluble APPα (sAPPα) and the APP α-C terminal fragment (αCTF). Here, α-secretase cleaves APP within the Aβ region, thus preventing amyloidogenic processing by BACE1.31 Compound 2 was earlier shown to reduce Aβ production by inhibiting the subsequent γ-secretase cleavage of βCTF.4 However, we have recently shown that 1 and compound 3a reduce production of Aβ primarily through shifting the β-cleavage to a nonamyloidogenic cleavage.5 Treatment with these compounds produced multiple long CTFs, including a 16 kDa CTF consisting of the C-terminal 141 amino acids of APP (CTF-141) which may have been produced by lysosomes, where it was most abundant.5 To examine whether compound 3a analogues also affect APP cleavage through either or both BACE1 and γ-secretase cleavage of APP, we examined their effects on production of Aβ peptides in N2a cells transfected with human APP-FL (full length) or with APP-C99 (βCTF), as described previously for Gleevec and analogues.20 We used 3a and analogues at 10 μM concentration. We also used BACEi MK8931 (0.5 μM) and GSi DAPT (1 μM) as controls. The results are shown in Figure 3C, D and SI Figure S-3. We found that the analogues of 3a, including compounds 5b and 5f, reduce production of both Aβ40 and Aβ42 peptides in APP-FL cells, but only of Aβ42 peptides in APP-C99 cells. None of the 3a analogues reduced Aβ40 peptides in APP-C99 cells. These results suggest that compounds 5b and 5f modulate BACE1 cleavage of APP, and in that respect are similar to Gleevec and 3a.5
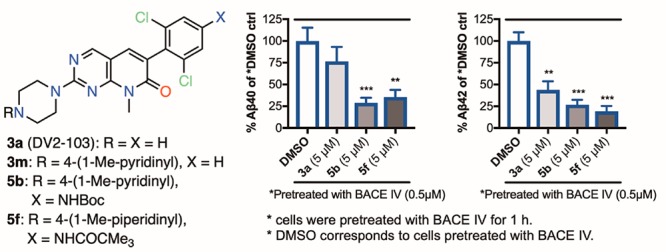
Earlier, we showed that a combination of BACE IV and 1 or its analogues reduce Aβ peptide secretion more potently than the sum of inhibitions produced by BACEIV and 1 used alone at equivalent concentrations.5,20 By using a combination of 3a or analogues and a general BACEi, we hope to reduce an effective dose, and thereby the toxicity, of the latter. This led us to further examine a combination of the PD-173955 analogues/BACE-1 inhibitor, BACE IV, and determine whether one of the potent PD-173955 analogues could also mediate a synergistic effect. We performed experiments by preincubating N2a695 cells with DMSO or 0.5 μM BACE IV alone for 1 h before further incubating with fresh media containing 5 μM compound 3a (or its analogues 5b or 5f) only. Indeed, we found that the 3a analogues 5b or 5f reduced both Aβ40 and Aβ42 production significantly compared to BACE IV-pretreated DMSO control alone (Figure 4A,B). This study has prompted us to develop and perform a larger screening study, in which brain-permeable 1 and 3a analogues and BACEi’s can be evaluated in combination.
Figure 4.

PD-173955 analogues reduce Aβ production potently in N2a695 cells preincubated with BACE IV. Two sets of experiments were performed using cells treated with DMSO alone or BACE IV for 1 h, before treatment with the freshly prepared media containing compounds 3a, 5b, and 5f. Shown on the left are (A) Aβ40 and (B) Aβ42 levels measured using human Aβ ELISA kits as percent of DMSO control for both experiments, and on the right are the BACE IV-pretreated experiment as percent of BACE IV-pretreated DMSO (depicted as *DMSO) control. All experiments were performed in duplicate or triplicate, and the results shown here are average of three independent experiments. Statistical significance: *P < 0.05%; **P < 0.01%; ***P < 0.001%.
In the current study, we have developed and evaluated kinase inactive analogues of 2 that possess B and C rings identical to 3a(3,5) and the parent compound 2, and there has been greater emphasis on A and D rings (Figure 1). While it would be interesting to also perform modifications of B/C rings and determine the impacts of such changes on drug activity, we have already identified 3a analogues, including 3m, 5b, 5c, 5f, or 5l, which can be further modified and used, especially in combination with a BACEi for reducing Aβ levels. Because Aβ peptides start accumulating in AD brain years before the onset of dementia,32 we anticipate that such compounds and combinations could be developed best for AD prevention. In a follow up study, we will develop and evaluate 3a analogues as single agents as well as in combination with a BACEi to determine their effects in an AD prevention model.
Conclusion
Kinase inactive analogues of PD173955, including compounds 3m, 5b, 5c and 5f, were developed as potent inhibitors of Aβ peptide production. These compounds function mainly through modulating the BACE1 cleavage of APP. The potential action mechanism of compounds 1 and 3a were discussed in an earlier study,5 which suggested that the compounds alter trafficking of APP away from recycling endosomes (where most Aβ is formed) to lysosomes where APP is degraded, thus bypassing the cell’s primary Abeta producing (amyloidogenic) pathway. The effects of the PD173955 derivatives on APP metabolism, especially those of 3a, are essentially identical to the effects of 1 and 3a on APP metabolism. Therefore, we believe that the mechanism of action of the derivatives is the same as that noted for 1 and 3a. The kinase inactive analogues of PD173955 can be combined with a BACEi to further reduce production of Aβ peptides. Additional modifications of 3a analogues, especially in rings B and C, and identification of compounds mediating a synergistic effect when combined with a BACEi remain the subject of future studies. We also plan to assess the effects of long CTFs (e.g., C141) and other APP fragments on cellular viability.
Experimental Procedures
Synthesis and characterization as well as screening and evaluation of PD-173955 analogues are described in the Supporting Information.
Statistical Analysis
The data are presented as means ± SEM. Data were analyzed by Student’s t test for single comparison and one-way ANOVA for multiple comparisons, and those showing P value < 0.05 were considered significant.
Acknowledgments
We are thankful to Drs. Sagi Vasudenva Naidu and Saumya Roy for conducting preliminary studies at the Scripps Research Institute, La Jolla, CA and to Ms. Mondana Ghias and Emily Chang for performing ELISA assays of some compounds. We are also thankful to Dr. Victor Bustos of the Rockefeller University for helpful discussions and to the Rockefeller Screening and Proteomics centers and Memorial Sloan Kettering Spectroscopic center for the 1H NMR and MS spectral data of the compounds. Funding support from JPB foundation (Grant #322 and 839) is duly acknowledged.
Glossary
Abbreviations
- Aβ
beta amyloid, amyloid beta
- Abl
Abelson murine leukemia viral oncogene homologue 1
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- BACE1
β-secretase
- BACEi
β-secretase inhibitor
- CTF
carboxy terminal fragment
- ELISA
enzyme-linked immunosorbent assay
- FL
full length
- GS
γ-secretase or gamma secretase
- GSi
γ-secretase inhibitor
- mCPBA
m-chloroperbenzoic acid
- MS
mass spectrometry
- MSD
Meso Scale Discovery
- Pd(PPh3)4
Tetrakistriphenyl-phosphine palladium(II)
- PTLC
preparative thin-layer chromatography.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00213.
Author Contributions
A.S., W.J.N., and S.C.S. designed the experiments, A.S., K.G., E.M., W.J.N., and S.C.S. performed the experiments, and A.S., W.J.N., and S.C.S. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Capdeville R.; Buchdunger E.; Zimmermann J.; Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discovery 2002, 1, 493–502. 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- Wisniewski D.; Lambek C. L.; Liu C.; Strife A.; Veach D. R.; Nagar B.; Young M. A.; Schindler T.; Bornmann W. G.; Bertino J. R.; Kuriyan J.; Clarkson B. Characterization of potent inhibitors of the Bcr-Abl and the c-Kit receptor tyrosine kinases. Cancer Res. 2002, 62, 4244–4255. [PubMed] [Google Scholar]
- Wu D.; Mand M. R.; Veach D. R.; Parker L. L.; Clarkson B.; Kron S. J. A solid-phase Bcr-Abl kinase assay in 96-well hydrogel plates. Anal. Biochem. 2008, 375, 18–26. 10.1016/j.ab.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer W. J.; Dou F.; Cai D.; Veach D.; Jean S.; Li Y.; Bornmann W. G.; Clarkson B.; Xu H.; Greengard P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12444–12449. 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer W. J.; Bettayeb K.; Sinha S. C.; Flajolet M.; Greengard P.; Bustos V. Gleevec shifts APP processing from a beta-cleavage to a nonamyloidogenic cleavage. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 1389–1394. 10.1073/pnas.1620963114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Shepherd J. D.; Murphy M. P.; Golde T. E.; Kayed R.; Metherate R.; Mattson M. P.; Akbari Y.; LaFerla F. M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Portelius E.; Bogdanovic N.; Gustavsson M. K.; Volkmann I.; Brinkmalm G.; Zetterberg H.; Winblad B.; Blennow K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010, 120, 185–193. 10.1007/s00401-010-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu T.; Liu Q.; Chen Y.-X.; Zhao Y.-F.; Li Y.-M. Aβ42 and Aβ40: similarities and differences. J. Pept. Sci. 2015, 21, 522–529. 10.1002/psc.2789. [DOI] [PubMed] [Google Scholar]
- Zhou Z.-d.; Chan C. H.-s.; Ma Q.-h.; Xu X.-h.; Xiao Z.-c.; Tan E.-k. The roles of amyloid precursor protein (APP) in neurogenesis: Implications to pathogenesis and therapy of Alzheimer disease. Cell Adh Migr 2011, 5, 280–292. 10.4161/cam.5.4.16986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B.; Vassar R.; Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99. 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow V. W.; Mattson M. P.; Wong P. C.; Gleichmann M. An overview of APP processing enzymes and products. NeuroMol. Med. 2010, 12, 1–12. 10.1007/s12017-009-8104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coimbra J. R. M.; Marques D. F. F.; Baptista S. J.; Pereira C. M. F.; Moreira P. I.; Dinis T. C. P.; Santos A. E.; Salvador J. A. R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178–178. 10.3389/fchem.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D. B.; May P. C.; Dean R. A.; Siemers E. R. Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2009, 10, 1657–1664. 10.1517/14656560903044982. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Lessons from a failed γ-Secretase Alzheimer trial. Cell 2014, 159, 721–726. 10.1016/j.cell.2014.10.016. [DOI] [PubMed] [Google Scholar]
- Imbimbo B. P.; Giardina G. A. M. G. γ–Secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr. Top. Med. Chem. 2011, 11, 1555–1570. 10.2174/156802611795860942. [DOI] [PubMed] [Google Scholar]
- Mullard A. BACE inhibitor bust in Alzheimer trial. Nat. Rev. Drug Discovery 2017, 16, 155. 10.1038/nrd.2017.45. [DOI] [PubMed] [Google Scholar]
- Cheret C.; Willem M.; Fricker F. R.; Wende H.; Wulf-Goldenberg A.; Tahirovic S.; Nave K. A.; Saftig P.; Haass C.; Garratt A. N.; Bennett D. L.; Birchmeier C. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013, 32, 2015–2028. 10.1038/emboj.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryeom S. W. The cautionary tale of side effects of chronic Notch1 inhibition. J. Clin. Invest. 2011, 121, 508–509. 10.1172/JCI45976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descamps O.; Spilman P.; Zhang Q.; Libeu C. P.; Poksay K.; Gorostiza O.; Campagna J.; Jagodzinska B.; Bredesen D. E.; John V. AβPP-selective BACE inhibitors (ASBI): novel class of therapeutic agents for Alzheimer’s disease. J. Alzheimer's Dis. 2013, 37, 343–355. 10.3233/JAD-130578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W.; Netzer W. J.; Sinha A.; Gindinova K.; Chang E.; Sinha S. C. Development of Gleevec analogues for reducing production of β-amyloid peptides through shifting β-cleavage of amyloid precursor proteins. J. Med. Chem. 2019, 62, 3122–3134. 10.1021/acs.jmedchem.8b02007. [DOI] [PubMed] [Google Scholar]
- Yan R. Physiological functions of the beta-site amyloid precursor protein cleaving enzyme 1 and 2. Front. Mol. Neurosci. 2017, 10, 97. 10.3389/fnmol.2017.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Lu D.; Wei H.-X.; Gu Y.; Selkoe D. J.; Wolfe M. S.; Augelli-Szafran C. E. Part 3: Notch-sparing γ-secretase inhibitors: SAR studies of 2-substituted aminopyridopyrimidinones. Bioorg. Med. Chem. Lett. 2016, 26, 2138–2141. 10.1016/j.bmcl.2016.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B.; Bornmann W. G.; Pellicena P.; Schindler T.; Veach D. R.; Miller W. T.; Clarkson B.; Kuriyan J. Crystal Structures of the Kinase Domain of c-Abl in Complex with the Small Molecule Inhibitors PD173955 and Imatinib (STI-571). Cancer Res. 2002, 62, 4236–4243. [PubMed] [Google Scholar]
- Klutchko S. R.; Hamby J. M.; Boschelli D. H.; Wu Z.; Kraker A. J.; Amar A. M.; Hartl B. G.; Shen C.; Klohs W. D.; Steinkampf R. W.; Driscoll D. L.; Nelson J. M.; Elliott W. L.; Roberts B. J.; Stoner C. L.; Vincent P. W.; Dykes D. J.; Panek R. L.; Lu G. H.; Major T. C.; Dahring T. K.; Hallak H.; Bradford L. A.; Showalter H. D. H.; Doherty A. M. 2-Substituted aminopyrido[2,3-d]pyrimidin-7(8H)-ones. Structure–activity relationships against selected tyrosine kinases and in vitro and in vivo anticancer activity. J. Med. Chem. 1998, 41, 3276–3292. 10.1021/jm9802259. [DOI] [PubMed] [Google Scholar]
- Palmer B. D.; Smaill J. B.; Rewcastle G. W.; Dobrusin E. M.; Kraker A.; Moore C. W.; Steinkampf R. W.; Denny W. A. Structure–activity relationships for 2-anilino-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorg. Med. Chem. Lett. 2005, 15, 1931–1935. 10.1016/j.bmcl.2005.01.079. [DOI] [PubMed] [Google Scholar]
- Maluenda I.; Navarro O. Recent developments in the Suzuki-Miyaura reaction: 2010–2014. Molecules 2015, 20, 7528–7557. 10.3390/molecules20057528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl P.; Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 2009, 1, 8. 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jester B. W.; Cox K. J.; Gaj A.; Shomin C. D.; Porter J. R.; Ghosh I. A coiled-coil enabled split-luciferase three-hybrid system: applied toward profiling inhibitors of protein kinases. J. Am. Chem. Soc. 2010, 132, 11727–11735. 10.1021/ja104491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. L.; Vassar R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. 10.1074/jbc.R800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R.; Knopman D. S.; Jagust W. J.; Shaw L. M.; Aisen P. S.; Weiner M. W.; Petersen R. C.; Trojanowski J. Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.