

Abstract
Self-assembled nanofibers are ubiquitous in nature and serve as inspiration for the design of supramolecular hydrogels. A multicomponent approach offers the possibility of enhancing the tunability and functionality of this class of materials. We report on the synergistic multicomponent self-assembly involving a peptide amphiphile (PA) and a 1,3:2,4-dibenzylidene-d-sorbitol (DBS) gelator to generate hydrogels with tunable nanoscale morphology, improved stiffness, enhanced self-healing, and stability to enzymatic degradation. Using induced circular dichroism of Thioflavin T (ThT), electron microscopy, small-angle neutron scattering, and molecular dynamics approaches, we confirm that the PA undergoes self-sorting, while the DBS gelator acts as an additive modifier for the PA nanofibers. The supramolecular interactions between the PA and DBS gelators result in improved bulk properties and cytocompatibility of the two-component hydrogels as compared to those of the single-component systems. The tunable mechanical properties, self-healing ability, resistance to proteolysis, and biocompatibility of the hydrogels suggest future opportunities for the hydrogels as scaffolds for tissue engineering and drug delivery vehicles.
Introduction
Multicomponent supramolecular self-assembly is ubiquitous in natural processes leading to the formation of highly ordered and complex architectures.1 The functionality of many biomacromolecules such as cytoskeleton actin and actin-binding proteins found in living organisms depends on the coassembly of multiple building blocks into one-dimensional nanostructures.2 These supramolecular assemblies result from cooperative and synergistic noncovalent interactions, which provide adaptive, dynamic, and responsive properties.
Multicomponent self-assembly is currently the focus of fundamental research aimed at dissecting the code embedded within natural supramolecular architectures3−5 to engineer innovative materials based on predictive molecular interactions.6−8 Such design strategies have been developed using well-defined molecular building blocks including β-sheet fibrillizing peptides, peptide amphiphiles (PAs), engineered polypeptides, and low-molecular-weight species.8−10 However, many of these artificial nanostructures are homotypic, consisting of one class of building block and thereby limiting the level of structural and functional complexities, diversities, and tunabilities. In contrast, multicomponent self-assembly offers the possibility to generate a wider range of more complex structures, enhance modularity, and provide spatiotemporal control of self-assembly.2,11 This approach has been used to harness synergistic properties as a result of using two different interacting molecular building blocks such as peptide–peptide,12−14 protein–peptide,15−17 PA–polysaccharide,18 protein–protein,19,20 and protein/peptide–DNA.21 The structures and properties emerging from these systems are opening new opportunities for the rational design of more complex and functional materials.6
Multicomponent self-assembly is a versatile strategy to design supramolecular hydrogels with molecular complexity, enhanced interfacial areas between nanofibers, tunable mechanical properties, in situ creation of new materials, and diverse morphology.22 These properties make such hydrogels suitable for a wide range of applications in drug delivery,23 tissue engineering,24 nanoreactor design, and optoelectronic materials.25 Specifically, multicomponent gels can have advantages compared to individual component systems. For example, multicomponent gels can exhibit optoelectronic properties that cannot be accessed using the single-building blocks.26 There are also examples where cellular behaviors can be improved using multicomponent hydrogels.27,28 It is also possible to prepare systems with unusual behavior such as delayed gelation3 or two-stage rheological control.29,30 Moreover, multicomponent self-assembly breaks the molecular homogeneity imposed by traditional single-building block hydrogels.
PAs represent a fascinating class of self-assembling building blocks for designing bioactive hydrogels.31−34 These amphiphilic molecules consist of at least one lipid chain attached to a peptide backbone and are able to self-assemble into high-aspect-ratio cylindrical nanofibers defined by the hydrophobic collapse of the alkyl region and electrostatic interactions between the charged head groups.35 PAs have been used in supramolecular coassembling systems with oppositely charged and structurally distinct molecules to develop sac-like structures,18 dynamic tubular materials,16 and hydrogels.17 However, the combination of PAs and other gelators with distinct molecular structures exhibiting similar charge has not been explored. Therefore, supramolecular self-assembly of a well-known PA molecule with another molecule with a well-characterized self-assembly mechanism is of considerable interest in designing a new breed of multicomponent hydrogels.
1,3(R):2,4(S)-dibenzylidene-d-sorbitol (DBS) is a well-known sugar-based low-molecular-weight gelator (LMWG) with a high capacity to self-assemble into fibrillar networks in organic solvents and water.36 The effectiveness of DBS as a gelator is due to intermolecular hydrogen bond interactions between the sugar units combined with π–π stacking and solvophobic interactions between the aromatic rings, which characterize its ‘butterfly-like’’ wing structure.37 In the past century, DBS has been widely used in industrial applications as a cosmetic additive, in dental composites, and as a polymer nucleation/clarification agent.37 Recently, Smith and colleagues reported the development of DBS derivatives including DBS-COOH38 and DBS-CONHNH239 that are capable of self-assembly in water into self-supported hydrogels by pH-switching and heat–cool transitions, respectively. Using molecular dynamics (MD) simulations, Knani and Alperstein determined that the carboxylic acid and acylhydrazone groups on these molecules play an important role in modifying both their solubility and capacity to self-assemble into fibrillar networks in aqueous systems.40 Hydrogels based on DBS-COOH and DBS-CONHNH2 have potential applications in environmental cleanup,39,41 drug encapsulation and delivery,42 electrocatalysis,43 cell culture,44 and as nanoreactors.45 Furthermore, these gelators have been hybridized with polymers to produce robust38,46 and multidomain,30,47 hydrogels, and more recently, DBS-COOH/DBS-CONHNH2 coassembled hydrogels48 but their combination with LMWGs of other types to design multicomponent hydrogels has not been investigated.
The combination of LMWGs from different classes remains relatively rare and offers considerable potential for the emergence of new functions, which can be exploited to develop materials with innovative properties. In this study, we report on the supramolecular design of multicomponent hydrogels based on a well-known PA molecule and DBS-COOH using a slow acidification protocol to fabricate hydrogels with emergent properties. The resulting hydrogels exhibit tunable stiffness, self-healing capacity, enhanced stability to enzymatic degradation, and tunable bioactivity. To the best of our knowledge, this is the first example of these two LMWGs being combined.
Materials and Methods
Preparation of Hydrogels
Both PA-E3(32) and DBS-COOH(38) were synthesized as previously reported. For single-component hydrogels, PA-E3 and DBS-COOH solutions (10 mM) were individually prepared in phosphate buffer saline (pH 7.4, 200 μL), and aliquots of NaOH (aq) (0.5 M, 10 μL) were added to dissolve the gelators (pH = 9). The solutions were then transferred to vials containing glucono-δ-lactone (GdL) (4 mg), followed by a thorough shaking to dissolve the GdL granules. The vials were left overnight for gelation to occur. The multicomponent hydrogels were prepared by mixing aliquot solutions of PA-E3 and DBS-COOH at various molar ratios expressed as percentages (100/0, 80/20, 50/50, 20/80, and 0/100) such that the final total concentration of the mixtures was 10 mM in each case. The solutions were then transferred to vials containing GdL, shaken, and left overnight for gelation to occur.
Thioflavin T (ThT) Fluorescence Assay
Aliquots of solutions (0.4 mM, 20 μL) of PA-E3, DBS-COOH, and PA-E3/DBS-COOH mixtures were added to GdL (0.2 mg). Aliquots of ThT (0.4 mM, 20 μL) were then added to the gelator solutions. Each sample was mixed by pipetting up and down three times, loaded into a 20 μL sample flow cell, sealed with paraffin wax, and immediately transferred onto an inverted laser scanning confocal microscope (LSCM) (Leica laser scanning confocal TCS SP2) with a 63-oil immersion objective, using excitation and emission wavelengths of 458 and 468 nm, respectively. Images of the self-assembled nanofibers were acquired after incubating the sols for 6 h. The flow cells were kept humid in Petri dishes to prevent evaporation. The hydrogels prepared using an identical protocol but without the ThT were used as control and did not exhibit any fluorescent signal.
Circular Dichroism (CD) Measurement for ThT Interactions
Circular dichroism (CD) was measured with a Chirascan circular dichroism spectrometer (Applied Photophysic Limited, U.K.) using a quartz cell with a 1 mm path length and the following parameters: data pitch, 0.5 nm; scanning mode, continuous; scanning speed, 100 nm/min; bandwidth, 2 nm; and accumulation, 5. All CD data are presented as ellipticity and recorded in millidegree (mdeg). Samples for CD measurements were prepared by adding GdL (0.4 mg/mL) to PA-E3, DBS-COOH, and equimolar PA-E3/DBS-COOH solutions (200 μM). Then, an aliquot of ThT (200 μM) was added to the gelator solutions, and the mixture was loaded into a quartz cell. CD spectra were obtained at a 2 min interval from 190 to 500 nm at a speed of 50 nm/min.
Transmission Electron Microscopy (TEM) Characterization
Solutions (1 mM, 40 μL) of PA-E3, DBS-COOH, and PA-E3/DBS-COOH mixtures were added to GdL (0.2 mg), shaken thoroughly, and incubated for 10 h. Samples were then mounted on holey carbon-coated copper grids that were preplasma-treated (Agar Scientific, Stansted, U.K.). The grids were immersed in ultrapure water for 30 s to remove excess and unadsorbed samples. The grids were then immersed in a solution of uranyl acetate (2%) for 30 s, and excess uranyl acetate solution was removed using filter paper. Grids were allowed to dry in a desiccator for 24 h at room temperature. Images were acquired on a JEOL 1230 transmission electron microscope fitted with a Morada CCD camera and operated at an acceleration voltage of 80 kV.
Atomic Force Microscopy (AFM) Characterization
Solutions of PA-E3 (1 mM, 40 μL, pH 11), DBS-COOH (1 mM, 40 μL, pH 11), and PA-E3/DBS-COOH mixtures were mixed with GdL (0.4 mg), and the mixtures were dropped onto freshly cleaved mica surface so that they self-assemble on the substrate. The samples were air-dried at room temperature for 24 h. AFM characterization of nanofibers was performed on an Asylum Research MFP-3D atomic force microscope (Santa Barbara, CA) without treatment. Ultrasharp silicon nitride tips (NSC15 noncontact silicon cantilevers, MikroMasch, Spain) were used. Typical scan settings involved the use of an applied piezo deflection voltage of 0.6–0.7 V at a scan rate of 0.7 Hz. All images were processed using Igor Pro software.
Small-Angle Neutron Scattering (SANS) Characterization
The required concentration (10 mM) of PA-E3 and DBS-COOH and various mixtures of PA-E3 and DBS-COOH were prepared in D2O (400 μL). The suspensions were dissolved by adding aliquots of NaOD (0.5 M, 10 μL), and the solutions were mixed with GdL (5 mg). After a thorough shaking until GdL granules fully dissolved, the mixtures were transferred into 1 mm path length UV spectrophotometer grade quartz cuvettes (Hellma). Gelation of the solutions was allowed to proceed overnight. Small-angle neutron scattering (SANS) measurements were performed on the fixed-geometry, time-of-flight LOQ diffractometer (ISIS Neutron and Muon Source, Oxfordshire, U.K.). A white beam of radiation with neutron wavelengths spanning 2.2–10 Å was enabled access to Q [Q = 4π sin(θ/2)/λ] range of 0.004–0.4 Å–1 with a fixed-sample detector distance of 4.1 m. The cuvettes were mounted in aluminum holders. The time taken for each measurement was approximately 30 min. All scattering data were normalized for the sample transmission, the background was corrected using a quartz cell filled with D2O, and the linearity and efficiency of the detector response were corrected using the instrument-specific software. Data were fitted using the appropriate models between 0.00485⟨Q⟩0.49132 Å–1 for consistency. The scattering length density (SLD) for D2O was set to 6.3 × 10–6 Å–2; SLD for scattering structures was set to 2.197 × 10–6 Å–2 for DBS-COOH only and 1.31 × 10–6 Å–2 for PA-E3 only. SLDs for mixtures of PA-E3 and DBS-COOH were based on compositional average—these values estimated using the NIST calculator with a (assumed) density of 1.3. Data for the PA-E3 hydrogel fit flexible elliptical cylinder model, while a power law was used to fit the data for DBS-COOH hydrogels, which suggests that the structures are large and outside the effective range of the SANS. For the PA-E3/DBS-COOH admixtures, a combination of the flexible elliptical cylinder model with a power law was used to fully fit the data. Details of the parameters for all of the fittings are presented in Table S1.
Rheological Kinetics
Rheological measurements were performed using a Discovery Hybrid Rheometer, Rheo-DHR3 (TA Instruments). All data were collected at 25 °C. For each sample, immediately following the addition of GdL (4 mg/mL) to solutions of the gelators (10 mM, pH 9), 100 μL of the mixture was added to the center of the bottom plate, and the upper geometry (parallel top plate with a 20 mm diameter) was lowered to a gap of 50 μm. A time sweep was performed where the storage and loss moduli were monitored for 3 h at a constant frequency of 1 Hz and a 0.5% strain at 25 °C. Light mineral oil was applied to the perimeter of the parallel plate to prevent evaporation of water over the course of the experiment.
Characterization of Nanofiber Growth by TEM
Solutions (0.5 mM, 100 μL) of PA-E3, DBS-COOH, and PA-E3/DBS-COOH mixtures were added to GdL (0.2 mg/mL) and shaken thoroughly. An aliquot (50 μL) of each sample was added onto a flat sheet of parafilm. Holey carbon-coated copper grids (Agar Scientific, Stansted, U.K.) that were preplasma-treated were carefully placed on top of each drop. The samples were kept in a temperature and humidity chamber to prevent evaporation. The grids were taken off at various time intervals, excess solution was removed using filter paper, and the grids were then immersed in a solution of uranyl acetate (2%) for 30 s and excess uranyl acetate solution removed using filter paper. Grids were allowed to dry in a desiccator for 24 h at room temperature. Images were acquired on a JEOL 1230 transmission electron microscope fitted with a Morada CCD camera and operated at an acceleration voltage of 80 kV.
Molecular Dynamics Simulations
The simulation was conducted using Material Studio 8.0 software (by Biovia). The dynamic atomistic simulation was performed according to the following steps:
Step 1: Building Cubic Cells
Five simulation cubic boxes (about 20 Å edge) were constructed using amorphous cell module for each of the following: DBS-COOH, PA-E3, and their mixtures at various ratios: 1:5, 1:1, and 5:1 (DBS-COOH/PA-E3).
Step 2: Molecular Dynamics Simulation
Dynamics simulation was performed at 300 K. The cells were subjected to 100 000 dynamic steps of 1 fs each at constant mole number, pressure, and temperature (NPT ensemble) to determine their density. This stage was followed by a constant mole number, volume, and temperature (NVT ensemble) refinement stage of 100 000 dynamic steps and a data collection stage of additional 400 000 NVT steps. All MD simulations were conducted using Forcite module with COMPASS II force field. The electrostatic term was considered using Ewald and the van der Waals term using atom-based summation methods with an accuracy of 10–3 kcal/mol. The repulsive cutoff for van der Waals term was chosen as 12.5 Å. For NPT molecular dynamics simulations, Nose thermostat and Berendsen barostat were chosen.
Step 3: Analysis
The resulting dynamic trajectories were analyzed using Forcite module analysis tools. The following properties were calculated:
Cohesive Energy Density (CED) and Solubility Parameter
Cohesive energy is the energy required to break the interactions between molecules. Generally, it is measured as the heat of vaporization of a liquid. The cohesive energy density (CED) corresponds to the cohesive energy per unit volume. The solubility parameter is the square root of the CED and is a measure of the ability of materials to dissolve each other.
Enthalpy of Mixing
CED values can be used to calculate the enthalpy of mixing (per unit volume) using the following equation
| 1 |
where Ecoh is the CED of constituents a, b, or the blend (ab); and Φa and Φb are the volume fractions of the two components in the blended system.
The enthalpy of mixing is the released or taken-up heat upon mixing of two substances. Generally, a negative Gibbs free energy of mixing is a necessary condition for substances to form a miscible phase. The entropy change during mixing may be negligible when polymers are involved, and miscibility can be determined from the enthalpy change of mixing instead of Gibbs free energy. An exothermic enthalpy of mixing is indicative of a miscible blend.
Radial Distribution Function (RDF)
Radial distribution function (also referred to as pair correlation function) gives a measure of the probability that, given the presence of an atom at the origin of an arbitrary reference frame, there will be an atom with its center located in a spherical shell of infinitesimal thickness at a distance r from the reference atom. RDF may serve as a tool to estimate intermolecular interactions like hydrogen bonding.
Amplitude Sweeps, Critical Strain, and Self-Recovery of Hydrogels
A parallel plate geometry (parallel top plate with an 8 mm diameter) was used with a 150 μm gap distance to perform frequency and amplitude sweeps. PA-E3 (10 mM), DBS-COOH (10 mM), and various stoichiometric mixtures of PA-E3/DBS-COOH hydrogels were prepared by adding GdL (10 mg/mL) to the gelator solutions. The hydrogels were left to form overnight before the measurements. Frequency scans were performed from 1 to 10 Hz under a strain of 0.5%. At this strain, the frequency sweeps were performed within the linear viscoelastic region, where the storage modulus (G′) and loss modulus (G″) are independent of the strain amplitude. Critical strain values were determined from the point where G′ values start to decrease with strain. The recovery properties of the hydrogels in response to applied shear forces were investigated using the following procedure: 0.1% (100 s), 100% (200 s), 0.1% (200 s), 100% (200 s), (400 s) with the applied shear force, expressed in terms of strain (%) and duration (s) in parentheses. For the strain recovery, 10 mM of PA-E3 was mixed with 10 mM of DBS-COOH to prepare PA-E3/DBS-COOH hydrogels.
Proteolytic Stability of the Hydrogels
Hydrogels of PA-E3 (10 mM), DBS-COOH (10 mM), and PA-E3 (5 mM)/DBS-COOH (5 mM) were prepared as previously described above. The hydrogels were immersed in phosphate-buffered saline (PBS) for 1 h and then incubated at 37 °C for 5 days in proteinase K solutions (5 mg/mL in PBS). Hydrogels were then prepared for scanning electron microscopy (SEM) by freeze-drying. SEM micrographs of the xerogels were acquired on Inspect F50 (FEI Comp, the Netherlands) after sputter-coating with gold (10 nm thick). Also, mass spectra of the mixtures of 0.1 mM PA-E3/proteinase K (5 mg/mL) and PA-E3/DBS-COOH/proteinase K (5 mg/mL) were acquired on liquid chromatography–mass spectrometer, comprising a 1100 Series LC and SL Ion Trap MSD (Agilent Technologies, U.K.). Weight loss measurements were performed by incubating each hydrogel in 500 mL of proteinase K solution (5 mg/mL) or phosphate buffer saline (PBS 1×) at 37 °C at various time intervals. The solutions with or without the enzyme were replaced with the freshly prepared solutions after each weight measurement. Then, the weights of hydrogels were recorded to determine the weight loss compared to the initial weights of the hydrogel. We computed the weight loss using equation (Wf – Wi)/Wi × 100, where Wi and Wf represent initial and final hydrogel weights, respectively.
Cellular Behaviors on PA-E3, DBS-COOH, and PA-E3/DBS-COOH Hydrogels
PA-E3, DBS-COOH, and PA-E3/DBS-COOH hydrogels were prepared as previously discussed above. The hydrogels were cross-linked with CaCl2 (50 mM) and washed with PBS until the hydrogels became neutral to pH strips (pH 7). Human-adipose-derived stem cells (hADSCs, 5000 cells/mL) were cultured on the hydrogels and incubated for 4 days at 37 °C and 5% CO2. Using a live/dead assay kit (Life Technologies, U.K.), we investigated biocompatibility of the hydrogels. The assay was performed by incubating the hydrogels with calcein AM (10 mM) and ethidium homodimer-1 (1 mM) for 30 min. Imaging was performed on an inverted confocal laser scanning microscope (CLSM, Leica, Germany).
Results and Discussion
Self-Assembly and Formation of Hydrogels
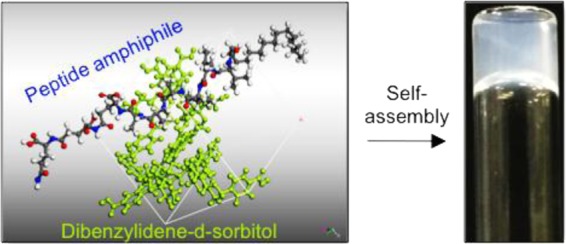
The PA used in this study is the negatively charged peptide CH3-(CH2)14-CONH-VVVAAAEEE-CONH2 (PA-E3), palmitoylated at the N-terminus (Figure 1a). This PA is known to assemble into micrometer-long nanofibers by calcium ion coordination.31,49 Conversely, DBS-COOH (Figure 1a) has been shown to assemble into nanofibers that are microns in length as a result of pH-switching.38 Aqueous solutions (10 mM) of gelator DBS-COOH and gelator PA-E3 were prepared, and the aliquots of each were mixed to provide a series of solutions with various stoichiometric ratios of PA-E3 to DBS-COOH (100/0, 80/20, 50/50, 20/80, and 0/100%) such that a constant total concentration (10 mM) was maintained in all mixtures. Stable hydrogels were formed by slow acidification of the basic solutions by adding GdL (10 mg/mL). GdL undergoes base-catalyzed hydrolysis to gluconic acid as previously described. The slow hydrolysis allows significantly more controlled and homogeneous gels to be formed as compared to the use of mineral acids.50 Consequently, the gradual decrease in pH below the pKa of the terminal carboxylic acid (pKa ∼4–5) of DBS-COOH and PA-E3(30,51) allows the slow protonation of the carboxylate groups (Figure 1b). This protonation directs the gradual self-assembly of both PA-E3 and DBS-COOH over a period of 4 h, leading to the kinetically controlled formation of homogeneous self-supported hydrogels (Figure 1c).
Figure 1.

Molecular information and sol-gel phase transition. (a) Molecular structure of PA-E3 and conformational perspective of the molecular structure of DBS-COOH (the numbering indicates alcohol groups on the sugar backbone). (b) Schematic representation of supramolecular self-sorting of PA-E3 and DBS-COOH by pH-switching. (c) Photographs of hydrogels of PA-E3, DBS-COOH, and PA-E3/DBS-COOH mixtures. Laser scanning confocal micrographs of nanofibers of (d) PA-E3, (e) PA-E3/DBS-COOH (4:1), (f) PA-E3/DBS-COOH (1:1), (g) PA-E3/DBS-COOH (1:4), and (h) DBS-COOH. Induced circular dichroism spectra of ThT during the self-assembly of (i) PA-E3, (j) DBS-COOH, and (k) PA-E3/DBS-COOH (1:1). Arrows indicate increasing CD intensity as a function of gelator self-assembly. Data acquired at 2 min interval.
Characterization of Assembly
Hydrogels were formed for all tested materials including PA-E3, DBS-COOH, and their combinations. The hydrogels of PA-E3 alone were optically transparent, while those of DBS-COOH were translucent. Moreover, there was an increasing degree of translucency with an increasing concentration of DBS-COOH in the two-component hydrogels, suggesting an increasing DBS-COOH nanofiber density in the materials. At the nanoscale, fluorescent microscopy using ThT was used to further confirm the self-assembly of PA-E3, DBS-COOH, and their mixtures. As revealed by LSCM, nanofibers of individually assembled PA-E3 (200 μM) and DBS-COOH (200 μM) containing an equimolar concentration of ThT (200 μM) fluoresced when excited at 488 nm (Figure 1d,h). Similarly, the nanofibers of PA-E3/DBS-COOH mixtures (200 μM) also exhibited strong fluorescence (Figure 1e–g) when excited at the same wavelength, which results from the integration of ThT within the hydrogel nanostructures. However, the fluorescence is more intense on DBS-COOH (Figure 1h) and PA-E3/DBS-COOH (Figure 1e–g) nanofibers than on the PA-E3 nanofibers (Figure 1d). We observed no fluorescence from the DBS gelator solution itself (Supporting Information Figure S2). It is well established that ThT is a molecular rotor and its fluorescence is viscosity-dependent.52,53 No fluorescence was observed pre-gelation, as the ThT is not incorporated into viscous environments. However, upon gelation, hydrophobic rigid fibers are formed and, incorporation of ThT into these, results in an increase in fluorescence.52 Therefore, our systems support the idea that the reason for the characteristic increase in the ThT fluorescence intensity following its incorporation into the DBS-COOH and the two-component (PA-E3/DBS-COOH) nanofibers than that into the PA-E3 alone nanofibers is due to the rigidity of the nanofibers, which prevents the rotation of the benzothiazole ring relative to the aminobenzene ring in the excited state.53 Previous studies have demonstrated that molecular gelator nanofibers are able to sequester cationic dyes such as methylene blue (MB) from an aqueous system.41 We have also demonstrated this with DBS-COOH using MB (data not shown). Therefore, we reasoned that DBS-COOH nanofibers sequester the cationic ThT dye in a similar fashion, which can also explain the observed increased fluorescence intensity.
We also confirmed by circular dichroism (CD) spectroscopy that the achiral ThT molecule acquired a chiral signature following its interaction with the self-assembled PA-E3, DBS-COOH, and PA-E3/DBS-COOH nanofibers in a differential and time-dependent manner (Supporting Information Figure S3). Such chirality transfer is believed to result from a ‘sergeants-and-soldiers’’ type of interaction54 between ThT and the self-assembled nanofibers. Upon incorporation into the PA-E3 nanofibers during self-assembly, ThT exhibited a negative band at 385 nm, which disappeared within 30 min of self-assembly, while another negative band emerged at 470 nm as the self-assembly progressed (Figure 1i and Supporting Information Figure S3a). On the other hand, ThT exhibited a bisignate Compton effect with positive and negative bands at 370 and 385 nm, respectively (Figure 1j and Supporting Information Figure S3b). Interestingly, the CD spectrum of ThT upon incorporation into the PA-E3/DBS-COOH nanofibers combines the distinct chiral signatures of both PA-E3 and DBS-COOH, as well as the emergence of a new band at 450 nm (Figure 1k and Supporting Information Figure S3c). This result suggests that ThT is able to discriminate between different chiral environments in the multicomponent hydrogels. Also, with this approach, we could infer that the two building blocks constituting our multicomponent hydrogels exhibited self-sorting self-assembly.
Characterization of Nanostructures
Transmission electron microscopy (TEM) revealed that the PA-E3 solution (0.1 mM, pH 10) on its own has preformed micellar structures that are ∼200 nm long. Such pre-gelation assembly of PAs into cylindrical micellar nanostructures has been previously reported to take place as a result of the hydrophobic collapse of the alkyl moieties in an aqueous environment.55−57 On the other hand, while such structures were not observed in the solution of pure DBS-COOH (Supporting Information Figure S4), they were observed in the equimolar mixture of PA-E3 and DBS-COOH. Upon gelation triggered by GdL hydrolysis, the PA-E3 solution further assembled into typical PA nanofibers that are microns in length and ∼7 nm in diameter (Figure 2ai, top panel), while the nanofibers of DBS-COOH (0.1 mM) on its own self-assembled into bundles of ribbons that are several microns in length and ∼80 nm in width as previously reported46 (Figure 2av). Atomic force microscopy (AFM) was used to confirm these structures (Figure 2bi,bv, middle panel). Interestingly, nanofibers of the PA-E3/DBS-COOH hydrogels (0.1 mM) appeared to have different geometries compared to those of the single-component hydrogel systems and be more entangled (Figure 2aii–iv, top panel). Furthermore, the density of this nanofiber network seemed to increase with increasing DBS-COOH concentration. Such entanglement is possibly due to the formation of interconnected self-sorting networks.58 The nanofiber-bundling phenomenon is more pronounced with the PA-E3/DBS-COOH hydrogels than in the hydrogels of DBS-COOH alone (Figure 2bii–iv, middle panel). It is important to mention that the difference in the appearance of the nanofiber images acquired with the two techniques (TEM and AFM) is probably due to different sample preparation procedures. In the case of AFM, the solutions were allowed to self-assemble on the substrates, while with TEM, images were acquired on preformed partial hydrogels.
Figure 2.

Nanostructure characterization. Nanostructures of nanofibers of (i) PA-E3, (ii) PA-E3/DBS-COOH (4:1), (iii) PA-E3/DBS-COOH (1:1), (iv) PA-E3/DBS-COOH (1:4), and (v) DBS-COOH at a 0.1 mM gelator concentration revealed by (a) TEM and (b) AFM. (c) SANS data for gels of (i) PA-E3, (ii) PA-E3/DBS-COOH (4:1), (iii) PA-E3/DBS-COOH (1:1), (iv) PA-E3/DBS-COOH (1:4), and (v) DBS-COOH at a 1 mM gelator concentration. In all cases, the fits to the data are shown as the red line through the open symbols.
To further characterize the internal structuring of the hydrogels without drying and staining effects that accompany imaging techniques, synchrotron small-angle neutron scattering (SANS) measurements were performed on the hydrogels. SANS data for the hydrogels of PA-E3 (10 mM) fitted best to a flexible elliptical cylinder model after the initial fixing of the background and setting the length to 2000 Å (an arbitrary value greater than the resolution of the technique) (Figure 2k, lower panel). Other parameters to fit the data for PA-E3 include a radius of 26.9 ± 0.1 Å, an axis ratio of 1.64 ± 0.02, and a Kuhn length of 134.0 ± 2.0 Å. On the other hand, data for the hydrogels of DBS-COOH fitted to a power law only with a power-law exponent of 2.72 ± 0.01, which implies that the structures are relatively large and outside the effective range of the SANS (Figure 2o). To fully fit the SANS data for the two-component hydrogels (10 mM), we needed to combine the flexible elliptical cylinder model with a power law (Figure 2l–n), as previously demonstrated.59 With this approach, we obtained a radius of 27.5 ± 0.3 Å, an axis ratio of 1.53 ± 0.03 Å, and a Kuhn length of 111.6 ± 2.1 Å for the 4:1 molar ratio PA-E3/DBS-COOH hydrogel; a radius of 28.4 ± 0.7 Å, an axis ratio of 1.36 ± 0.06, and a Kuhn length of 96.3 ± 3.2 Å for the 1:1 molar ratio PA-E3/DBS-COOH hydrogel; and a radius of 27.9 ± 1.1 Å, an axis ratio of 1.52 ± 0.10, and a Kuhn length of 114.4 ± 9.21 Å for the 1:4 molar ratio PA-E3/DBS-COOH hydrogel. These results indicate that the nanoscale flexible elliptical cylinders are similar across the data set in terms of radius, axis ratio, and Kuhn length. The small differences in the fitted parameters can be attributed to the minimal perturbation effect of DBS-COOH on the PA-E3 fiber formation during the self-assembly process,60 suggesting that PA-E3 fiber assembly still occurs in the presence of DBS-COOH. The exponents for the power law are 2.47 ± 0.05, 2.76 ± 0.04, and 2.95 ± 0.07 for the hydrogels with 4:1, 1:1, and 1:4 molar ratios of PA-E3/DBS-COOH, respectively. For convenient comparison, the data for the fittings are summarized in Supporting Information Table S1. These power-law exponents suggest that there is an increasing nanofiber network formation with increasing concentration of DBS-COOH, in agreement with the LSCM (Figure 1e–g), TEM (Figure 2b–d, top panel), and AFM (Figure 2g–i, middle panel) images. However, it is important to mention that the radius of the nanofibers is such that the scattering at mid- to high-Q range is dominated by the flexible elliptical model as opposed to the power law. We confirmed this dominant flexible elliptical model by plotting the intensity at Q of 0.06151 Å−1 against % PA-E3 in the mixture to obtain a straight line fit as opposed to a nonlinear fit expected if DBS-COOH is entrained within the nanofibers of PA-E3 (Supporting Information Figure S5). Linking this all together, we propose that self-sorting dominates, leading to fibers that consist of PA-E3 only and others that consist of DBS-COOH only. Since the PA-E3 assembles first, it will do so in the presence of unassembled DBS-COOH, and hence the assembly of PA-E3 may be affected by the presence of the additive. The DBS-COOH then assembles to give the self-sorted system, which is known to play an important role in determining bulk properties of hydrogels.61
Kinetics of Self-Assembly
Micelles Act as Nuclei for Faster Assembly on PA-Containing Systems
Understanding the kinetics of self-assembly is important not only for gaining insight into the molecular mechanism but also for the design and modulation of properties of multicomponent hydrogel systems. In this study, we decided to use dynamic oscillatory rheology to monitor the self-assembly kinetics of PA-E3, DBS-COOH, and the equimolar PA-E3/DBS-COOH mixture. According to the time-sweep experiment within the linear viscoelastic region (LVR) of the hydrogels, PA-E3 displayed the expected rapid self-assembly (Figure 3a), whereas DBS-COOH exhibited a delay of about 20 min prior to self-assembly (Figure 3b). The PA-E3 system exhibits a two-stage increase in G′ and G″. By analogy to other works,62 this can be associated with interactions between initially formed fibers and a decrease of charge in the nanofibers. We stress, however, that given the difficulty to deconvolute the relative effects provided by fiber bundling, cross-linking, and reduction of charges in the fibers, interpretation of the rheology data must be taken with care. Interestingly, with the inclusion of DBS-COOH, the equimolar mixture of PA-E3/DBS-COOH exhibited a slightly shorter delay of about 10 min prior to self-assembly upon the addition of GdL (Figure 3c). This suggests that the two-component system displayed a different self-assembly mechanism to pure DBS-COOH. It is well known that PA-E3 tends to form cylindrical micellar nanostructures in water due to desolvation of the apolar palmitoyl group63 and electrostatic repulsion between the negatively charged head groups.64 These structures are expected to provide nucleation sites and thereby reduce the entropic cost of PA self-assembly in both the single- and two-component systems. This hypothesis is based on a nonclassical nucleation theory, in which nucleation and growth are much faster in a heterogeneous system comprising nuclei than the homogeneous counterpart having no preformed nuclei.65,66 Therefore, as expected, unlike the DBS-COOH system on its own, both PA-E3 and the equimolar mixture of PA-E3/DBS-COOH underwent faster self-assembly.
Figure 3.

Kinetics of self-assembly into nanofibers. Evolution of nanofibers is monitored during the sol–hydrogel phase transition by recording storage (G′) and loss (G″) modulus values as a function of time for 10 mM of (a) PA-E3, (b) DBS-COOH, and (c) equimolar mixture of PA-E3/DBS-COOH at 25 °C, and self-assembly is initiated by the addition of GdL (10 mg/mL); frequency = 10 Hz, strain = 0.5%. (d) TEM images showing self-assembly of PA-E3, DBS-COOH, and PA-E3/DBS-COOH into nanofibers at various time intervals.
The self-assembly of PA-E3, DBS-COOH, and the equimolar PA-E3/DBS-COOH mixtures into a solid-like gel, as revealed by G′, reached a plateau within 115, 92, and 63 min, respectively (Figure 3a–c), implying that the gels were supersaturated at the plateau region. Beyond the gelation point, tan δ also decreases with time for the self-assembly of PA-E3, DBS-COOH, and PA-E3/DBS-COOH mixtures and then levels off, representing the completion of self-assembly (Supporting Information Figure S6). The observed time difference for the full assembly of PA-E3, DBS-COOH, and PA-E3/DBS-COOH equimolar mixtures can be attributed to their distinct self-assembly events. Bearing in mind that PA-E3 already assumed a pregelation nanostructure, there is a high tendency for PA-E3 to undergo an assembly whereby there is a gradual increase in the number and length of the nanostructures until an insoluble sample-spanning network of nanofibers is formed.67 Given the sigmoidal rheographs of PA-E3 and that of the PA-E3/DBS-COOH mixture, we suggest that the self-assembly mechanism of PA-E3 is consistent with such isodesmic assembly with three set-lag phases: initial rearrangement/bundling of the cylindrical micelles, elongation, and supersaturation (Supporting Information Figure S1). On the other hand, DBS-COOH alone exhibits a different cooperative self-assembly (nucleation-growth) mechanism via an initial formation and growth of nuclei, above which a critical point gelation occurs.68 The initiation of nucleation by DBS-COOH results in a delayed emergence of the assembly for about 20 min following a prolonged lag phase (Supporting Information Figure S1). However, once assembly is initiated, it then rapidly proceeds in a cooperative manner. Similar to the assemblies of PA-E3, DBS-COOH assembly has three sets of lag phases but with a different initial event—nucleation, elongation, and supersaturation. Interestingly, the PA-E3/DBS-COOH assembly shows the fastest establishment (∼63 min) of an overall gel network. This system (PA-E3/DBS-COOH) combines the initial nuclei of the PA-E3 system, reducing the lag phase, with the cooperative network assembly of DBS-COOH, hence optimizing both the initial nucleation step and the assembly of the full-sample-spanning network (Supporting Information Figure S1). This is a clear example of the dual benefits of this two-component assembly system.
The Avrami exponents, n, for the temporal changes in the structure of PA-E3 and DBS-COOH and the equimolar mixture of PA-E3/DBS-COOH as a result of gradual pH-switching that drives self-assembly are 2.0, 2.2, and 2.7, respectively (Supporting Information Figures S7–S9). With these values, we deduced that DBS-COOH and PA-E3 self-assembled into a two-dimensionanl nanostructure, while the Avrami exponent obtained for the equimolar PA-E3/DBS-COOH mixture suggests the formation of a three-dimensional nanostructure or networks. It is worth noting that these Avrami exponents for DBS-COOH are higher than those previously determined by NMR and CD methods.38 This reflects the fact that determining Avrami exponents by rheology will indicate the formation of a sample-spanning network in two/three dimensions, whereas the other methods report directly on one-dimensionally nanofiber assembly. These results indicate a greater degree of three-dimensionality in the presence of DBS-COOH, which is in agreement with the earlier discussion about the ability of this gelator to enhance nanofiber aggregation and sample-spanning nanofiber network formation.
Remarkably, the slow self-assembly kinetics that result from using low gelator concentrations (0.01 mM, pH 9) and the in situ protonation resulting from GdL hydrolysis facilitated real-time observations of the growth of the nanofibers by TEM. As shown in Figure 3d, at the onset of self-assembly (t = 0), PA-E3 already exists in a preassembled micellar form, while DBS-COOH exists in a free and monomeric form at time t = 0 min. There are also micellar nanostructures with similar dimensions in the equimolar solutions of PA-E3/DBS-COOH. Interestingly, within 15 min of adding GdL, DBS-COOH self-assembled into ∼200 nm long nanofibers, while the PA-E3 and PA-E3/DBS-COOH micelles grew into ∼600 nm long nanofibers. Further into the incubation period (45 and 60 min), the nanofibers grew into entangled, interpenetrated, and long nanofiber networks in all cases. DBS-COOH and PA-E3/DBS-COOH nanofibers are clearly more bundled than PA-E3 and DBS-COOH alone, in agreement with the kinetic observations from the rheological study described above. The hierarchical nanofiber growth further confirms the nucleation–elongation–supersaturation mechanism we proposed for the stepwise increase in the G′ values observed in Figures 3a–c and 4, which also follows the kinetic model of the step growth of β-amyloid peptide nanofibers.69 It is important to mention that the differences between nanofibers of PA-E3, DBS-COOH, and PA-E3/DBS-COOH are not sufficiently significant given the need to air-dry the samples.
Figure 4.

Mechanism of self-assembly. Proposed mechanisms of self-assembly of (a) PA-E3, (b) DBS-COOH, and (c) PA-E3/DBS-COOH when triggered with GdL in aqueous systems. The green fluorescent images represent ThT fluorescent micrographs of self-assembled nanofibers.
All-Atom Molecular Dynamics (MD) Simulations of Self-Assembly
It has been established that the structures, self-assembling driving forces, surface charge, and hydrophobicity of building blocks in a multicomponent self-assembling system are key to determining whether they will coassemble or self-sort.70−72 Therefore, molecular dynamics simulations were performed using Material Studio 8.0 software from Biovia Software Inc. (San Diego, CA)73 to understand how the structural differences, driving forces, and hydrophilicity of DBS-COOH and PA-E3 dictate the type of internal nanostructuring in these two-component hydrogels. According to solubility parameter calculations, the theoretical solubility parameters of DBS-COOH and PA-E3 are 24.36 (J/cm3)1/2 and 23.13 (J/cm3)1/2, respectively, which implies that DBS-COOH is slightly more polar than PA-E3. Upon mixing the two components, the cohesive energy density (CED) decreases as the ratio of PA-E3 increases (Supporting Information Figure S10). Moreover, the enthalpy of mixing (ΔHmix) values of DBS-COOH and PA-E3 was negative and became more negative with a decreasing amount of PA-E3, indicating mixing of the two components (Supporting Information Figure S11).
Molecules of PA-E3 and DBS-COOH were constructed and optimized using DMOL3 quantum mechanical module. Self-assembly of PA-E3 and DBS-COOH and mixtures of both were studied by dynamics simulation using Forcite module with COMPASS II force field. The results after 500 ps dynamic steps (Figure 5b–e) indicate that the introduction of some molecules of DBS-COOH into the simulation box of PA-E3 does not disturb the PA-E3 self-assembly, suggesting self-sorting of PA-E3. However, DBS-COOH molecules did not self-sort and instead bind to the surface of the PA-E3 molecules through multiple hydrogen-bonding interactions. At a higher DBS-COOH/PA-E3 ratio, the bound DBS-COOH molecules within close proximity self-assemble, thereby facilitating interfacial interaction with the PA-E3 (Figure 5d). This interaction might explain the tendency of the two-component PA-E3/DBS-COOH system to form bundled nanofibers with improved mechanical properties. Radial distribution function (RDF) was used to estimate the intermolecular (hydrogen bond) interaction distance between molecules of the individual components (PA-E3 and DBS-COOH), as well as their mixtures. It was found that the most significant intermolecular interactions that underpin PA-E3 self-assembly are formed between H-carboxyl and O-carbonyl groups (interaction distance of 1.7 Å and intensity of 5 (Figure 5e)). On the other hand, the dominant intermolecular interactions between DBS-COOH molecules are O-carbonyl/H-O6 and O6/H-O6 with interaction distances of 1.8 and 1.7 Å, respectively (Supporting Information Table S2). Interestingly, when PA-E3 molecules self-assemble in the presence of DBS-COOH, the intensity of the interaction distance between H-carboxyl and O-carbonyl groups, as well as H-amide and O-carbonyl of PA-E3, remains constant (Figure 5e,f), suggesting that the presence of DBS-COOH does not affect the self-assembly of PA-E3. However, by measuring the interaction distance between O-carbonyl/H-O6, as well as O6/H-O6 of DBS-COOH at 1:1 (DBS-COOH/PA-E3) molar ratio, we observed no interaction formed between DBS-COOH molecules (Figure 5g,h), thus indicating that DBS-COOH molecules preferentially interact with the self-assembled PA-E3 structure at this molar ratio. When the molar ratio of DBS-COOH increases to 5:1 (DBS-COOH/PA-E3), a significant interaction is formed between DBS-COOH molecules (Figure 5g,h). In all cases, intermolecular hydrogen bond (H-bond) interactions are formed between PA-E3 and DBS-COOH by several groups but the most significant interaction is formed between O-carbonyl of DBS-COOH and H-carboxyl of PA-E3 with an interaction distance of 1.7 Å (Supporting Information Table S2). The interactions that the H-amide of PA-E3 forms with various groups of DBS-COOH are less significant, and in all cases, the interaction intensity decreases with the decrease of PA-E3 concentration (Figure 5i–l). DBS-COOH does not affect the interactions between PA-E3 as revealed by RDF results, suggesting that PA-E3 undergoes self-sorting. However, the presence of PA-E3 causes DBS-COOH to alter its self-assembly fashion, which might corroborate our findings with SANS and dynamic time-sweep rheology.
Figure 5.

Molecular dynamics simulation. Periodic cubic cell containing (a) 1 molecule of DBS-COOH (green) and 5 molecules of PA-E3, (b) 3 molecules of DBS-COOH (green) and 3 molecules of PA-E3, (c) 5 molecules of DBS-COOH (green) and 1 molecule of PA-E3, and (d) 12 molecules of purely DBS-COOH after a 500 ps dynamics simulation. Intermolecular interaction intensities between (e) H-carboxyl and O-carbonyl of PA-E3 as pure component and in the presence of DBS-COOH at various molar ratios, (f) H-amide and O-carbonyl of PA-E3 as pure component and in the presence of DBS-COOH at various molar ratios, (g) O-carbonyl of DBS-COOH and H-O6 of DBS-COOH as pure component and in the presence of PA-E3 at various molar ratios, (h) O6 of DBS-COOH and H-O6 of DBS-COOH as pure component and in the presence of PA-E3 at various molar ratios, (i) O-carbonyl of DBS-COOH and H-carboxyl of PA-E3 at various molar ratios, (j) O-carbonyl of DBS-COOH and H-amide of PA-E3 at various molar ratios, (k) O6 of DBS-COOH and H-amide of PA-E3, and (l) O5 of DBS-COOH and H-amide of PA-E3.
In summary, the interactions mediating the self-assembly can be discussed on the basis of the energetics of molecular interactions. The interactions that formed the self-assembled structures between PA-E3 and DBS-COOH will be a balance between all possible energies. The PA-E3 self-assembly is driven by the hydrophobic collapse of the hydrocarbon tail, while the polar heads are displayed on the periphery of the nanofibers. Therefore, the energy associated with the assembly of PA-E3 is too large for DBS-COOH to fully disrupt, but then DBS-COOH can form some adventitious interactions with parts (polar head group) of PA-E3 that are not responsible for self-assembly to provide some favorable energetics. In this way, small amounts of DBS-COOH interact with the assembling PA-E3 but without disrupting it. As the concentration of DBS increases, self–self interactions become more important for DBS-COOH because there are more potential DBS-COOH binding partners, resulting in interactions between DBS-COOH molecules becoming more energetically significant.
Viscoelastic and Self-Recovery Properties of Hydrogels
Given the increasing hydrogel translucency that is observed with the growing concentration of DBS-COOH in the PA-E3/DBS-COOH hydrogels (Figure 1b) as a result of higher nanofiber density, we used dynamic oscillatory rheometry to quantify its effect on the bulk properties (stiffness) of the hydrogels. Based on amplitude-sweep measurements, hydrogels for PA-E3 (10 mM) and DBS-COOH (10 mM) exhibited storage moduli (G′) of ∼9.5 and ∼27 kPa, respectively (Figure 6a). Interestingly, G′ values increased linearly with an increasing percentage of DBS-COOH in the PA-E3/DBS-COOH hydrogels (Figure 6a and Supporting Information Figure S12), indicating a synergistic effect. Also, from the amplitude-sweep measurements, the critical strain (i.e., the strain at which the hydrogels break) for DBS-COOH and PA-E3 were 0.8 (Supporting Information Figure S13) and 2.7% (Supporting Information Figure S14), respectively. The critical strain values for the two-component hydrogels range between 1 and 2% (Supporting Information Figures S15–S18) and slightly decrease as the amount of PA-E3 in the hydrogels decreases, suggesting that PA-E3 is able to promote the formation of resilient hydrogels. Previous studies have shown that π–π stacking between DBS gelators confers enhanced rigidity on their self-assembled structures.40 This rigid structure formation suggests a reason for higher G′ values (also lower values of tan δ) of DBS-COOH hydrogels and the PA-E3/DBS-COOH than those of the nonaromatic PA-E3 hydrogels (Supporting Information Figure S13). It is important to note that the total concentration of the gelators remains fixed (10 mM) in all multicomponent hydrogels; so, differences cannot be assigned to total gelator loading.
Figure 6.

Mechanics and self-healable properties. (a) Plots of G′ against the percentage of DBS-COOH in the hydrogels. (b)–(d) show the self-recovery of PA-E3, DBS-COOH and the equimolar concentration of PA-E3 and DBS-COOH, respectively. In all cases, the hydrogels were subjected to a constant frequency of 10 Hz and strain of 0.1% (100 s), 100% (100 s), 0.1% (200 s), 100% (100 s), 0.1% (200 s), 100% (100 s), and 0.1% (400 s).
Self-assembly through noncovalent bonds such as π–π stacking, hydrogen bonding, and host–guest interactions holds great potential for constructing self-healable hydrogels due to their intrinsic reversibility.74−76 For example, Stevens and co-workers have harnessed the ability of noncovalent β-sheet cross-links to fabricate self-healable hybrid hydrogels owing to their ability to reassemble and recover fully after being strained to failure.77 Given the structural dissimilarity between the aromatic sugar gelator (DBS-COOH) and the β-sheet forming PA gelator (PA-E3), we wanted to dissect the impact of the interplay of the π–π stacking and hydrogen bonding that DBS-COOH provides and the noncovalent β-sheet cross-links that PA-E3 offers to the self-healing ability of the PA-E3/DBS-COOH hydrogel.
To test this hypothesis, the thixotropic properties of the hydrogels after high shear loads were measured in dynamic time-sweep experiment under strain amplitudes within (0.1%) and beyond (100%) the linear viscoelastic region (LVR) of the hydrogels (Supporting Information Figure S12 for the LVR). Under high shear load, the hydrogels undergo an internal breakage as indicated by the significant decrease and inversion of G′ and G″ values. This signifies that the liquid-like behavior of the hydrogels dominates under high strain amplitude, which indicates that the hydrogels have been broken. After three cycles of strain amplitude, hydrogels of PA-E3 displayed full (100%) recovery (Figure 6b), while DBS-COOH hydrogels only recovered 62% (Figure 6c) and hydrogels with a 1:1 molar ratio of PA-E3/DBS-COOH exhibited 98% recovery (Figure 6d). The two-component gel, therefore, exhibited enhanced stiffness relative to individual building blocks as a result of the DBS-COOH aromatic structure while maintaining a high level of recovery compared to that of the DBS-COOH hydrogels due to the PA-E3 noncovalent β-sheet cross-links. This result demonstrates how the two distinct gelators cooperate in the multicomponent hydrogels. Also, these data reveal how interactions of self-assembling building blocks at the nanoscale and the spatiotemporal arrangement of the nanofibers can translate into material bulk properties at the macroscale level.78,79
Proteolytic Stability and Effects on Cell Morphology
To further demonstrate the synergistic material properties that can emerge from the multicomponent PA-E3/DBS-COOH hydrogels, we assessed the material’s resistance to proteolysis. PAs have been widely used in vivo as therapeutic agents in regenerative medicine. However, the peptide bonds with l-chirality are known to quickly degrade after systemic delivery.80 Instability of alkylated l-peptides to proteolysis has also been reported.81 Therefore, d-enantiomers with better resistance to proteolysis have previously been used as a substitute or a protective corona for the l-enantiomers by coassembly.82−84 Covalent attachment of polymers to the peptide surface is another approach that has previously been explored.80 For example, Stupp and co-workers employed a supramolecular coassembly strategy to develop proteolytically stable PA nanofibers consisting of an antitumor PA in combination with its PEGylated analogue.85 The use of a nonpeptidic low-molecular-weight gelator as a protective corona for l-peptides against premature proteolysis is another approach that has not been explored in the field of biomaterial development. Our self-assembled system bearing well-defined structures of a lipidated l-peptide and acetalized d-sugar would provide an alternative approach to extend the functionality of PAs.
To investigate proteolytic stability, PA-E3/DBS-COOH and control hydrogels interacted with the aqueous solution of proteinase K (50 mg/mL). Digestions were assessed using scanning electron microscopy (SEM) to characterize the morphology of PA-E3, DBS-COOH, and PA-E3/DBS-COOH hydrogels. Unlike the nanostructure of PA-E3 hydrogels submerged in PBS (Figure 7a), Figure 7d shows that the PA-E3 hydrogel nanostructures were digested and transformed into aggregates of nanoparticles after incubating with proteinase K for 5 days. This structural transformation is reminiscent of a report by Hamley and co-workers using chymotrypsin to induce nanofiber–nanoparticle transformation in a PA solution.81 As expected, DBS-COOH hydrogel nanostructures (Figure 7b) remained intact after treatment with proteinase K (Figure 7e), suggesting that DBS-COOH lacks the specific motifs for enzymatic cleavage. Indeed, equimolar mixtures of PA-E3/DBS-COOH hydrogels were resistant to enzymatic digestions as the nanostructures remained intact (Figure 7f) when compared with the nanostructure of the hydrogels that were not treated with proteinase K (Figure 7c), suggesting that DBS-COOH acted as a protective corona around PA-E3 nanofibers in the two-component hydrogels, making it impossible for the enzyme to access peptide bonds in PA-E3. To further elucidate the enzymatic degradation of the PA-E3 by proteinase K, liquid chromatography–mass spectrometry (LC–MS) was employed. As shown in Supporting Information Figure S19a,aii, an LC–MS spectrum for the PA-E3/proteinase K mixture reveals multiple peaks with m/z values of 1177.80, 1155.70, 1078.70, 847.5, 846.5, 817.7, 520.2, and 418.3 Da. The sharp peaks with m/z values 1177.80 and 1155.70 Da correspond to [M + Na] + and [M + H] +, respectively, where M is the calculated mass of PA-E3. The remaining peaks are the masses of the PA-E3 fragments following proteolysis. Also, the ultraviolet–visible (UV) chromatogram displayed two broad bands after digestion (Supporting Information Figure S19c). Interestingly, only two peaks with m/z values 1177.80 and 1155.70 Da, which correspond to [M + Na] + and [M + H] +, respectively, were observed on the spectrum of PA-E3/DBS-COOH/proteinase K mixture (Supporting Information Figure S19b), suggesting that DBS-COOH indeed acted as a protective corona for PA-E3 in the multicomponent hydrogels, limiting the proteolysis of PA-E3. Impacts of this morphological transformation on the macroscopic hydrogels were also assessed by a weight loss measurement approach. After incubating the DBS-COOH and DBS-COOH/PA-E3 hydrogels in enzyme solution, as well as PA-E3 hydrogels in PBS for 22 days, the weight ratio remained constant. In contrast, the weight ratio of PA-E3 hydrogels in enzyme solution drastically reduced within 22 days of incubation (Figure 7g). These results further support our argument that the macroscopic properties of multicomponent hydrogels are dependent on the nanoscale composition.
Figure 7.

Biological functionalities. Stability of nanofibers of (a) DBS-COOH, (b) PA-E3, (c) 1:1 molar ratio of DBS-COOH and PA-E3 submerged in PBS solution. (d) DBS-COOH, (e) PA-E3, and (f) 1:1 molar ratio of DBS-COOH and PA-E3 after incubation with proteinase K for 5 days, as revealed by SEM. (g) Degradation profile of PA-E3, DBS-COOH, and PA-E3/DBS-COOH hydrogels after incubation with proteinase K at 37 °C for 22 days. PA-E3 hydrogels were also incubated in PBS under similar conditions. LSCM images of human-adipose-derived stem cells seeded on hydrogels of (h) PA-E3, (i) PA-E3/DBS-COOH (4:1), (j) PA-E3/DBS-COOH (1:1), (k) PA-E3/DBS-COOH (1:4), (l) DBS-COOH, and (m) cells plated on TCP for 4 days. Scale bar: 300 μm.
To investigate the response of cells to the multicomponent hydrogels, we cultured human-adipose-derived stem cells (ADSCs) on the hydrogels prepared with various stoichiometric ratios of PA-E3 and DBS-COOH. After 4 days of incubation, the cells were stained with live/dead dyes. Cells stained with the green dye (calcein AM) were alive, while those stained with the red dye (ethidium homodimer) were dead. As shown in Figure 7h, the cells on the PA-E3 hydrogels assumed a round morphology after 4 days in culture. In contrast, hydrogels of DBS-COOH were fully covered with live cells. Also, the cells exhibited an elongated morphology (cell spreading) (Figure 7l) similar to cells cultured on tissue culture plastic (TCP) (Figure 7m). For the multicomponent hydrogels with 4:1 and 1:1 ratios of PA-E3/DBS-COOH, there was a mixture of both round and elongated cells. However, the cells seeded on hydrogels with a 1:4 ratio of PA-E3 and DBS-COOH formed a network of elongated morphology (Figure 7k). Evidently, cells were more viable on DBS-COOH hydrogels than those on PA-E3 hydrogels. The multicomponent hydrogels are intermediate between the two—clearly, DBS-COOH can impart some of cell compatibility to these hybrid materials. The tunable morphology of the cells also shows how molecular composition and mechanical properties of supramolecular hydrogels can potentially be used to influence cell growth and potentially, in the future, decide cell phenotypes. We hope to characterize cell differentiation in detail in the future.
Conclusions
We report on the synergistic properties arising from the self-sorting of a PA- and a DBS-based gelator by slow acidification. Compared to the individual components, the resulting multicomponent hydrogels exhibited a series of properties including tunable nanoscale morphology, improved stiffness (endowed by DBS gelator), enhanced self-healing (endowed by the PA), stability to enzymatic degradation (provided by the DBS gelator), and cell compatibility (provided by the DBS gelator). Induced circular dichroism of ThT, SANS, and molecular dynamics simulations demonstrated self-sorting with the DBS-COOH initially acting as an additive adsorbed on the surface of the PA-E3 nanofibers through multiple hydrogen-bonding interactions. The adsorbed sugar molecules within close proximity then interact with each other, thereby facilitating interfacial interactions between nanofibers. We reason that these interfacial interactions impart the nanofiber network with enhanced stiffness, as well as resistance to enzymatic degradation. With regard to the assembly mechanism, the PA-E3 exhibits different assembly kinetics than DBS-COOH, with the PA-E3 benefitting from the presence of multiple cylindrical micellar nuclei, avoiding the lag phase inherent in the DBS-COOH. However, the assembly of the DBS gelator is more cooperative and rapid once nucleation has occurred. Interestingly, the multicomponent system benefits from both advantages of the PA and DBS systems and is the fastest to establish a full-sample-spanning gel network. Finally, human-adipose-derived stem cells could be cultured on the DBS gelator and the two-component hydrogels with a sufficiently high concentration of DBS-COOH, opening new opportunities for developing complex hydrogels for regenerative medicine. In summary, we have developed and characterized a PA-E3/DBS-COOH hybrid system that expands the scope and capacity of both PA-E3 and DBS-COOH with each of the two LMWG systems endowing the resulting hybrid gel with its own unique capabilities.
Acknowledgments
This work was supported by the ERC Starting Grant (STROFUNSCAFF) and the UK Regenerative Medicine Platform (UKRMP2) Acellular/Smart Materials. D.J.A. thanks the EPSRC for a fellowship (EP/L021978/1). D.K.S. gratefully thanks the EPSRC for funding support (EP/P03361X/1). The experiment at the ISIS Neutron and Muon Source was allocated beam time under Xpress access proposal 1890153 and collected on SANS2D. This work was benefitted from the SasView software, originally developed by the DANSE project under NSF award DMR-0520547. The authors thank Dr. Vicente Araullo-Peters and Dr. Giulia Mastroianni at Nanovision and School of Biological and Chemical Sciences (SBCS), QMUL, for technical supports. The authors thank Dr. Stephen King and Dr. Sarah Rogers from ISIS for SANS experiment.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.9b01882.
Overview of multicomponent self-assembly; ThT fluorescent microscopy for sol samples; full-range CD spectra for ThT–gelator interactions; TEM images of gelator sols; SANS fitting parameters; tan δ values for gelator self-assembly kinetics; Avrami equations and plots; molecular dynamics computation; amplitude sweeps and critical strain for hydrogels; and UV–TIC spectra for hydrogel digestions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bai Y.; Luo Q.; Liu J. Protein self-assembly via supramolecular strategies. Chem. Soc. Rev. 2016, 45, 2756–2767. 10.1039/C6CS00004E. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Mata A. Multicomponent self-assembly as a tool to harness new properties from peptides and proteins in material design. Chem. Soc. Rev. 2018, 47, 3721–3736. 10.1039/C8CS00121A. [DOI] [PubMed] [Google Scholar]
- Colquhoun C.; Draper E. R.; Eden E. G. B.; Cattoz B. N.; Morris K. L.; Chen L.; McDonald T. O.; Terry A. E.; Griffiths P. C.; Serpell L. C.; et al. The effect of self-sorting and co-assembly on the mechanical properties of low molecular weight hydrogels. Nanoscale 2014, 6, 13719–13725. 10.1039/C4NR04039B. [DOI] [PubMed] [Google Scholar]
- Smith M. M.; Smith D. K. Self-sorting multi-gelator gels—mixing and ageing effects in thermally addressable supramolecular soft nanomaterials. Soft Matter 2011, 7, 4856–4860. 10.1039/c1sm05316g. [DOI] [Google Scholar]
- Onogi S.; Shigemitsu H.; Yoshii T.; Tanida T.; Ikeda M.; Kubota R.; Hamachi I. In situ real-time imaging of self-sorted supramolecular nanofibres. Nat. Chem. 2016, 8, 743–752. 10.1038/nchem.2526. [DOI] [PubMed] [Google Scholar]
- Raeburn J.; Adams D. J. Multicomponent low molecular weight gelators. Chem. Commun. 2015, 51, 5170–5180. 10.1039/C4CC08626K. [DOI] [PubMed] [Google Scholar]
- Liyanage W.; Vats K.; Rajbhandary A.; Benoit D. S. W.; Nilsson B. L. Multicomponent dipeptide hydrogels as extracellular matrix-mimetic scaffolds for cell culture applications. Chem. Commun. 2015, 51, 11260–11263. 10.1039/C5CC03162A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makam P.; Gazit E. Minimalistic peptide supramolecular co-assembly: expanding the conformational space for nanotechnology. Chem. Soc. Rev. 2018, 47, 3406–3420. 10.1039/C7CS00827A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Wang Z.; Zhang X. Amphiphilic building blocks for self-assembly: From amphiphiles to supra-amphiphiles. Acc. Chem. Res. 2012, 45, 608–618. 10.1021/ar200226d. [DOI] [PubMed] [Google Scholar]
- Zayed J. M.; Nouvel N.; Rauwald U.; Scherman O. A. Chemical complexity—supramolecular self-assembly of synthetic and biological building blocks in water. Chem. Soc. Rev. 2010, 39, 2806–2816. 10.1039/b922348g. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Lovrak M.; Liu Q.; Maity C.; le Sage V. A. A.; Guo X.; Eelkema R.; van Esch J. H. Hierarchically compartmentalized supramolecular gels through multilevel self-sorting. J. Am. Chem. Soc. 2019, 141, 2847–2851. 10.1021/jacs.8b09596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y.; Waltman A.; Han H.; Collier J. H. Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano 2016, 10, 9274–9286. 10.1021/acsnano.6b03409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalily M. A.; Bakan G.; Kucukoz B.; Topal A. E.; Karatay A.; Yaglioglu H. G.; Dana A.; Guler M. O. Fabrication of supramolecular n/p-nanowires via coassembly of oppositely charged peptide-chromophore systems in aqueous media. ACS Nano 2017, 11, 6881–6892. 10.1021/acsnano.7b02025. [DOI] [PubMed] [Google Scholar]
- Redondo-Gómez C.; Abdouni Y.; Becer C. R.; Mata A. Self-assembling hydrogels based on a complementary host–guest peptide amphiphile pair. Biomacromolecules 2019, 20, 2276–2285. 10.1021/acs.biomac.9b00224. [DOI] [PubMed] [Google Scholar]
- Hudalla G. A.; Modica J. A.; Tian Y. F.; Rudra J. S.; Chong A. S.; Sun T.; Mrksich M.; Collier J. H. A Self-adjuvanting supramolecular vaccine carrying a folded protein antigen. Adv. Healthcare Mater. 2013, 2, 1114–1119. 10.1002/adhm.201200435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inostroza-Brito K. E.; Collin E.; Siton-Mendelson O.; Smith K. H.; Monge-Marcet A.; Ferreira D. S.; Rodríguez R. P.; Alonso M.; Rodríguez-Cabello J. C.; Reis R. L.; et al. Co-assembly, spatiotemporal control and morphogenesis of a hybrid protein–peptide system. Nat. Chem. 2015, 7, 897–904. 10.1038/nchem.2349. [DOI] [PubMed] [Google Scholar]
- Hedegaard C. L.; Collin E. C.; Redondo-Gómez C.; Nguyen L. T. H.; Ng K. W.; Castrejón-Pita A. A.; Castrejón-Pita J. R.; Mata A. Hydrodynamically guided hierarchical self-assembly of peptide–protein bioinks. Adv. Funct. Mater. 2018, 28, 1703716 10.1002/adfm.201703716. [DOI] [Google Scholar]
- Capito R. M.; Azevedo H. S.; Velichko Y. S.; Mata A.; Stupp S. I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Matthaei J. F.; Baneyx F. A Self-assembling two-dimensional protein array is a versatile platform for the assembly of multicomponent nanostructures. Biotechnol. J. 2018, 13, e11800141 10.1002/biot.201800141. [DOI] [PubMed] [Google Scholar]
- An B.; Wang X.; Cui M.; Gui X.; Mao X.; Liu Y.; Li K.; Chu C.; Pu J.; Ren S.; et al. Diverse supramolecular nanofiber networks assembled by functional low-complexity domains. ACS Nano 2017, 11, 6985–6995. 10.1021/acsnano.7b02298. [DOI] [PubMed] [Google Scholar]
- Ni R.; Chau Y. Structural mimics of viruses through peptide/DNA co-assembly. J. Am. Chem. Soc. 2014, 136, 17902–17905. 10.1021/ja507833x. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Redondo-Gómez C.; Mata A.. Multicomponent Self-assembly: Supramolecular Design of Complex Hydrogels for Biomedical Applications. In Self-assembling Biomaterials; Woodhead Publishing, 2018; pp 371–397. [Google Scholar]
- Li Y.; Liu Y.; Ma R.; Xu Y.; Zhang Y.; Li B.; An Y.; Shi L. A g-quadruplex hydrogel via multicomponent self-assembly: formation and zero-order controlled release. ACS Appl. Mater. Interfaces 2017, 9, 13056–13067. 10.1021/acsami.7b00957. [DOI] [PubMed] [Google Scholar]
- Wong Po Foo C. T. S.; Lee J. S.; Mulyasasmita W.; Parisi-Amon A.; Heilshorn S. C. Two-component protein-engineered physical hydrogels for cell encapsulation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 22067–22072. 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla A. M.; Draper E. R.; Nolan M. C.; Brasnett C.; Seddon A.; Mears L. L. E.; Cowieson N.; Adams D. J. Self-sorted oligophenylvinylene and perylene bisimide hydrogels. Sci. Rep. 2017, 7, 8380 10.1038/s41598-017-08644-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross E. R.; Sproules S.; Schweins R.; Draper E. R.; Adams D. J. Controlled tuning of the properties in optoelectronic self-sorted gels. J. Am. Chem. Soc. 2018, 140, 8667–8670. 10.1021/jacs.8b05359. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Smith A. M.; Das A. K.; Hodson N. W.; Collins R. F.; Ulijn R. V.; Gough J. E. Self-assembled peptide-based hydrogels as scaffolds for anchorage-dependent cells. Biomaterials 2009, 30, 2523–2530. 10.1016/j.biomaterials.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Alakpa E. V.; Jayawarna V.; Lampel A.; Burgess; Karl V.; West; Christopher C.; Bakker; Sanne C. J.; Roy S.; Javid N.; Fleming S.; Lamprou; Dimitris A.; et al. Tunable supramolecular hydrogels for selection of lineage-guiding metabolites in stem cell cultures. Chem 2016, 1, 298–319. 10.1016/j.chempr.2016.07.001. [DOI] [Google Scholar]
- Draper E. R.; Eden E. G. B.; McDonald T. O.; Adams D. J. Spatially resolved multicomponent gels. Nat. Chem. 2015, 7, 848–852. 10.1038/nchem.2347. [DOI] [PubMed] [Google Scholar]
- Cornwell D. J.; Daubney O. J.; Smith D. K. Photopatterned multidomain gels: multi-component self-assembled hydrogels based on partially self-sorting 1,3:2,4-dibenzylidene-D-sorbitol derivatives. J. Am. Chem. Soc. 2015, 137, 15486–15492. 10.1021/jacs.5b09691. [DOI] [PubMed] [Google Scholar]
- McClendon M. T.; Stupp S. I. Tubular hydrogels of circumferentially aligned nanofibers to encapsulate and orient vascular cells. Biomaterials 2012, 33, 5713–5722. 10.1016/j.biomaterials.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata A.; Palmer L.; Tejeda-Montes E.; Stupp S. I.. Nanotechnology in Regenerative Medicine: Methods and Protocols; Humana Press: Totowa, NJ, 2012; Vol. 811, pp 39–49. [DOI] [PubMed] [Google Scholar]
- Chin S. M.; Synatschke C. V.; Liu S.; Nap R. J.; Sather N. A.; Wang Q.; Álvarez Z.; Edelbrock A. N.; Fyrner T.; Palmer L. C.; et al. Covalent-supramolecular hybrid polymers as muscle-inspired anisotropic actuators. Nat. Commun. 2018, 9, 2395 10.1038/s41467-018-04800-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata A.; Hsu L.; Capito R.; Aparicio C.; Henrikson K.; Stupp S. I. Micropatterning of bioactive self-assembling gels. Soft Matter 2009, 5, 1228–1236. 10.1039/b819002j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks M. P.; Sato K.; Palmer L. C.; Stupp S. I. Supramolecular assembly of peptide amphiphiles. Acc. Chem. Res. 2017, 50, 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn K. K.; Oh H.; Hashemipour R.; Weiss R. G.; Raghavan S. R. Insights into organogelation and its kinetics from Hansen solubility parameters. Toward a priori predictions of molecular gelation. Soft Matter 2014, 10, 2632–2640. 10.1039/c3sm52297k. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Vieira V. M. P.; Cornwell D. J.; Whitelaw N. K.; Smith D. K. 1,3:2,4-Dibenzylidene-D-sorbitol (DBS) and its derivatives–efficient, versatile and industrially-relevant low-molecular-weight gelators with over 100 years of history and a bright future. Soft Matter 2015, 11, 4768–4787. 10.1039/C5SM00845J. [DOI] [PubMed] [Google Scholar]
- Cornwell D. J.; Okesola B. O.; Smith D. K. Hybrid polymer and low molecular weight gels–dynamic two-component soft materials with both responsive and robust nanoscale networks. Soft Matter 2013, 9, 8730–8736. 10.1039/c3sm51967h. [DOI] [Google Scholar]
- Okesola B. O.; Smith D. K. Versatile supramolecular pH-tolerant hydrogels which demonstrate pH-dependent selective adsorption of dyes from aqueous solution. Chem. Commun. 2013, 49, 11164–11166. 10.1039/c3cc45969a. [DOI] [PubMed] [Google Scholar]
- Knani D.; Alperstein D. Simulation of DBS, DBS-COOH, and DBS-CONHNH2 as Hydrogelators. J. Phys. Chem. A 2017, 121, 1113–1120. 10.1021/acs.jpca.6b11130. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Smith D. K. Applying low-molecular weight supramolecular gelators in an environmental setting – self-assembled gels as smart materials for pollutant removal. Chem. Soc. Rev. 2016, 45, 4226–4251. 10.1039/C6CS00124F. [DOI] [PubMed] [Google Scholar]
- Howe E. J.; Okesola B. O.; Smith D. K. Self-assembled sorbitol-derived supramolecular hydrogels for the controlled encapsulation and release of active pharmaceutical ingredients. Chem. Commun. 2015, 51, 7451–7454. 10.1039/C5CC01868D. [DOI] [PubMed] [Google Scholar]
- Okesola B. O.; Suravaram S. K.; Parkin A.; Smith D. K. Selective extraction and in situ reduction of precious metal salts from model waste to generate hybrid gels with embedded electrocatalytic nanoparticles. Angew. Chem. Int. Ed. 2016, 55, 183–187. 10.1002/ange.201507684. [DOI] [PubMed] [Google Scholar]
- Vieira V. M. P.; Lima A. C.; de Jong M.; Smith D. K. Commercially relevant orthogonal multi-component supramolecular hydrogels for programmed cell growth. Chem. - Eur. J. 2018, 24, 15112–15118. 10.1002/chem.201803292. [DOI] [PubMed] [Google Scholar]
- Slavík P.; Kurka D. W.; Smith D. K. Palladium-scavenging self-assembled hybrid hydrogels – reusable highly-active green catalysts for Suzuki–Miyaura cross-coupling reactions. Chem. Sci. 2018, 9, 8673–8681. 10.1039/C8SC04561E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira V. M. P.; Hay L. L.; Smith D. K. Multi-component hybrid hydrogels – understanding the extent of orthogonal assembly and its impact on controlled release. Chem. Sci. 2017, 8, 6981–6990. 10.1039/C7SC03301J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornwell D. J.; Okesola B. O.; Smith D. K. Multidomain hybrid hydrogels: Spatially resolved photopatterned synthetic nanomaterials combining polymer and low-molecular-weight gelators. Angew. Chem., Int. Ed. 2014, 53, 12461–12465. 10.1002/anie.201405098. [DOI] [PubMed] [Google Scholar]
- Piras C. C.; Smith D. K. Sequential assembly of mutually-interactive supramolecular hydrogels and fabrication of multi-domain materials. Chem. - Eur. J. 2019, 25, 11318–11326. 10.1002/chem.201902158. [DOI] [PubMed] [Google Scholar]
- Webber M. J.; Tongers J.; Newcomb C. J.; Marquardt K.-T.; Bauersachs J.; Losordo D. W.; Stupp S. I. Supramolecular nanostructures that mimic VEGF as a strategy for ischemic tissue repair. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 13438–13443. 10.1073/pnas.1016546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams D. J.; Butler M. F.; Frith W. J.; Kirkland M.; Mullen L.; Sanderson P. A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter 2009, 5, 1856–1862. 10.1039/b901556f. [DOI] [Google Scholar]
- Stendahl J. C.; Rao M. S.; Guler M. O.; Stupp S. I. Intermolecular forces in the self-assembly of peptide amphiphile nanofibers. Adv. Funct. Mater. 2006, 16, 499–508. 10.1002/adfm.200500161. [DOI] [Google Scholar]
- Raeburn J.; Chen L.; Awhida S.; Deller R. C.; Vatish M.; Gibson M. I.; Adams D. J. Using molecular rotors to probe gelation. Soft Matter 2015, 11, 3706–3713. 10.1039/C5SM00456J. [DOI] [PubMed] [Google Scholar]
- Stsiapura V. I.; Maskevich A. A.; Kuzmitsky V. A.; Uversky V. N.; Kuznetsova I. M.; Turoverov K. K. Thioflavin T as a molecular rotor: fluorescent properties of thioflavin T in solvents with different viscosity. J. Phys. Chem. B 2008, 112, 15893–15902. 10.1021/jp805822c. [DOI] [PubMed] [Google Scholar]
- Langeveld-Voss B. M. W.; Waterval R. J. M.; Janssen R. A. J.; Meijer E. W. Principles of “Majority Rules” and “Sergeants and Soldiers” applied to the aggregation of optically active polythiophenes: Evidence for a multichain phenomenon. Macromolecules 1999, 32, 227–230. 10.1021/ma981349y. [DOI] [Google Scholar]
- Deshmukh S. A.; Solomon L. A.; Kamath G.; Fry H. C.; Sankaranarayanan S. K. R. S. Water ordering controls the dynamic equilibrium of micelle–fibre formation in self-assembly of peptide amphiphiles. Nat. Commun. 2016, 7, 12367 10.1038/ncomms12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K.; Ji W.; Palmer L. C.; Weber B.; Barz M.; Stupp S. I. Programmable assembly of peptide amphiphile via noncovalent-to-covalent bond conversion. J. Am. Chem. Soc. 2017, 139, 8995–9000. 10.1021/jacs.7b03878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantakitti F.; Boekhoven J.; Wang X.; Kazantsev R. V.; Yu T.; Li J.; Zhuang E.; Zandi R.; Ortony J. H.; Newcomb C. J.; et al. Energy landscapes and functions of supramolecular systems. Nat. Mater. 2016, 15, 469–476. 10.1038/nmat4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerkle L. E.; Rowan S. J. Supramolecular gels formed from multi-component low molecular weight species. Chem. Soc. Rev. 2012, 41, 6089–6102. 10.1039/c2cs35106d. [DOI] [PubMed] [Google Scholar]
- Draper E. R.; Wallace M.; Schweins R.; Poole R. J.; Adams D. J. Nonlinear effects in multicomponent supramolecular hydrogels. Langmuir 2017, 33, 2387–2395. 10.1021/acs.langmuir.7b00326. [DOI] [PubMed] [Google Scholar]
- Fleming S.; Debnath S.; Frederix P. W. J. M.; Hunt N. T.; Ulijn R. V. Insights into the coassembly of hydrogelators and surfactants based on aromatic peptide amphiphiles. Biomacromolecules 2014, 15, 1171–1184. 10.1021/bm401720z. [DOI] [PubMed] [Google Scholar]
- Chen J.-Y.; Yuan B.; Li Z.-Y.; Tang B.; Gupta A.; Bhosale S.-V.; Li J.-L. Synergistic coassembly of two structurally different molecular gelators. Langmuir 2016, 32, 12175–12183. 10.1021/acs.langmuir.6b03527. [DOI] [PubMed] [Google Scholar]
- Chen L.; Morris K.; Laybourn A.; Elias D.; Hicks M. R.; Rodger A.; Serpell L.; Adams D. J. Self-assembly mechanism for a naphthalene–dipeptide leading to hydrogelation. Langmuir 2010, 26, 5232–5242. 10.1021/la903694a. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; Newcomb C. J.; Meijer E. W.; Stupp S. I. Pathway selection in peptide amphiphile assembly. J. Am. Chem. Soc. 2014, 136, 8540–8543. 10.1021/ja503882s. [DOI] [PubMed] [Google Scholar]
- Fu I. W.; Markegard C. B.; Chu B. K.; Nguyen H. D. The role of electrostatics and temperature on morphological transitions of hydrogel nanostructures self-assembled by peptide amphiphiles via molecular dynamics simulations. Adv. Healthcare Mater. 2013, 2, 1388–1400. 10.1002/adhm.201200400. [DOI] [PubMed] [Google Scholar]
- Yu R.; Lin N.; Yu W.; Liu X. Y. Crystal networks in supramolecular gels: formation kinetics and mesoscopic engineering principles. CrystEngComm 2015, 17, 7986–8010. 10.1039/C5CE00854A. [DOI] [Google Scholar]
- Abbah S. A.; Delgado L. M.; Azeem A.; Fuller K.; Shologu N.; Keeney M.; Biggs M. J.; Pandit A.; Zeugolis D. I. Harnessing hierarchical nano- and micro-fabrication technologies for musculoskeletal tissue engineering. Adv. Healthcare Mater. 2015, 4, 2488–2499. 10.1002/adhm.201500004. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Nieuwenhuizen M. M. L.; de Greef T. F. A.; van der Schoot P.; Schenning A. P. H. J.; Meijer E. W. How to distinguish isodesmic from cooperative supramolecular polymerisation. Chem. - Eur. J. 2009, 16, 362–367. 10.1002/chem.200902415. [DOI] [PubMed] [Google Scholar]
- Hirst A. R.; Coates I. A.; Boucheteau T. R.; Miravet J. F.; Escuder B.; Castelletto V.; Hamley I. W.; Smith D. K. Low-Molecular-Weight Gelators: Elucidating the principles of gelation based on gelator solubility and a cooperative self-assembly model. J. Am. Chem. Soc. 2008, 130, 9113–9121. 10.1021/ja801804c. [DOI] [PubMed] [Google Scholar]
- Hsieh M.-C.; Lynn D. G.; Grover M. A. Kinetic model for two-step nucleation of peptide assembly. J. Phys. Chem. B 2017, 121, 7401–7411. 10.1021/acs.jpcb.7b03085. [DOI] [PubMed] [Google Scholar]
- Kubota R.; Liu S.; Shigemitsu H.; Nakamura K.; Tanaka W.; Ikeda M.; Hamachi I. Imaging-based study on control factors over self-sorting of supramolecular nanofibers formed from peptide- and lipid-type hydrogelators. Bioconjugate Chem. 2018, 29, 2058–2067. 10.1021/acs.bioconjchem.8b00260. [DOI] [PubMed] [Google Scholar]
- Wu A.; Isaacs L. Self-Sorting: The Exception or the Rule?. J. Am. Chem. Soc. 2003, 125, 4831–4835. 10.1021/ja028913b. [DOI] [PubMed] [Google Scholar]
- Draper E. R.; Adams D. J. How should multicomponent supramolecular gels be characterised?. Chem. Soc. Rev. 2018, 47, 3395–3405. 10.1039/C7CS00804J. [DOI] [PubMed] [Google Scholar]
- Biovia Software Inc. 2019, http://accelrys.com/products/collaborative-science/biovia-materials-studio/ (accessed Jan 23, 2019).
- Nakahata M.; Takashima Y.; Yamaguchi H.; Harada A. Redox-responsive self-healing materials formed from host–guest polymers. Nat. Commun. 2011, 2, 511 10.1038/ncomms1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Du S.; Liu Y.; Zhao H.; Chen C.; Li Z.; Lin J.; Zhang Y.; Zhang J.; Wan X. Tunable, luminescent, and self-healing hybrid hydrogels of polyoxometalates and triblock copolymers based on electrostatic assembly. Chem. Commun. 2014, 50, 1447–1450. 10.1039/C3CC48732F. [DOI] [PubMed] [Google Scholar]
- Yuan D.; Xu B. Heterotypic supramolecular hydrogels. J. Mater. Chem. B 2016, 4, 5638–5649. 10.1039/C6TB01592A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke D. E.; Pashuck E. T.; Bertazzo S.; Weaver J. V. M.; Stevens M. M. Self-Healing, Self-Assembled β-Sheet Peptide–Poly(γ-glutamic acid) Hybrid Hydrogels. J. Am. Chem. Soc. 2017, 139, 7250–7255. 10.1021/jacs.7b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M. C.; Fuentes Caparrós A. M.; Dietrich B.; Barrow M.; Cross E. R.; Bleuel M.; King S. M.; Adams D. J. Optimising low molecular weight hydrogels for automated 3D printing. Soft Matter 2017, 13, 8426–8432. 10.1039/C7SM01694H. [DOI] [PubMed] [Google Scholar]
- Yan C.; Altunbas A.; Yucel T.; Nagarkar R. P.; Schneider J. P.; Pochan D. J. Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. 10.1039/c0sm00642d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle M.; Bernkop-Schnürch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- Dehsorkhi A.; Hamley I. W.; Seitsonen J.; Ruokolainen J. Tuning self-assembled nanostructures through enzymatic degradation of a peptide amphiphile. Langmuir 2013, 29, 6665–6672. 10.1021/la401025r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland L. L.; Twomey J. D.; Vogel S.; Hsieh A. H.; Yu Y. B. Enhancing biocompatibility of d-oligopeptide hydrogels by negative charges. Biomacromolecules 2013, 14, 406–412. 10.1021/bm301598g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X.; Zhou J.; Shi J.; Xu B. Supramolecular hydrogelators and hydrogels: from soft matter to molecular biomaterials. Chem. Rev. 2015, 115, 13165–13307. 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanekamp R. J.; Welch J. J.; Nilsson B. L. Proteolytic stability of amphipathic peptide hydrogels composed of self-assembled pleated β-sheet or coassembled rippled β-sheet fibrils. Chem. Commun. 2014, 50, 10133–10136. 10.1039/C4CC04644G. [DOI] [PubMed] [Google Scholar]
- Toft D. J.; Moyer T. J.; Standley S. M.; Ruff Y.; Ugolkov A.; Stupp S. I.; Cryns V. L. Coassembled cytotoxic and pegylated peptide amphiphiles form filamentous nanostructures with potent antitumor activity in models of breast cancer. ACS Nano 2012, 6, 7956–7965. 10.1021/nn302503s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.