

Abstract

C-terminal Src kinase (CSK) functions as a negative regulator of T cell activation through inhibitory phosphorylation of LCK, so inhibitors of CSK are of interest as potential immuno-oncology agents. Screening of an internal kinase inhibitor collection identified pyridazinone lead 1, and a series of modifications led to optimized compound 13. Compound 13 showed potent activity in biochemical and cellular assays in vitro and demonstrated the ability to increase T cell proliferation induced by T cell receptor signaling. Compound 13 gave extended exposure in mice upon oral dosing and produced a functional response (decrease in LCK phosphorylation) in mouse spleens at 6 h post dose.
Keywords: CSK inhibitor, kinase inhibitor, immuno-oncology, Metabolite ID
The development of antibodies targeting CTLA-4, PD-1, and PD-L1 to activate the immune system holds great promise for the treatment of cancer. Although these treatments offer substantial therapeutic benefit, as reflected by their recent approval for the treatment of melanoma, nonsmall cell lung cancer, renal cell carcinoma, and a number of other indications, there remains a population of patients that do not respond to currently available immuno-oncology agents. There is consequently a critical need for additional agents to help extend the benefits of immunotherapy to additional patients and indications.1−3
In T cells, the SRC-family kinase LCK plays a key role in initiating the proximal T cell receptor pathway by phosphorylating the ξ and CD3 chains of the T cell receptor, as well as downstream kinases such as ZAP-70.4 LCK is in turn negatively regulated by phosphorylation on Tyr505 by c-terminal SRC kinase (CSK), which causes LCK to adopt its closed, inactive conformation.5−7 Inhibition of CSK could therefore augment T cell activation in response to antigen recognition by the T cell receptor by retaining LCK in an activated form. Indeed, siRNA knockdown of CSK in primary T cells and Jurkat cells increases LCK signaling and response to T cell receptor stimulation.8 Studies in transgenic mice expressing a variant of CSK engineered to be sensitive to chemical inhibition have demonstrated that this inhibition increases the response of T cells to weak antigens.9 In addition, both CSK and LCK have been shown to associate with PD-1, and PD-1 activation has been shown to reduce ZAP-70 phosphorylation, indicating that CSK inhibition could synergize with anti-PD-1/PD-L1 therapy.10,11 To date, there have been no reports of potent small molecule CSK inhibitors.12 The development of suitable tool molecules to evaluate the potential to activate T cells through CSK inhibition is therefore of great interest.
Screening of an internal kinase inhibitor collection identified compound 1 (Figure 1) as a starting point for optimization. Despite its modest potency in a CSK HTRF binding assay (Table 1), this compound increased ZAP-70 phosphorylation in Jurkat cells and had no measurable LCK activity. As the ultimate aim of inhibiting CSK was the enhancement of LCK activity, selectivity for inhibition of CSK over LCK was considered highly important. Although the two proteins are only 44% identical in their kinase domains, only two of the residues facing the ATP-binding pocket differ (Leu251 and Ala381 in Lck), complicating attempts to obtain selective inhibitors. The initial goals for optimization were to improve potency in the binding and cellular assays and to improve the low metabolic stability in human and mouse liver microsomes, while maintaining selectivity against LCK.
Figure 1.

Structures of CSK inhibitors 1–14.
Table 1. Characterization of CSK Inhibitors 1–6a,b.
| Cpd | CSK IC50 (nM) | LCK IC50 (nM) | ZAP-70 EC50 (nM) (Ymax) | HLM/MsLM %Rem. |
|---|---|---|---|---|
| 1 | 5600 | >50000 | 7900(73%) | 2/33 |
| 2 | 79 | 24000 | 5700(180%) | 65/15 |
| 3 | 80 | 31000 | 4050(350%) | 68/9 |
| 4 | 70 | 43000 | 2000(350%) | 53/78 |
| 5 | 430 | 34000 | 2700(280%) | 72/1 |
| 6 | 8 | 26000 | 420(270%) | 61/59 |
For assay conditions and replicate numbers, see the Supporting Information
HLM: Human liver microsomes. MsLM: Mouse liver microsomes.%Rem: Percent remaining at 10 min.
Since structural information regarding the binding of 1 to CSK was not available at the outset of the project, initial optimization efforts focused on introducing conformational biases as a means to enhance potency. The azetidinylmethyl group in 1 was replaced with a piperidine (compound 2) based on the hypothesis that this would reduce conformational flexibility and favor the necessary orientation of the urea group. This resulted in a substantial increase in CSK potency and improvement in human metabolic stability, although the increase in cellular potency was modest and mouse metabolic stability remained low.
An attempt to bias the conformation of the biaryl junction to favor a nonplanar orientation by introduction of a methyl group at C4′′ of the pyridazinone (3) resulted in a similar profile, as did replacement of the C3′ chloro substituent on the phenyl ring with a larger ethyl group (4). Movement of the C4′′ methyl to C5′′ (5) caused a slight drop in potency; incorporation of methyl groups at both positions (6) gave a moderate increase in both binding and cellular potency. Interestingly, compounds containing both the C3′ ethyl and C4′′ methyl groups (4 and 6) showed substantially increased mouse metabolic stability, whereas compounds containing only one of the two groups (3 and 5) did not.
Unfortunately, efforts to further increase potency by straightforward modification of compound 6 proved unfruitful, so attempts were made to replace the pyridazinone with a series of other groups that had the potential to maintain a similar pattern of hydrogen bonding interactions. This approach was validated by compound 7, which was designed based on the hypothesis that N1′′ and the C2′′ hydrogen of the pyrazolopyridine would occupy a similar location to the C6′′ carbonyl and N1′′ hydrogen of pyridazinone 6. This compound had an IC50 value of 5 nM in the CSK HTRF assay and EC50 of 49 nM in the cellular ZAP-70 assay (Table 2); since compounds in this series often had IC50 values below 3 nM in the CSK HTRF assay (the lower limit of the assay), they were also tested in a Caliper assay to give a more precise measurement of their potency.
Table 2. Characterization of CSK Inhibitors 7–14a,b.
| CSK IC50 (nM) |
|||||
|---|---|---|---|---|---|
| Cpd | HTRF | Caliper | LCK IC50 (nM) | ZAP-70 EC50 (nM) (Ymax) | HLM/MsLM %Rem |
| 7 | 5 | 4 | 300 | 49(250%) | 84/24 |
| 8 | <3 | 4 | 120 | 42(190%) | 72/10 |
| 9 | 26 | 21 | 3100 | 1400(240%) | 71/21 |
| 10 | <3 | 2 | 26 | 28(220%) | 80/2 |
| 11 | 42 | 13 | 42 | >20000 | NT/59 |
| 12 | <3 | 4 | 230 | 88(370%) | 85/9 |
| 13 | <3 | 4 | 260 | 41(360%) | 91/100 |
| 14 | 4 | 6 | 110 | 56(210%) | 85/99 |
For assay conditions and replicate numbers, see the Supporting Information
NT: not tested.
Investigation of the C3′ chloro substituent revealed that replacement with a methyl group (compound 8) maintained potency, while its removal (9) resulted in a decrease in both biochemical and cellular potency. This result suggested that this group plays an important role through direct interaction with CSK and/or influence on the conformation of the C4′ amide bond. An attempt to improve potency by introduction of a methoxy group at C6′′ (10) had minimal effect on CSK potency but did cause an undesired increase in LCK potency. Despite this, compound 10 maintained activity in the cellular assay, indicating that even a relatively low level of selectivity for CSK over LCK may be sufficient in a cellular context.
Despite the exciting levels of potency shown by these compounds, their poor mouse metabolic stability prevented their use in animal studies. In an attempt to remove potential sites of metabolism on the substituted piperidine, compound 11, in which this substituent has been truncated to a methyl group, was prepared. Although 11 did show an increase in mouse metabolic stability, it also had lower CSK potency and did not show cellular activity.
In order to ascertain the location of its metabolic vulnerabilities, metabolism of compound 8 by mouse liver microsomes was studied and oxidation of the tert-butyl group was identified as the sole metabolite (Figure 2).13
Figure 2.

Sites of metabolism of compounds 8 and 12.
This knowledge spurred attempts to block metabolism of the tert-butyl group by replacing it with a series of groups containing polar and electron-withdrawing substituents. Although this yielded compounds that maintained the high level of potency shown by 8, these compounds did not show the expected increase in metabolic stability, as typified by cyano amide 12. Analysis of the metabolism of compound 12 revealed a new site of metabolism, oxidation of the indazole ring. Attempts were made to block this by fluorination of the indazole ring; the C-5 fluoro derivative 13 showed a dramatic improvement in metabolic stability combined with good biochemical and cellular potency. Interestingly, compound 14, which contains the fluoroindazole but maintains the tert-butyl urea that had proved to be metabolically labile in other analogs, also showed improved metabolic stability. This suggests that the role of the fluorine on the indazole extends beyond simply preventing oxidation of the indazole through local steric or electronic effects, possibly by creating an unfavorable interaction with the metabolizing enzyme.
In an attempt to understand the binding mode of these chemotypes, efforts were made to obtain crystal structures with several compounds. Although no usable crystal structures were obtained with CSK, a structure of 11 in complex with LCK was obtained (Figure 3). The structure reveals that 11 sits in the ATP binding site and makes hydrogen bonds from N1′′ of the pyrazolopyridine ring to the hinge backbone NH of Met319 and from the amide NH at C4′ to the side chain hydroxyl oxygen of Thr316. In addition, the distance between C7′′ of the pyrazolopyridine and the backbone carbonyl oxygen of Met319 is 3.1 Å and the distance between C2′′ of the backbone oxygen of Glu317 is 3.5 Å, indicating the presence of favorable binding interactions between these residues and 11. The amide carbonyl oxygen at C3 of the indazole ring makes a hydrogen bond with the side chain of Lys273.
Figure 3.

X-ray structure of LCK and 11. Hydrogen bonds and favorable interactions are denoted with dashes. The carbons and ribbon representation of LCK are colored green except for the loop between β-strand3 and helix-C which is colored blue. The carbons of 11 are colored pink, oxygen is red, and nitrogen is blue. Residues in the binding pocket that differ between LCK and CSK have red labels. Bottom right: Close-up of the surface of LCK around the indazole. The surface of LCK is colored green except for the loop between the β-strand3 and helix-C which is colored blue.
The C3′ methyl of the phenyl ring points into a small pocket below β-strand 3 and contributes to the nonplanarity of the phenyl ring relative to the amide. This likely explains the decrease in potency observed in compound 9 as compared to 7 and 8. The indazole ring forms an edge-to-face interaction with Phe285 and has a lipophilic interaction with Met280. Comparison of the structure of LCK with 11 bound to the apo structure (Figure 4) shows that 11 binds in an inactive DFG-out conformation, with significant distortion of helix C.
Figure 4.
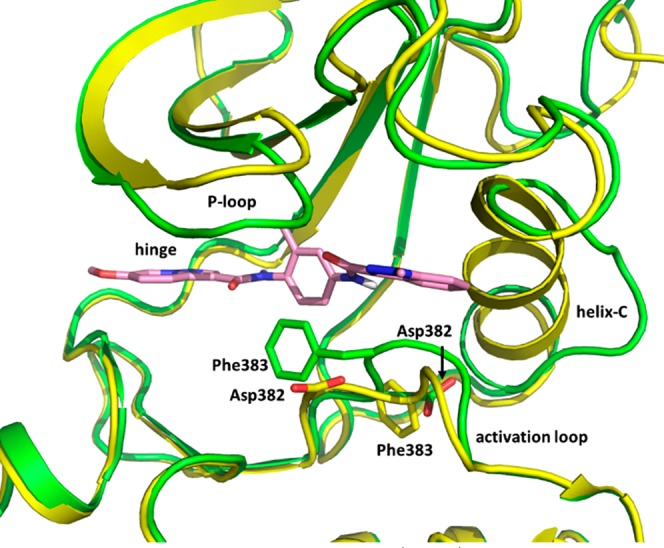
X-ray structures of LCK and 11 (green) superposed with apo LCK (yellow, PDB code 3LCK) highlighting the residues of the DFG motif. The carbons of 11 are colored pink.
It is noteworthy that the N1 methyl group in compound 11 points toward the loop between β-strand 3 and α-helix C. Analysis of the protein surface in this area (Figure 1 inset) shows relatively little space to accommodate substituents larger than this methyl group, and compounds containing a substituted piperidine at this position show increased selectivity for CSK relative to 11. This region of LCK shares little homology with CSK, so this increased selectivity is likely attributable to an increased ability of the corresponding region of CSK to make favorable interactions with substituents larger than methyl. The N1 substituent is not in proximity to Leu251 or Ala381, the two residues in the binding pocket that are different in CSK (corresponding to Ile201 and Ser331, respectively), so the sequence differences in the loop between β-strand 3 and α-helix C are likely the major contributor to selectivity between LCK and CSK.
Illustrative syntheses of compounds 8 and 4 are shown in Schemes 1 and 2. Alkylation of indazole 15 with mesylate 16 followed by ester hydrolysis gave carboxylic acid 17. Acylation of bis-aniline 18 with this acid occurred primarily at the less hindered amine to give 19, which was then acylated a second time with acid 20.
Scheme 1. Synthesis of Compound 8.

Reagents and conditions: (a) 16, Cs2CO3, DMF, 80 °C, 39%; (b) LiOH, MeOH, H2O, quant.; (c) 18, PyBOP, Hünig’s base, DMF, 79%; (d) 20, PyBOP, Hünig’s base, THF, rt to 60 °C, 47%; (e) HCl, dioxane, quant.; (f) tert-butyl isocyanate, Hünig’s base, DMF, 41%.
Scheme 2. Synthesis of Compound 4.

Reagents and conditions: (a) PdCl2(dppf)·DCM, bis(pinacolato)diboron, KOAc, dioxane, 100 °C, 39%; (b) 23, PdCl2(dppf)·DCM, Na2CO3, DMF, H2O, 35%; (c) 17, PyBOP, Hünig’s base, DMF, 67%; (d) HCl, dioxane, quant.; (e) tert-butyl isocyanate, Hünig’s base, DCM, 67%.
Removal of the Boc group and installation of the tert-butyl urea with tert-butyl isocyanate completed the synthesis of compound 8. For the preparation of 4 (Scheme 2), bromide 21 was converted to boronic ester 22, which was then coupled to choloropyridazinone 23 to give 24. Coupling with acid 17 was effected with PyBOP; the choice of coupling reagents was important, as reagents such as HATU had a tendency to form adducts with unprotected pyridazinones such as 24 and its coupled product. Synthesis of 4 was completed by removal of the Boc group and formation of the urea with tert-butyl isocyanate.
Based on their favorable potency and metabolic stability profiles, pyridazinones 4 and 6 and pyrazolopyridines 13 and 14 were subjected to further in vitro profiling (Table 3). All compounds showed high protein binding. Compounds 13 and 14 showed long T1/2 values in the presence of both human and mouse liver microsomes, while the T1/2 of compound 4 was moderate for mouse and short for human, and the T1/2 of compound 6 was low for both species. Pyrazolopyridine 13 was chosen for advancement into mouse PK/PD studies due to its high level of potency and metabolic stability; 4 was chosen to represent the pyridazinone series due to its greater metabolic stability relative to 6.
Table 3. Further Profiling of Compounds 4, 6, 13, and 14a,b.
| Cpd | HLM T1/2 (min) | MsLM T1/2 (min) | Human PB %Free | Mouse PB %Free | FSI(100) |
|---|---|---|---|---|---|
| 4 | 10 | 31 | 0.4% | 0.9% | 5.5 |
| 6 | 14 | 12 | 0.5% | 1.0% | 3.0 |
| 13 | >120 | >120 | <0.1% | 0.3% | 6.7 |
| 14 | >120 | 88 | 0.4% | <0.2% | 8.9 |
PB: protein binding.
FSI(100): percent of 238 kinases tested in an internal panel with an IC50 value within 100-fold of the compound’s CSK HTRF IC50.
At a 100 mg/kg oral dose, compound 4 showed a relatively high initial exposure (Table 4) with a T1/2 of 1.9 h and negligible exposure at 24 h. In contrast, compound 13 had an extended absorption period with a maximum concentration at 7 h, a 5.3 h T1/2, and substantial exposure even at 24 h after dosing. Spleens from the mice were harvested, and the level of LCK Y505 phosphorylation was measured. Mice treated with 4 showed an 87% decrease in LCK phosphorylation compared to vehicle-treated mice at 3 h, when plasma concentration was 8.7 μM. The decrease for mice treated with 13 was 88% at 6 h (close to the Tmax of 7 h). Thus, both 4 and 13 show the anticipated effect of CSK inhibition upon oral dosing in mice.
Table 4. PK and PD Parameters for Compounds 4 and 13a.
| Cpd | Cmax (μM) | Tmax (h) | C24h (μM) | AUC (μM·h) | T1/2 (h) | ΔLCK pY505 |
|---|---|---|---|---|---|---|
| 4 | 17.3 | 0.3 | 0.001 | 51.5 | 1.9 | –87% |
| 13 | 19.3 | 7 | 1.3 | 221.5 | 5.3 | –88% |
ΔLCK pY505: percent change in LCK pY505 compared to vehicle, t = 3 h for 4, t = 6 h for 13. C57bl/6 mice were used.
To test whether compound 13 was able to activate T cells in a context-appropriate manner, an assay was utilized in which Chinese hamster ovary (CHO) cells were mixed with CD4+ T cells.14 Two types of cells were used: parental CHO cells and CHO cells expressing OKT, a cognate antibody for the T cell receptor. These cells were treated with compound 13 and monitored for IL-2 secretion and proliferation. The results from this experiment are summarized in Figure 5. As expected, the parental CHO cells induced a minimal response which was not increased by compound 13. CHO cells expressing OKT induced a stronger response, which was increased in a dose-dependent fashion by 13. At 333 nM of compound 13, IL-2 secretion was increased by 5.7-fold; under the conditions employed, proliferation reached a maximum increase of 2-fold at 37 nM. Taken together, these results confirm the hypothesis that CSK inhibition can enhance the response of T cells to antigen stimulation.
Figure 5.

Top: CD4+ T cell IL-2 secretion in response to 13. Bottom: CD4+ T cell proliferation in response to 13.
In summary, we have developed a series of small molecule inhibitors of CSK that can serve as in vitro and in vivo tools to evaluate the potential of this target as an immuno-oncology therapy. Switching from a pyridazinone to pyrazolopyridine hinge binder gave a substantial increase in cellular potency. Metabolite identification studies enabled the strategic blocking of metabolic soft spots by the exchange of a tert-butyl urea for a cyano amide and fluorination of an indazole, culminating in the discovery of compound 13. Compound 13 reduced inhibitory LCK phosphorylation in vivo upon oral dosing and showed the ability to enhance T cell activation in response to antigen stimulation. Notably, this increase was not observed in the absence of an antigen. These findings support further evaluation of the potential of CSK inhibition to enhance antitumor immune response. The molecules described in this manuscript provide suitable tools to evaluate the relationship between the extent and duration of CSK inhibition and the efficacy and tolerability of treatment in animal models, an important consideration given the toxicity observed in knockout mice.15,16
Acknowledgments
The authors wish to acknowledge the contributions of the CSK Discovery Working Group.
Glossary
Abbreviations
- CHO
Chinese hamster ovary
- CTLA-4
cytotoxic T lymphocyte-associated protein 4
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HTRF
homogeneous time-resolved fluorescence
- LCK
lymphocyte-specific protein tyrosine kinase
- PD
pharmacodynamics
- PD-1
programmed cell death protein 1
- PD-L1
programmed death-ligand 1
- PK
pharmacokinetics
- PyBOP
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- ZAP-70
zeta-chain-associated protein kinase 70
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00354.
Experimental procedures and characterization data for the compounds described (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
This work was funded by Bristol-Myers Squibb Company.
The authors declare the following competing financial interest(s): The authors of this manuscript are employees of Bristol-Myers Squibb.
Supplementary Material
References
- Lonberg N.; Korman A. J. Masterful antibodies: Checkpoint blockade. Cancer Immunol. Res. 2017, 5, 275–281. 10.1158/2326-6066.CIR-17-0057. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Toogood P. L. Small molecule immuno-oncology therapeutic agents. Bioorg. Med. Chem. Lett. 2018, 28, 319–329. 10.1016/j.bmcl.2017.12.044. [DOI] [PubMed] [Google Scholar]
- Thill P. A.; Weiss A.; Chakraborty A. K. Phosphorylation of a tyrosine residue on Zap70 by Lck and its subsequent binding via an SH2 domain may be a key gatekeeper of T cell receptor signaling in vivo. Mol. Cell. Biol. 2016, 36, 2396–2402. 10.1128/MCB.00165-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M.; Nada S.; Yamanishi Y.; Yamamoto T.; Nakagawa H. CSK: a Protein-tyrosine kinase involved in regulation of src family kinases. J. Biol. Chem. 1991, 266, 24249–24252. [PubMed] [Google Scholar]
- Mustelin T.; Tasken K. Positive and negative regulation of T-cell activation through kinases and phosphatases. Biochem. J. 2003, 371, 15–27. 10.1042/bj20021637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M. Regulation of the Src family kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. 10.7150/ijbs.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang T.; Abrahamsen H.; Myklebust S.; Enserink J.; Prydz H.; Mustelin T.; Amarzguioui M.; Tasken K. Knockdown of C-terminal Src kinase by siRNA-mediated RNA interference augments T cell receptor signaling in mature T cells. Eur. J. Immunol. 2004, 34, 2191–2199. 10.1002/eji.200425036. [DOI] [PubMed] [Google Scholar]
- Manz B. N.; Tan Y. X.; Courtney A. H.; Rutaganira F.; Palmer E.; Shokat K. M.; Weiss A. Small molecule inhibition of Csk alters affinity recognition by T cells. eLife 2015, 4, e08088 10.7554/eLife.08088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard K.-A.; Fitz L. J.; Lee J. M.; Benander C.; George J. A.; Wooters J.; Qiu Y.; Jussif J. M.; Carter L. L.; Wood C. R.; Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3 signalosome and downstream signaling to PKC. FEBS Lett. 2004, 574, 37–41. 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- Yokosuka T.; Takamatsu M.; Kobayashi-Imanishi W.; Hashimoto-Tane A.; Azuma M.; Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a report of a CSK inhibitor with an IC50 of 1.9 μM in an ELISA assay, see:; Kilimnik A.; Kostjukova M. N.; Pyatkin I. H.; Pronin A. M.; Strelnikova S. R.; Fedotov Y. A.; Kolesnikov A. V. Novel small-molecule inhibitors of C-terminal Src Kinsase (Csk). Cell. Mol. Biol. Lett. 2003, 8, 588. [Google Scholar]
- Paiva A. A.; Klakouski C.; Li S.; Johnson B. M.; Shu Y.-Z.; Josephs J.; Zvyaga T.; Zamora I.; Shou W. Z. Development, optimization, and implementation of a centralized metabolic soft spot assay. Bioanalysis 2017, 9, 541–552. 10.4155/bio-2016-0299. [DOI] [PubMed] [Google Scholar]
- For application of a similar assay, see: Englehardt J. J.; Selby M. J.; Korman A. J.; Feingersh M. D.; Stevens B. L.. Anti-Icos agonist antibodies and uses thereof. WO 2018/187613 A2, October 11, 2018.
- For additional studies on a different series of CSK inhibitors from this group, see: Wang C.; Cao C.; Berman-Booty L.; Eraslan R.; Sanjuan M.; Vite G.; Hunt J.; Fink B.; Wee S.. Targeting CSK kinase activity to enhance antitumor immunity [abstract]. Proceedings of the AACR Special Conference on Tumor Immunology and Immunotherapy; 2017. Oct 1–4; AACR, Boston, MA. Philadelphia (PA).; Cancer Immunol. Res. 2018, 6 ( (9 Suppl), ), Abstract nr B02. [Google Scholar]
- Berman-Booty L. D.; Eraslan R.; Hanumegowda U.; Cantor G. H.; Bounous D. I.; Janovitz E. B.; Jones B. K.; Buiakova O.; Hayward M.; Wee S. Systemic loss of C-terminal Src kinase expression elicits spontaneous suppurative inflammation in conditional knockout mice. Vet. Pathol. 2018, 55, 331–340. 10.1177/0300985817747330. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.