

Abstract
Recent decades have seen an alarming increase in the incidence of cardia gastric adenocarcinoma (CGA) while noncardia gastric adenocarcinoma (NCGA) has decreased. In 2012, 260 000 CGA cases (age‐standardised rate (ASR); 3.3/100 000) and 691 000 NCGA cases (ASR; 8.8/100 000) were reported worldwide. Compared with women, men had greater rates for both the subsites, especially for CGA. Recently, four molecular subtypes of GC have been proposed by the Cancer Genome Atlas (TCGA) and the Asian Cancer Research Group (ACRG); however, these classifications do not take into account predisposing germline variants and their possible interaction with somatic alterations in carcinogenesis. The etiology of adenocarcinoma of the cardia and the gastroesophageal junction (GEJ) is not known. It is thought that CGA is distinct from adenocarcinomas located in the esophagus or distal stomach, both epidemiologically and biologically. Moreover, CGA is often identified in the advanced stage having a poor prognosis. Therefore, understanding the risk and the role of predisposing factors in etiology of CGA can inform clinical practice and counseling for risk reduction. In this paper, we showed that GC family history, lifestyle, demographics, gastroesophageal reflux disease, Helicobacter pylori infection, and multiple genetic and epigenetic risk factors as well as several predisposing conditions may underlie susceptibility to CGA. However, several genome‐wide association studies (GWASs) should be conducted to identify novel high‐penetrance genes and pathways as well as causal germline variants predisposing to CGA. They must include different ethnic groups, especially from high‐incidence countries for CGA, because some risk loci are ancestry‐specific. In parallel, statistical methods can be developed to identify cancer predisposition genes (CPGs) from tumor sequencing data. It is also necessary to find novel long noncoding RNAs related to the risk of CGA. Taken altogether, new cancer risk prediction models, including all genetic and nongenetic factors influencing risk, should be developed to facilitate risk assessment, disease prevention, and early diagnosis and intervention of CGA in the future.
Keywords: cardia gastric adenocarcinoma, Helicobacter pylori, risk biomarkers
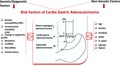
The etiology of adenocarcinoma of the cardia and the gastroesophageal junction (GEJ) is not known and is doubted. It is thought that cardia gastric adenocarcinoma (CGA) is distinct from adenocarcinomas located in the esophagus or distal stomach, both epidemiologically and biologically. Moreover, CGA is often identified in the advanced stage having a poor prognosis. In this paper, we would like to ascertain the possible role of GC family history, lifestyle, demographics, gastroesophageal reflux disease, Helicobacter pylori infection, and multiple genetic and epigenetic risk factors as well as several predisposing conditions in susceptibility to CGA.
1. INTRODUCTION
Gastric cancer (GC) is the fifth common cancer (6.8%) in the world and the third leading cause of death related to cancer (8.8%) worldwide.1 In fact, the complicated interaction between Helicobacter pylori (H pylori) infection and genetic, epigenetic, and environmental factors results in GC.2 Gastric adenocarcinoma is the prominent type of GC, which is classified into two major histological subtypes of intestinal and diffuse adenocarcinoma according to Lauren's classification, reflecting its pathogenesis.3 There are two GC subtypes, cardia (occurring in the 1‐cm (cm) proximal and 2‐cm distal area of the esophago‐gastric junction) gastric adenocarcinoma (CGA) and noncardia (distal: involving the distal and middle parts of the stomach) gastric adenocarcinoma (NCGA).4 In 2012, 260 000 CGA cases (age‐standardised rate (ASR) 3.3 per 100 000) and 691 000 NCGA cases (ASR 8.8) were reported all over the world. The greatest regional rates of both GC subsites were in Eastern/Southeastern Asia (in men, ASRs: 8.7 and 21.7 for CGA and NCGA, respectively). NCGA was observed more commonly than CGA with a mean ratio of 2:1 in most countries, but in some populations, the rates of NCGA incidence were less than the global mean.5 Ardabil Province in Northwest of Iran has the highest CGA rates in the world. In Ardabil, over one‐third of the GC occurs in the cardia region of the stomach having only 5%‐10% of the whole stomach, and the ASRs for CGA are 26.4 and 8.6 for males and females, respectively.6
The etiology of adenocarcinoma of the cardia and the gastroesophageal junction (GEJ) is not known and is doubted. It is thought that CGA is distinct from adenocarcinomas located in the esophagus or distal stomach, both epidemiologically and biologically.7 Moreover, CGA is often identified in the advanced stage having a poor prognosis. In this paper, we would like to ascertain the possible role of GC family history, lifestyle, demographics, gastroesophageal reflux disease, H pylori infection, and multiple genetic and epigenetic risk factors as well as several predisposing conditions in susceptibility to CGA. Therefore, understanding risk and the role of these factors in etiology of CGA can inform clinical practice and counseling for risk reduction.
2. FAMILY HISTORY
Most GCs are sporadic; however, nearly 10% represents familial aggregation with an unclear molecular basis. Hereditary cancers constitute less than 3% of all stomach cancers and are recessed into the three autosomal dominant syndromes: hereditary diffuse GC (HDGC), familial intestinal GC, and gastric adenocarcinoma and proximal polyposis of the stomach.8 HDGC is the most commonly known familial GC and is characterized by CDH1 deletion. However, it is rare, not taking into account a large proportion of family clustering.9 The incidence rate of HDGC in the cardia and noncardia subsites of the stomach is also not clear.
Family history of GC raises the risk of its development, with risks ranging from 1.3 to 3.0 for the first‐degree relatives of GC cases. GC development under 50 years of age is probably followed by family history.10 People with a positive paternal family history were at higher risk of GC compared to positive maternal family history.11 Coexistence of two risk factors including a positive family history and infection with a CagA‐positive H pylori isolate could increase more than 16‐fold risk of NCGA and eightfold total risk of CGA.12 Thus, identifying inherited parameters among subjects with GC family histories is an important step for due diagnosis and management of the disease.
3. DEMOGRAPHIC AND BEHAVIORAL FACTORS
The GC incidence increases with age. The median age for GC diagnosis is 70.13 Compared with women, men had greater rates for both the subsites, especially for CGA (male‐to‐female ratio 3:1).5 This marked difference is likely to be due to endogenous factors, such as reproductive hormones, different prevalence of central obesity between two sexes, or different premenopausal iron status. However, it cannot be explained by different smoking histories.14 Estrogen—the female sex hormone—is a suppressor of the inflammatory response and cytokine production in certain tissues, thus likely having similar effects in the upper gastrointestinal (GI) tract. In addition, lower body iron stored during their reproductive years in females might change the degree of DNA damage caused by chronic inflammation. Male predominance of upper GI adenocarcinomas is also related to the intestinal subtype rather than tumor subsite because of delayed development of this subtype in females before 50‐60 years.15
A meta‐analysis study revealed that smoking was associated with CGA and the relative risk (RR) was 1.87. RR rose from 1.3 for the lowest intake to 1.7 for about 30 cigarettes per day.16 Risks of CGA were higher than those of NCGA in former, moderate, and high‐intensity cigarette smokers.17 It also relates opium use to a higher risk of GC18 with an augmented CGA risk (OR = 2.8).19 The obesity prevalence, indicated by body mass index (BMI ≥30 kg/m2), has increased over the past two decades. Fat is metabolically active and generates many compounds that move in the body. These products (eg, insulin‐like growth factor and leptin) are related to malignancies, probably via inducing pro‐growth changes in the cycle of a cell, declined cell death, and pro‐neoplastic cellular variations.20 Meta‐analysis showed that risen BMI correlated with the CGA risk (CGA, summary relative risk, SRRs = 1.21 and 1.82 for overweight and obesity, respectively, but not with NCGA (NCGA; SRRs = 0.93 and 1.00 for overweight and obesity, respectively.21 A meta‐analysis revealed a 21% decline in GC risk, in those having higher physical activity compared to the least active ones. This risk decline was reported for both NCGA (37% risk reduction) and CGA (20% risk reduction).22
4. GASTROESOPHAGEAL REFLUX DISEASE
Gastroesophageal reflux disease (GERD), troublesome and recurrent heartburn and regurgitation, is known as a primary risk factor for upper gastrointestinal cancers. Significant associations have been found between CGA and GERD, with two‐ to fourfolds of increased risk in many studies; however, not all studies confirm it.23, 24 The increase in the occurrence of CGA in the Western world was elaborated by increasing GERD incidence and obesity.25 CGA was related with gastric atrophy (OR = 3.92) and GERD symptoms (OR = 10.08), hence results show two different etiologies of CGA, one resulting from intense atrophic gastritis (intestinal or diffuse subtype) as NCGA and another from GERD (intestinal subtype).23, 26 Endoscopic screening of men with chronic GERD symptoms (≥5 years) who have at least two additional risk factors (eg age >50 years, central obesity, past or current history of smoking, White race, or family history of Barrett esophagus) is suggested by current guidelines.27 However, there are junctional cancers in patients who never had typical reflux diseases, largely explained by two entities of partial hiatus hernia and intrasphincteric reflux.28 Hiatal hernia (HH) is a significant independent risk factor for CGA and esophageal adenocarcinoma. HH in combination with reflux symptoms was strongly associated with the risk of esophageal adenocarcinomas (OR = 8.11). This association was more modest for CGA (OR = 2.93).29 It has also been shown that in the asymptomatic, moderately overweight population with no reflux, there are cardiac mucosal lengthening and proximal extension of gastric acid within the lower esophageal sphincter, thus likely causing the observed change in the cardiac mucosa. These changes may be related to the etiology of CGA and GEJ, often seen in people without a history of reflux disease.30, 31
5. Helicobacter pylori INFECTION
The main risk factor of intestinal metaplasia, chronic atrophic gastritis, and gastric adenocarcinoma is H pylori that colonizes the human stomach.32 Studies on Asian countries have revealed a higher positive association between H pylori infection and CGA, while some other studies of Western countries have reported no association or even inverse association.33, 34 The meta‐analysis provided evidence for a positive association between CGA and H pylori infection. For CGA, summary RR was 1.08 (95% CI 0.83‐1.40), greater in high‐risk (RR = 1.98; 95% CI 1.38‐2.83) than in low‐risk situations (RR = 0.78; 95% CI 0.63‐0.97).35 Individual antigen testing has revealed that CagA positivity is associated with an increased risk of CGA and NCGA, which is in line with other studies conducted in Asian populations.36 The vacA c1 genotype of H pylori has strongly increased the risk of CGA (OR = 14.11). H pylori vacA c1 genotype is also thought to be the primary bacterial biomarker for the prediction of CGA risk in Iranian males aged >55.37 In contrast, the vacA c2 genotype, particularly in combination with cagPAI genotypes (ie cagH, cagL, cagG, and orf17), showed strong inverse associations with the risk of CGA and non‐CGA, indicating a coordinated relationship between the vacA c2 and cagPAI genotypes.38
6. GENETIC RISK FACTORS
6.1. New molecular subtypes of GC
Recently, four molecular subtypes of GC have been determined by the Cancer Genome Atlas (TCGA) project, which include Epstein‐Barr virus (EBV), microsatellite instability (MSI), genomically stable (GS), and chromosomal instability (CIN).39 CIN subtype, which mostly occurs in the esophago‐gastric junction (EGJ)/cardia, represents at least 50% of GCs.40 It is related to intestinal‐type histology, showing elevated frequency in the EGJ/cardia, according to TCGA characterization (65%).41 Furthermore, the Asian Cancer Research Group (ACRG) has proposed other molecular classification, including mesenchymal subgroup (MSS/EMT), microsatellite instability subgroup (MSI), Microsatellite Stable TP53‐positive (MSS/TP53+, corresponding to EBV+ subtype by TCGA), and Microsatellite Stable TP53‐negative tumors (MSS/TP53−, corresponding to CIN subtype by TCGA). Microsatellite‐unstable tumors, which occur in the antrum, are hypermutated intestinal‐subtype tumors having the best prognosis and the lowest frequency of recurrence (22%) of the four subtypes. The mesenchymal‐like type, including diffuse‐subtype tumors, which have the tendency to occur at an earlier age, shows the worst prognosis and the highest recurrence frequency (63%) of the four subtypes.42
These classifications open new horizons for identification of relevant genomic subsets for precision oncology using highly complex methodologies, including genomic screening and molecular, epigenetic, and functional characterization. However, the two classifications have some limitations. They lack a prospective validation on a large scale, including patients from other geographic regions of the world. The differences between them are greater than similarities, which include differences in molecular mechanisms, relation to prognosis, and the distribution of Lauren's diffuse subtype among the four subgroups. Neither of them considers active and nonmalignant stromal cells. Stromal gene expression profiles may influence assignment to a specific subtype. On the other hand, novel stromal‐based signatures have been related to the dominant cancer phenotypes. Thus, the classification of GC can be improved from a tumor stroma perspective.43, 44, 45
Although these subtypes may be related to the prognosis of GC patients and determine the patient's benefits from adjuvant chemotherapy after large‐scale validation trials, they do not take into account predisposing inherited germline variants for cancer. Recent data have shown that somatic cancer genes also show recessive rare, damaging germline variants (RDGVs) that predispose to cancer via a two‐hit mechanism.46 This indicates a possible interaction of the germline variants with somatic driver alterations in carcinogenesis. For example, germline variants in RBFOX1, a gene encoding an RNA‐binding protein involved in splicing, increase the incidence of SF3B1 somatic mutation by eightfold. Similarly, 19p13.3 variants are associated with a fourfold increase in somatic mutation rate of the PTEN tumor suppressor gene.47 However, the impact of large‐scale tumor sequencing has been limited in identifying cancer predisposition genes (CPGs).
6.2. Single‐nucleotide polymorphisms in CGA
Single‐nucleotide polymorphisms (SNPs) are natural genetic changes occurring with different frequencies in various populations. Some SNPs may change the gene expression profile and influence function of the gene, leading to risen susceptibility risk to the range of some disorders, like cancer. There are many instances of polymorphic genes, which raise the susceptibility to GC.
6.2.1. PRKAA1
One SNP, rs10074991 in PRKAA1 at 5p13.1, reached genome‐wide significance for CGA. PRKAA1 protein is a catalytic subunit of AMP‐activated protein kinase (AMPK), crucial for the regulation of cellular energy metabolism. To respond to the decline of intracellular ATP levels, AMPK stimulates energy‐production pathways and prevents processes of energy consuming leading to the inhibition of biosynthesis of protein, carbohydrate, and lipid, and prevention of cell growth and proliferation.48
6.2.2. MUC1 and PLCE1
The glycoprotein Mucin 1 is aberrantly glycosylated and overexpressed in epithelial cancers, and plays an important role in disease progression.49 Phospholipase C epsilon‐1 (PLCE1) is a phospholipase C isoenzyme encoded by PLCE1 gene, it interacts with the proto‐oncogene Ras among other proteins. PLCE1‐related signaling network affects many critical carcinogenetic processes like metabolism, proliferation, survival, and tumor growth. In a genome‐wide association study (GWAS) conducted among Chinese people, positive correlations among SNPs in MUC1 and CGA and NCGA were similar. Two independent GWAS datasets in Chinese showed associations between multiple variants at 10q23, on gene PLCE1, and CGA risk.50, 51
6.2.3. NF‐κBs
NF‐κBs are stimulated in many cancers, the equivalent of “nonclassical oncogene.” The combined effect analysis revealed that when carrying the NFKBIA gene polymorphism site of rs696 (AA) and NFKB1 gene polymorphism site of rs3755867 (GG), the CGA incidence risk was more than the time the adverse genotype (OR = 5.22) was not carried.52
6.2.4. IL1B‐31C, IL1B‐511T, and IL1RN2
Non‐Asian populations also showed augmented risks among IL1B‐31C, IL1B‐511T, and IL1RN2 carriers for CGA, but this was not significant in Asian populations.53
6.2.5. P27 (kip1)
The p27kip1 expression is an early event in gastric tumorigenesis, and is regarded as a candidate molecular biomarker for early GC.54 P27 (kip1) polymorphisms may be associated with the CGA susceptibilities in North China.
6.2.6. MTHFR
The enzyme methylenetetrahydrofolate reductase (MTHFR) has an important role in the regulation of methionine and homocysteine concentrations in folate metabolism.55 Individuals with the MTHFR 677TT variant genotype possessed a twofold increased CGA risk (OR = 2.04).56
6.2.7. ADPRT
A study showed ORs of 2.17 and 1.61 for CGA in the ADPRT (Adenosine diphosphate ribosyl transferase) Ala/Ala or XRCC1 (X‐ray repair cross‐complementing 1) Gln/Gln genotype carriers, respectively, compared to noncarriers. Gene‐gene interaction of XRCC1 and ADPRT polymorphisms raised the OR of CGA in a hasty manner (OR for the combined XRCC1 Gln/Gln and ADPRT Ala/Ala genotypes was 6.43).57
6.2.8. COX‐2
COX‐2, a major enzyme converting arachidonate to prostaglandins, is not present in normal cells unless quickly stimulated by different carcinogens. The level of COX‐2 was considerably increased in gastrointestinal cancer.58 Multivariate logistic regression analysis showed that the −1195AA, −765GC, and 587Arg/Arg genotypes of COX‐2 were related with increased CGA risk (OR = 1.50, OR = 2.06, and OR = 1.67, respectively). These results showed that the functional polymorphisms of COX‐2, when interacting with smoking, have an influential impact on developing CGA.59
6.2.9. MDM2
Some epidemiological studies have found an association between murine double minute 2 (MDM2) SNP309 and the risk of different cancer types. TP53 induces intracellular expression of MDM2, whereas the latter induces the downregulation of TP53, the auto‐regulatory feedback loop between TP53 and MDM2. The relationship between MDM2 SNP309 and GC risk was meaningful, especially in CGA for the H pylori‐positive population group.60 Genotype analyses demonstrated that increased risk for development of CGA was correlated with the MDM2 309G and the P53 72Pro allele compared to the P53 72Arg allele and the MDM2 309T in an allele dose‐dependent manner.61
6.2.10. RANK
Overexpression of receptor activator of nuclear factor κ B (RANK) directly induces epithelial‐to‐mesenchymal transition and stem‐like phenotypes in tumor cells and normal mammary epithelial cells. The RANK/ RANKL/OPG system, mechanistically, affects tumor cell invasion and migration.62 RANK rs1805034 T>C correlates with susceptibility to CGA, which is more obvious in elderly patients, male patients, smokers, and patients with no alcohol consumption.63
6.2.11. PD‐1
Programmed cell death‐1 (PD‐1) is a major preventer of antitumor responses; it is a cogent candidate for genetic risk of subjects to many malignancies. Two ligands of PD‐1, programmed death‐1 ligand 1 (PD‐L1) and PD‐L2, inhibit activation and proliferation of T cells, leading to tumor escape from immune surveillance.64 A considerable increased risk of CGA related with the PD‐1 rs2227982 C>T polymorphism was observed among ever drinking subjects (TT vs CC: OR = 2.53, TT+CT vs CC: OR = 2.04).65 According to TCGA, PD‐L1 gene was frequently amplified in EBV‐positive GC, probably indicating the higher immunogenicity of this GC subclass. Amplification of a chromosomal region 9p24.1 (locus of PD‐L1 and PD‐L2) has been seen at 15% of EBV‐positive GC.66
6.2.12. MYT1
MYT/NZF family transcription factors include two major members, myelin transcription factor 1 (MYT1, or neural zinc finger 2 (NZF2)) and its homologue MYT1‐like (MYT1L or NZF1); each of them has six copies of a ZnF including a C2HC consensus sequence. MYT1 is also related with carcinoma.67 MYT1L rs17039396 variants could be a suitable prognostic indicator for GC, especially among the CGA.68
6.2.13. XPG
XPG gene (or ERCC5) affects the excision of an *24‐32 bp DNA segment having the bulky adduct in nucleotide excision repair (NER). The T/T genotype of XPG and rs751402 C/T SNP T allele was correlated with an increased CGA risk in younger subjects (≤61 years; OR = 1.33). The T/T genotype carriers must receive periodic upper gastrointestinal endoscopy to facilitate the early diagnosis and cure of CGA.69
6.2.14. MMP‐2
Matrix metalloproteinase‐2 (MMP‐2) is mainly responsible for regulating inflammatory response.70 People with the CC genotype of MMP‐2 had >threefold augmented risk (OR = 3.36) for development of CGA in comparison to those with the variant CT or TT genotype.71 MMP‐2 C−1306T polymorphism is a risk factor for CGA and the multifactor interactions among polymorphisms in FASL, MMP‐2, and FAS affect the CGA development.72 The detailed information regarding the genetic factors of CGA are indicated in Table 1.
Table 1.
Role of genetic factors in CGA
| Case/control | P‐value | OR (95% CI) | Ref. | |
|---|---|---|---|---|
| PRKAA1 (rs10074991) | 3042/7548 | 7.36 × 10−12 | 0.83 (0.79‐0.88) | [ 48] |
| MUC‐1 | [ 50] | |||
| rs4072037 (A>G) | 1213/3302 | 9.5 × 10−5 | 0.75 (0.62‐0.87) | |
| rs4460629 (C>T) | 1.3 × 10−4 | 0.74 (0.64‐0.86) | ||
| PLCE1 | [ 50, 51] | |||
| rs2274223 (A>G) | 2766/ 11013 | 1.7 × 10−39 | 1.55 (1.45‐1.66) | |
| rs2274223 (A>G) | 4.2 × 10−15 | 1.57 (1.40‐1.76) | ||
| rs3765524 (C>T) | 1213/3302 | 7.4 × 10−15 | 1.56 (1.40‐1.75) | |
| rs3781264 (T>C) | 1.1 × 10−13 | 1.60 (1.41‐1.81) | ||
| rs11187842 (C>T) | 7.1 × 10−12 | 1.63 (1.42‐1.87) | ||
| rs753724 (G>T) | 8.0 × 10−12 | 1.63 (1.42‐1.87) | ||
| NFKBIA (rs696 AA) | NA | <.05 | 5.22 (1.10, 24.92) | [ 52] |
| NFKB1 (rs3755867 GG) | ||||
| P27(kip1) V/V | 256/437 | <.05 | 2.56 (1.06‐4.78) | [ 54] |
| MTHFR‐ 677TT | 217/468 | <.05 | 2.04 (1.28‐3.26) | [ 56] |
| ADPRT (Ala/Ala) | 500/1000 | .017 | 2.17 (1.55‐3.04) | [ 57] |
| XRCC1 (Gln/Gln) | <.0001 | 1.61 (1.06 ‐2.44) | ||
| COX‐2 | [ 59] | |||
| 1195AA | 357/985 | .038 | 1.50 (1.05‐2.13) | |
| 765GC | .009 | 2.06 (1.29‐3.29) | ||
| 587Arg/Arg | .033 | 1.67 (1.04‐2.66) | ||
| MDM2 ‐309 | [ 60] | |||
| GG vs TT | 999/2322 | <.05 | 2.00 (1.61‐2.50) | |
| GT vs TT | 1.50 (1.20‐1.88) | |||
| RANK (rs1805034 T>C) | [ 63] | |||
| TC vs TT | 323/592 | .026 | NR | |
| CC vs TT | .0003 | NR | ||
| TC/CC vs TT | .0019 | NR | ||
| CC vs TT/TC | .002 | NR | ||
| PD‐1 (rs2227982 C>T) | [ 65] | |||
| TT vs CC | 330/608 | .028 | 2.53 (1.11‐5.79) | |
| TT+CT vs CC | .047 | 2.04 (1.01‐4.13) | ||
| MYT1L (rs17039396 GG) | 174/90 | .001 | NR | [ 68] |
| XPG (rs751402) | [ 69] | |||
| C/T | 212/216 | <.05 | 1.33 (1.00‐1.76) | |
| T/T | .05 | 1.77 (1.12‐3.30) | ||
| MMP2 −1306CC | 356/789 | <.05 | 3.36 (2.34‐4.97) | [ 71] |
| MMP‐2 −1306CC | ||||
| FASL‐ 844TT or TC | 344/324 | <.05 | 4.58 (2.07‐10.14) | |
| FAS‐ 1377AA | [ 72] |
Abbreviations: NA, not available; NR, not reported; SNP, single‐nucleotide polymorphism.
7. EPIGENETIC RISK FACTORS
Promoter CpG island hypermethylation is popular in human cancers and correlates with transcriptional silencing of the associated gene.73 RASSF1A is placed on 3p21.3 and regulates apoptosis, cell cycle, microtubule stability, and other physiological activities. Epigenetic silencing of RASSF1A gene expression through promoter hypermethylation affects CGA. The RASSF1A gene's promoter methylation increased the CGA risk significantly (OR = 7.50).74 The CpG island hypermethylation at the promoter region of HLTF has also been found in the colon and stomach cancers, manifesting that aberrant methylation of HLTF affects carcinogenesis. HLTF methylation may be present in gastric cardia dysplasia phases and may affect the CGA development in subjects with a family history of UGIC.75 The impact of TSP1 on cancer progression is still controversial and shows stimulatory and inhibitory effects. Epigenetic silencing of TSP1 gene via promoter hypermethylation can affect CGA.76 CAV1 may regulate multiple intracellular signaling pathways. CAV1 expression loss with aberrant promoter methylation was detected in some human cancers. The CpG island shore methylation of CAV1 possibly affects the CGA progression and is a prognostic methylation biomarker for CGA cases.77
The loss of p16 (INK4A) protein expression can be detected in 45% of cardiac, esophageal, and gastric adenocarcinoma and correlates with p16 (INK4A) gene hypermethylation. Methylation of CpG in the EBV‐positive class is even greater than that in the MSI class. Moreover, viral cancers have a unique pattern of downregulation‐related methylation of CDKN2A (p16). Hypermethylation of p16 (INK4A) is a common research outcome in CGA.78 The proximal promoter aberrant hypermethylation and MEG3 enhancer region were seen in tissues of CGA. Also, the enhancer region and proximal promoter hypermethylation and dysregulation of MEG3 and miR‐770 were correlated with a survival of poorer CGA patients.79 Aberrant hypermethylation‐mediated downregulation of C5orf66‐AS1 may play critical roles in CGA tumorigenesis and C5orf66‐AS1 can be a prognostic marker in the prediction of CGA patients' survival.80 Epigenetic silencing of Wnt‐antagonist gene expression via promoter hypermethylation can influence CGA.81
Being land of E‐cadherin gene, high methylation status of 5' CPG may be a mechanism in developing CGA.82 A recent study indicated that there were a lot of males with CGA characterized by higher GATA5 DNA methylation values.83 FBXO32 (atrogin‐1) is an Fbox protein family member and has one of the four subunits of the ubiquitin protein ligase complex, contributing to muscle atrophy.84 Aberrant hypermethylation of FBXO32 is a mechanism resulting in loss or downexpression of the gene in CGA. FBXO32 is assumed as a functional tumor suppressor, and FBXO32 gene reactivation may have a therapeutic potential, indicating its role as a prognostic marker for CGA cases.85 It is demonstrated that the loss of RKIP expression and hypermethylation can be regarded as a marker to anticipate clinical result of CGA. It is suggested that RKIP is a new candidate gene among metastasis suppressors.86 The detailed information regarding the epigenetic factors of CGA are indicated in Table 2.
Table 2.
Role of epigenetic factors in CGA
| Case/control | P‐value | OR (95% CI) | Ref. | |
|---|---|---|---|---|
| RASSF1A | 92/30 | <.001 | 7.50 (2.78‐20.23) | [ 74] |
| HLTF | 96/96 | <.05 | NR | [ 75] |
| TSP1 | 96/96 | <.001 | NR | [ 76] |
| CAV1 | 172/172 | <.001 | NR | [ 77] |
| p16INK4A | 50/50 | .002 | NR | [ 78] |
| MEG3 | 134/134 | <.001 | NR | [ 79] |
| C5orf66‐AS1 | 125/125 | <.001 | NR | [ 80] |
| Wnt‐antagonist genes | ||||
| sFRP1 | 94/94 | .000 | NR | [ 81] |
| sFRP 2 | .001 | NR | ||
| sFRP 4 | .000 | NR | ||
| sFRP 5 | .000 | NR | ||
| Wif‐1 | .000 | NR | ||
| Dkk3 | .000 | NR | ||
| E‐cadherin | 92/92 | <.001 | NR | [ 82] |
| GATA5 | 105/105 | <.05 | NR | [ 83] |
| FBXO32 | 139/139 | <.001 | NR | [ 85] |
| RKIP | 145/145 | .000 | NR | [ 86] |
| miR‐25/miR‐93/miR‐106b | ||||
| rs1534309 | 107/1284 | 5.38 × 10−3 | 0.56 (0.37‐0.86) | [ 87] |
| rs2070215 | .0421 | 1.37 (5 1.02‐1.85) | ||
Abbreviations: NA, not available; NR, not reported; SNP, single‐nucleotide polymorphism.
8. LONG NONCODING RNAs
Long noncoding RNAs (lncRNAs) are transcribed RNAs longer than 200 nt which lack an open reading frame of considerable length. lncRNAs are expressed at lower levels compared to mRNAs. lncRNAs’ ectopic expression influences the GC development.88 There are not many articles on the variations of lncRNAs and the risk of CGA development. Notable downregulation of LOC100130476 was observed in primary CGA tissues, and SGC‐7901 and BGC‐823 cell lines. LOC100130476 can function as a tumor inhibitor gene in carcinogenesis of CGA. Aberrant methylation at the CpG sites next to the transcription start site within exon 1 might be important for gene silencing. LOC100130476 ectopic expression is considered a new biomarker for the early diagnosis of GC.89 C5orf66‐AS1 was considerably downregulated in cell lines and CGA tissues, and the level of expression was correlated with lymph node metastasis, pathological differentiation, TNM stage, and distant metastasis or recurrence.90 Table 3 shows the results obtained from microarray analysis of lncRNAs in CGA.
Table 3.
Role of ncRNAs in promoting CGA
| Expression changes | Case/control | P‐value | Fold change (log2) | Ref. | |
|---|---|---|---|---|---|
| LncRNAs | |||||
| C5orf66‐AS1 | Downregulated | 125/125 | <.01 | NA | [ 80] |
| LOC100130476 | Downregulated | 121/121 | .013 | 1.907 (1.148‐3.166)a | [ 89] |
| ASHG19A3A028863 | Upregulated | 12/12 | <.05 | 169.6730934 | [ 90] |
| ASHG19A3A040903 | Upregulated | 41.90954829 | |||
| ASHG19A3A041865 | Upregulated | 39.16918169 | |||
| ASHG19A3A018727 | Upregulated | 28.88943866 | |||
| ASHG19A3A052295 | Upregulated | 24.55914831 | |||
| GUST‐20‐P1426265844 | Upregulated | 22.40102966 | |||
| ASHG19A3A041043 | Upregulated | 20.64951965 | |||
| ASHG19A3A033911 | Upregulated | 15.82403426 | |||
| ASHG19A3A026346 | Upregulated | 15.43079683 | |||
| ASHG19A3A007184 | Downregulated | 59.38580626 | |||
| ASHG19A3A018598 | Downregulated | 15.16286445 | |||
| ASHG19A3A038967 | Downregulated | 9.499758688 | |||
| ASHG19A3H0000023 | Downregulated | 9.473660683 | |||
| ASHG19A3A018662 | Downregulated | 9.338922844 | |||
| ASHG19A3A007413 | Downregulated | 8.588461452 | |||
| ASHG19A3A011053 | Downregulated | 7.817390602 | |||
| ASHG19A3A035937 | Downregulated | 7.2417301 | |||
| ASHG19A3A055173 | Downregulated | 5.954896947 | |||
| ASHG19A3A0001119 | Downregulated | 4.960711075 | |||
| Micro RNAs | |||||
| miR‐770 | Downregulated | 134/134 | <.01 | NR | [ 79] |
| miR‐141 | Downregulated | 41/41 | <.05 | NR | [ 91] |
| miR‐203a | Downregulated | 127/127 | .033 | 1.77 (1.046‐3.011)a | [ 92] |
| miR‐107 (rs2296616 TC/CC) | Upregulated | NA | NR | 1.49 (1.01‐2.20)b | [ 93] |
| miR‐3656 | Downregulated | 21/21 | 1.89E−16 | −3.29535 | [ 94] |
| miR‐378c | Downregulated | 8.96E−14 | −1.80765 | ||
| miR‐628‐3p | Downregulated | 2.23E−13 | −2.03238 | ||
| miR‐US33‐3p | Downregulated | 2.67E−13 | −2.25544 | ||
| miR‐148a‐3p | Downregulated | 2.67E−13 | −1.63085 | ||
| miR‐H10 | Downregulated | 4.43E−13 | −2.84551 | ||
| miR‐638 | Downregulated | 8.99E−13 | −1.55968 | ||
| miR‐483‐5p | Downregulated | 2.20E−12 | −1.35334 | ||
| miR‐675‐5p | Downregulated | 5.11E−12 | −1.70156 | ||
| miR‐1184 | Downregulated | 2.67E−11 | −1.00147 | ||
| miR‐299‐5p | Downregulated | 3.05E−11 | −1.66357 | ||
| miR‐4285 | Downregulated | 4.74E−11 | −1.06365 | ||
| miR‐3665 | Downregulated | 9.57E−11 | −1.95478 | ||
| miR‐H25 | Downregulated | 1.04E−10 | −1.61128 | ||
| miR‐H17 | Downregulated | 1.41E−10 | −1.53334 | ||
| miR‐3195 | Downregulated | 1.41E−10 | −1.28305 | ||
| miR‐518e‐5p | Downregulated | 1.41E−10 | −0.97021 | ||
| miR‐3196 | Downregulated | 7.06E−10 | −2.64801 | ||
| miR‐30d‐5p | Downregulated | 7.06E−10 | −0.74407 | ||
| miR‐3124‐5p | Downregulated | 2.21E−09 | −2.60563 | ||
| miR‐196a‐5p | Upregulated | 3.36E−14 | 4.111534 | ||
| miR‐135b‐5p | Upregulated | 2.67E−13 | 2.555514 | ||
| miR‐2355‐3p | Upregulated | 2.68E−13 | 1.517697 | ||
| miR‐4307 | Upregulated | 1.05E−09 | 2.371521 | ||
| miR‐1244 | Upregulated | 3.68E−09 | 2.409671 | ||
| miR‐892a | Upregulated | 1.05E−08 | 1.8554 | ||
| miR‐20a‐5p | Upregulated | 1.15E−08 | 1.501549 | ||
| miRPlusA1087 | Upregulated | 6.38E−08 | 2.115592 | ||
| miR‐93‐5p | Upregulated | 1.06E−07 | 1.5392 | ||
| miR‐455‐3p | Upregulated | 1.80E−07 | 1.568063 | ||
| miR‐105‐5p | Upregulated | 1.96E−07 | 1.755387 | ||
| miR‐764 | Upregulated | 2.58E−07 | 1.650002 | ||
| miR‐130b‐5p | Upregulated | 4.98E−07 | 1.660447 | ||
| miR‐506‐3p | Upregulated | 2.66E−06 | 1.605885 | ||
| miR‐454‐3p | Upregulated | 3.92E−06 | 1.515466 | ||
| miR‐142‐3p | Upregulated | 4.35E−06 | 1.524762 | ||
| miR‐3591‐3p | Upregulated | 1.19E−05 | 1.452323 | ||
| miR‐196b‐5p | Upregulated | 1.67E−05 | 1.682773 | ||
| miR‐3664‐5p | Upregulated | 4.36E−05 | 1.737875 | ||
| miR‐636 | Upregulated | 9.98E−05 | 1.557929 | ||
Abbreviations: NA, not available; NR, not reported.
OR (95% CI).
Hazard ratio (HR).
9. MICRORNAS
MicroRNAs (miRNAs) are single‐stranded small (20‐22 nt) ncRNAs which regulate gene expression and contribute to a broad spectrum of biological processes like cell proliferation, differentiation, apoptosis, endothelial cell migration, and angiogenesis.95 Some studies reported that miR‐141 was decreased and correlated with lymph node metastases in CGA and advanced TNM stage. Additionally, miR‐141 may stop cell proliferation and trigger apoptosis in adenocarcinoma gastric cell line. Also, miR‐141 may directly stop MACC1 through binding to its 3'‐UTR. It can affect the signaling pathways of MEK/ERK and p38 MAPK. It is a potential therapeutic goal for treating CGA cases.91 MEG3 and miR‐770 were notably downregulated in CGA patients and correlated with lymph node metastasis and TNM stage. The aberrant hypermethylation of the proximal promoter and MEG3 enhancer region was observed in CGA.79 Two tagSNPs of cluster 7.1 (miR‐25/miR‐93/miR‐106b) were found to be related with the GC cardia localization, rs2070215 (OR = 1.37) and rs1534309 (OR = 0.56).87 Significant downregulation and proximal promoter methylation of miR‐203b and miR‐203a in CGA were observed in CGA tissue. CGA cases in stage III and IV with decreased expression or hypermethylation of miR‐203a showed weak survival. MiR‐203b and miR‐203a may act as tumor suppressive miRNAs,miR‐203a reactivation may be regarded as a prognostic marker for CGA subjects.92 MiR‐107 is dysregulated in CGA pathogenesis, and the SNP rs2296616 may affect the process.93 It was found that four miRNAs (ie, miR‐3196, miR‐1244, miR‐135b‐5p, and miR‐628‐3p) were associated with differentiation of CGA. The miR‐196a‐5p was correlated with age of CGA onset. Survival analysis revealed that the miR‐135b‐5p expression level was correlated with survival of CGA.94 Table 3 presents the results obtained from microarray analysis of miRNAs in CGA.
10. CONCLUSION
CGA is a multi‐factorial ailment and most cases are sporadic, although familial cases have been reported. There is much difference between CGA and NCGA in terms of tumor features, distinct etiological factors, and biological behaviors. Lifestyle, H pylori infection, GERD, and multiple genetic, epigenetic, and environmental risk factors have been related to an increased risk of CGA. However, several GWASs, followed by a large‐scale GWAS meta‐analysis, should be conducted to identify novel high‐penetrance genes and pathways as well as causal germline variants predisposing to CGA. They must include different ethnic groups, especially from high‐incidence countries for CGA, because some risk loci are ancestry‐specific.96, 97 In parallel, statistical methods can also be developed to identify CPGs from tumor sequencing data. Then, it should be largely explored how the genetic germline variants and somatic alterations interact to develop CGA in populations with different ethnic backgrounds. A little experiment has also been done on the impact of lncRNAs on the carcinogenesis of the CGA. Therefore, next‐generation high‐throughput RNA‐sequencing techniques can enable us to find novel ncRNA biomarkers related to the risk of CGA. Taken altogether, new cancer risk prediction models, including all genetic and nongenetic factors influencing risk should be developed to facilitate risk assessment, disease prevention, and early diagnosis and intervention of CGA in the future.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
EA, SLN, and SZ provided direction in the preparation of the manuscript. EA and SLN performed primary literature research. EA and SLN wrote the first draft of manuscript. SZ, AY, and FP discussed and revised the manuscript. EA, AY, and FP managed the references. SLN approved the version to be published.
ACKNOWLEDGMENTS
This study was supported by the National Institute for Medical Research Development Grant No.958117. The supporter had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. There was no additional external funding received for this study.
Abdi E, Latifi‐Navid S, Zahri S, Yazdanbod A, Pourfarzi F. Risk factors predisposing to cardia gastric adenocarcinoma: Insights and new perspectives. Cancer Med. 2019;8:6114–6126. 10.1002/cam4.2497
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Chmiela M, Karwowska Z, Gonciarz W, Allushi B, Staczek P. Host pathogen interactions in Helicobacter pylori related gastric cancer. World J Gastroenterol. 2017;23:1521‐1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lauren P. The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma. An Attempt at a histo‐clinical classification. Acta Pathol Et Microbiolo Scand. 1965;64:31‐49. [DOI] [PubMed] [Google Scholar]
- 4. Odze RD, Riddell RH, Bosman FT, et al. Premalignant lesions of the digestive system In: Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. 4th edn Lyon, France: IARC; 2010. [Google Scholar]
- 5. Colquhoun A, Arnold M, Ferlay J, Goodman KJ, Forman D, Soerjomataram I. Global patterns of cardia and non‐cardia gastric cancer incidence in 2012. Gut. 2015;64:1881‐1888. [DOI] [PubMed] [Google Scholar]
- 6. Malekzadeh R, Sotoudeh M, Derakhshan M, et al. Prevalence of gastric precancerous lesions in Ardabil, a high incidence province for gastric adenocarcinoma in the northwest of Iran. J Clin Pathol. 2004;57:37‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang L‐D, Zheng S, Zheng Z‐Y, Casson AG. Primary adenocarcinomas of lower esophagus, esophagogastric junction and gastric cardia: in special reference to China. World J Gastroenterol. 2003;9:1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16:e60‐e70. [DOI] [PubMed] [Google Scholar]
- 9. McLean MH, El‐Omar EM. Genetics of gastric cancer. Nat Rev Gastroenterol Hepatol. 2014;11:664‐674. [DOI] [PubMed] [Google Scholar]
- 10. Hemminki K, Sundquist J, Ji J. Familial risk for gastric carcinoma: an updated study from Sweden. Br J Cancer. 2007;96:1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bakir T, Can G, Siviloglu C, Erkul S. Gastric cancer and other organ cancer history in the parents of patients with gastric cancer. Eur J Cancer Prev. 2003;12:183‐189. [DOI] [PubMed] [Google Scholar]
- 12. Chen M‐J, Wu D‐C, Ko Y‐C, Chiou Y‐Y. Personal history and family history as a predictor of gastric cardiac adenocarcinoma risk: a case‐control study in Taiwan. Am J Gastroenterol. 2004;99:1250. [DOI] [PubMed] [Google Scholar]
- 13. Howlader N, Noone A, Krapcho M, et al. SEER cancer statistics review, 1975‐2008. Bethesda, MD: National Cancer Institute; 2011. Based on November 2010 SEER data submission, posted to the SEER website, http://seer.cancer.gov/csr/1975_2008. Accessed June, 8 [Google Scholar]
- 14. Freedman ND, Derakhshan MH, Abnet CC, Schatzkin A, Hollenbeck AR, McColl KE. Male predominance of upper gastrointestinal adenocarcinoma cannot be explained by differences in tobacco smoking in men versus women. Eur J Cancer. 2010;46:2473‐2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derakhshan MH, Liptrot S, Paul J, Brown IL, Morrison D, McColl KE. Oesophageal and gastric intestinal‐type adenocarcinomas show the same male predominance due to a 17 year delayed development in females. Gut. 2009;58:16‐23. [DOI] [PubMed] [Google Scholar]
- 16. Ladeiras‐Lopes R, Pereira AK, Nogueira A, et al. Smoking and gastric cancer: systematic review and meta‐analysis of cohort studies. Cancer Causes Control. 2008;19:689‐701. [DOI] [PubMed] [Google Scholar]
- 17. Praud D, Rota M, Pelucchi C, et al. Cigarette smoking and gastric cancer in the Stomach Cancer Pooling (StoP) Project. Eur J Cancer Prev. 2018;27:124‐133. [DOI] [PubMed] [Google Scholar]
- 18. Sadjadi A, Derakhshan MH, Yazdanbod A, et al. Neglected role of hookah and opium in gastric carcinogenesis: a cohort study on risk factors and attributable fractions. Int J Cancer. 2014;134:181‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shakeri R, Malekzadeh R, Etemadi A, et al. Opium: an emerging risk factor for gastric adenocarcinoma. Int J Cancer. 2013;133:455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alemán JO, Eusebi LH, Ricciardiello L, Patidar K, Sanyal AJ, Holt PR. Mechanisms of obesity‐induced gastrointestinal neoplasia. Gastroenterology. 2014;146:357‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y, Liu L, Wang X, et al. Body mass index and risk of gastric cancer: a meta‐analysis of a population with more than ten million from 24 prospective studies. Cancer Epidemiol Biomark Prev. 2013;22:1395‐1408. [DOI] [PubMed] [Google Scholar]
- 22. Singh S, Varayil JE, Devanna S, Murad MH, Iyer PG. Physical activity is associated with reduced risk of gastric cancer: a systematic review and meta‐analysis. Cancer Prev Res. 2014;7:12‐22. [DOI] [PubMed] [Google Scholar]
- 23. Derakhshan MH, Malekzadeh R, Watabe H, et al. Combination of gastric atrophy, reflux symptoms and histological subtype indicates two distinct aetiologies of gastric cardia cancer. Gut. 2008;57:298‐305. [DOI] [PubMed] [Google Scholar]
- 24. Figueroa JD, Terry MB, Gammon MD, et al. Cigarette smoking, body mass index, gastro‐esophageal reflux disease, and non‐steroidal anti‐inflammatory drug use and risk of subtypes of esophageal and gastric cancers by P53 overexpression. Cancer Causes Control. 2009;20:361‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Islami F, Sheikhattari P, Ren J, Kamangar F. Gastric atrophy and risk of oesophageal cancer and gastric cardia adenocarcinoma—a systematic review and meta‐analysis. Ann Oncol. 2010;22:754‐760. [DOI] [PubMed] [Google Scholar]
- 26. Mukaisho K‐I, Nakayama T, Hagiwara T, Hattori T, Sugihara H. Two distinct etiologies of gastric cardia adenocarcinoma: interactions among pH, Helicobacter pylori, and bile acids. Front Microbiol. 2015;6:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Falk GW. Updated guidelines for diagnosing and managing barrett esophagus. Gastroenterol Hepatol (NY). 2016;12:449‐451. [PMC free article] [PubMed] [Google Scholar]
- 28. Lee YY, McColl KE. Pathophysiology of gastroesophageal reflux disease. Best Pract Res Clin Gastroenterol. 2013;27:339‐351. [DOI] [PubMed] [Google Scholar]
- 29. Wu AH, Tseng CC, Bernstein L. Hiatal hernia, reflux symptoms, body size, and risk of esophageal and gastric adenocarcinoma. Cancer. 2003;98:940‐948. [DOI] [PubMed] [Google Scholar]
- 30. Lee YY, Wirz AA, Whiting JG, et al. Waist belt and central obesity cause partial hiatus hernia and short‐segment acid reflux in asymptomatic volunteers. Gut. 2014;63:1053‐1060. [DOI] [PubMed] [Google Scholar]
- 31. Robertson EV, Derakhshan MH, Wirz AA, et al. Central obesity in asymptomatic volunteers is associated with increased intrasphincteric acid reflux and lengthening of the cardiac mucosa. Gastroenterology. 2013;145:730‐739. [DOI] [PubMed] [Google Scholar]
- 32. Watari J, Chen N, Amenta PS, et al. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World J Gastroenterol. 2014;20:5461‐5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Helicobacter and Cancer Collaborative Group . Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001;49:347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pourfarzi F, Whelan A, Kaldor J, Malekzadeh R. The role of diet and other environmental factors in the causation of gastric cancer in Iran–a population based study. Int J Cancer. 2009;125:1953‐1960. [DOI] [PubMed] [Google Scholar]
- 35. Cavaleiro‐Pinto M, Peleteiro B, Lunet N, Barros H. Helicobacter pylori infection and gastric cardia cancer: systematic review and meta‐analysis. Cancer Causes Control. 2011;22:375‐387. [DOI] [PubMed] [Google Scholar]
- 36. Shakeri R, Malekzadeh R, Nasrollahzadeh D, et al. Multiplex H. pylori serology and risk of gastric cardia and non‐cardia adenocarcinomas. Cancer Res. 2015;75:4876‐4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bakhti SZ, Latifi‐Navid S, Zahri S, Bakhti FS, Hajavi N, Yazdanbod A. Are Helicobacter pylori highly cytotoxic genotypes and cardia gastric adenocarcinoma linked? Lessons from Iran. Cancer Biomark. 2017;21:235‐246. [DOI] [PubMed] [Google Scholar]
- 38. Bakhti SZ, Latifi‐Navid S, Zahri S, Yazdanbod A Inverse association of Helicobacter pylori cagPAI genotypes with risk of cardia and non‐cardia gastric adenocarcinoma. Cancer Med. 2019;1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lim B, Kim JH, Kim M, Kim SY. Genomic and epigenomic heterogeneity in molecular subtypes of gastric cancer. World J Gastroenterol. 2016;22:1190‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chia NY, Tan P. Molecular classification of gastric cancer. Ann Oncol. 2016;27:763‐769. [DOI] [PubMed] [Google Scholar]
- 42. Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449‐456. [DOI] [PubMed] [Google Scholar]
- 43. Dunne PD, McArt DG, Bradley CA, et al. Challenging the cancer molecular stratification dogma: intratumoral heterogeneity undermines consensus molecular subtypes and potential diagnostic value in colorectal cancer. Clin Cancer Res. 2016;22:4095‐4104. [DOI] [PubMed] [Google Scholar]
- 44. Garattini SK, Basile D, Cattaneo M, et al. Molecular classifications of gastric cancers: novel insights and possible future applications. World J Gastrointest Oncol. 2017;9:194‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uhlik MT, Liu J, Falcon BL, et al. Stromal‐based signatures for the classification of gastric cancer. Cancer Res. 2016;76:2573‐2586. [DOI] [PubMed] [Google Scholar]
- 46. Park S, Supek F, Lehner B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nat Commun. 2018;9:2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carter H, Marty R, Hofree M, et al. Interaction landscape of inherited polymorphisms with somatic events in cancer. Cancer Discov. 2017;7:410‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu N, Wang Z, Song X, et al. Genome‐wide association study of gastric adenocarcinoma in Asia: a comparison of associations between cardia and non‐cardia tumours. Gut. 2016;65:1611‐1618. 10.1136/gutjnl-2015-309340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nath S, Mukherjee P. MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. 2014;20:332‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abnet CC, Freedman ND, Hu N, et al. A shared susceptibility locus in PLCE1 at 10q23 for gastric adenocarcinoma and esophageal squamous cell carcinoma. Nat Genet. 2010;42:764‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang LD, Zhou FY, Li XM, et al. Genome‐wide association study of esophageal squamous cell carcinoma in Chinese subjects identifies susceptibility loci at PLCE1 and C20orf54. Nat Genet. 2010;42:759‐763. [DOI] [PubMed] [Google Scholar]
- 52. Li D, Wu C, Cai Y, Liu B. Association of NFKB1 and NFKBIA gene polymorphisms with susceptibility of gastric cancer. Tumor Biol. 2017;39:1010428317717107 [DOI] [PubMed] [Google Scholar]
- 53. Persson C, Canedo P, Machado JC, El‐Omar EM, Forman D. Polymorphisms in inflammatory response genes and their association with gastric cancer: a HuGE systematic review and meta‐analyses. Am J Epidemiol. 2011;173:259‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guo W, Cui Y, Fang S, Li Y, Wang N, Zhang J. Association of polymorphisms of p21cip1 and p27kip1 genes with susceptibilities of esophageal squamous cell carcinoma and gastric cardiac adenocarcinoma. Ai zheng. 2006;25:194‐199. [PubMed] [Google Scholar]
- 55. He L, Shen Y. MTHFR C677T polymorphism and breast, ovarian cancer risk: a meta‐analysis of 19,260 patients and 26,364 controls. Onco Targets Ther. 2017;10:227‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miao X, Xing D, Tan W, Qi J, Lu W, Lin D. Susceptibility to gastric cardia adenocarcinoma and genetic polymorphisms in methylenetetrahydrofolate reductase in an at‐risk Chinese population. Cancer Epidemiol Prev Biomark. 2002;11:1454‐1458. [PubMed] [Google Scholar]
- 57. Miao X, Zhang X, Zhang L, et al. Adenosine diphosphate ribosyl transferase and X‐ray repair cross‐complementing 1 polymorphisms in gastric cardia cancer. Gastroenterology. 2006;131:420‐427. [DOI] [PubMed] [Google Scholar]
- 58. Hein DW, Doll MA, Gray K, Rustan TD, Ferguson RJ. Metabolic activation of N‐hydroxy‐2‐aminofluorene and N‐hydroxy‐2‐acetylaminofluorene by monomorphic N‐acetyltransferase (NAT1) and polymorphic N‐acetyltransferase (NAT2) in colon cytosols of Syrian hamsters congenic at the NAT2 locus. Can Res. 1993;53:509‐514. [PubMed] [Google Scholar]
- 59. Zhang X‐M, Zhong R, Liu L, et al. Smoking and COX‐2 functional polymorphisms interact to increase the risk of gastric cardia adenocarcinoma in Chinese population. PLoS ONE. 2011;6:e21894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen W, Wu Q, Ren H. Meta‐analysis of associations between MDM2 SNP309 polymorphism and gastric cancer risk. Biomed Rep. 2014;2:105‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang M, Guo Y, Zhang X, et al. Interaction of P53 Arg72Pro and MDM2 T309G polymorphisms and their associations with risk of gastric cardia cancer. Carcinogenesis. 2007;28:1996‐2001. [DOI] [PubMed] [Google Scholar]
- 62. Palafox M, Ferrer I, Pellegrini P, et al. RANK induces epithelial–mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Can Res. 2012;72:2879‐2888. [DOI] [PubMed] [Google Scholar]
- 63. Zhang W‐B, Gu H‐Y, Shi Y‐J, et al. RANK rs1805034 T>C polymorphism is associated with susceptibility to gastric cardia adenocarcinoma in a Chinese population. Oncol Res Treat. 2015;38:503‐510. [DOI] [PubMed] [Google Scholar]
- 64. Blank C, Gajewski TF, Mackensen A. Interaction of PD‐L1 on tumor cells with PD‐1 on tumor‐specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tang W, Chen Y, Chen S, Sun B, Gu H, Kang M. Programmed death‐1 (PD‐1) polymorphism is associated with gastric cardia adenocarcinoma. Int J Clin Exp Med. 2015;8:8086. [PMC free article] [PubMed] [Google Scholar]
- 66. Derks S, Liao X, Chiaravalli AM, et al. Abundant PD‐L1 expression in Epstein‐Barr Virus‐infected gastric cancers. Oncotarget. 2016;7:32925‐32932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stevens SJ, Van Ravenswaaij‐Arts C, Janssen JW, et al. MYT1L is a candidate gene for intellectual disability in patients with 2p25. 3 (2pter) deletions. Am J Med Genet A. 2011;155:2739‐2745. [DOI] [PubMed] [Google Scholar]
- 68. Zhang Y, Zhu H, Zhang X, et al. Clinical significance of MYT1L gene polymorphisms in Chinese patients with gastric cancer. PLoS ONE. 2013;8:e71979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou R‐M, Niu C‐X, Wang N, et al. XPG gene polymorphisms and the risk of gastric cardia adenocarcinoma. Genet Test Mol Biomark. 2016;20:432‐437. [DOI] [PubMed] [Google Scholar]
- 70. Jakubowska K, Pryczynicz A, Januszewska J, et al. Expressions of matrix metalloproteinases 2, 7, and 9 in carcinogenesis of pancreatic ductal adenocarcinoma. Dis Markers. 2016;2016:9895721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Miao X, Yu C, Tan W, et al. A functional polymorphism in the matrix metalloproteinase‐2 gene promoter (‐1306C/T) is associated with risk of development but not metastasis of gastric cardia adenocarcinoma. Cancer Res. 2003;63:3987‐3990. [PubMed] [Google Scholar]
- 72. Liu L, Wu C, Wang Y, et al. Association of candidate genetic variations with gastric cardia adenocarcinoma in Chinese population: a multiple interaction analysis. Carcinogenesis. 2010;32:336‐342. [DOI] [PubMed] [Google Scholar]
- 73. Jones PA, Laird PW. Cancer‐epigenetics comes of age. Nat Genet. 1999;21:163. [DOI] [PubMed] [Google Scholar]
- 74. Zhou SL, Cui J, Fan ZM, et al. Polymorphism of A133S and promoter hypermethylation in Ras association domain family 1A gene (RASSF1A) is associated with risk of esophageal and gastric cardia cancers in Chinese population from high incidence area in northern China. BMC Cancer. 2013;13:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guo W, Dong Z, Guo Y, Chen Z, Kuang G, Yang Z. Aberrant methylation of the CpG island of HLTF gene in gastric cardia adenocarcinoma and dysplasia. Clin Biochem. 2011;44:784‐788. [DOI] [PubMed] [Google Scholar]
- 76. Guo W, Dong Z, He M, et al. Aberrant methylation of thrombospondin‐1 and its association with reduced expression in gastric cardia adenocarcinoma. J Biomed Biotechnol. 2010;2010:721485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guo Y‐L, Zhu T‐N, Guo W, et al. Aberrant CpG island shore region methylation of CAV1 is associated with tumor progression and poor prognosis in gastric cardia adenocarcinoma. Arch Med Res. 2016b;47:460‐470. [DOI] [PubMed] [Google Scholar]
- 78. Sarbia M, Geddert H, Klump B, Kiel S, Iskender E, Gabbert HE. Hypermethylation of tumor suppressor genes (p16INK4A, p14ARF and APC) in adenocarcinomas of the upper gastrointestinal tract. Int J Cancer. 2004;111:224‐228. [DOI] [PubMed] [Google Scholar]
- 79. Guo W, Dong Z, Liu S, et al. Promoter hypermethylation‐mediated downregulation of miR‐770 and its host gene MEG3, a long non‐coding RNA, in the development of gastric cardia adenocarcinoma. Mol Carcinog. 2017;56:1924‐1934. [DOI] [PubMed] [Google Scholar]
- 80. Guo W, Lv P, Liu S, et al. Aberrant methylation‐mediated downregulation of long noncoding RNA C5orf66‐AS1 promotes the development of gastric cardia adenocarcinoma. Mol Carcinog. 2018;57(7):854‐865. 10.1002/mc.22806 [DOI] [PubMed] [Google Scholar]
- 81. Guo Y, Guo W, Chen Z, Kuang G, Yang Z, Dong Z. Hypermethylation and aberrant expression of Wnt‐antagonist family genes in gastric cardia adenocarcinoma. Neoplasma. 2011;58:110. [DOI] [PubMed] [Google Scholar]
- 82. Guo W, Dong Z, Guo Y, Kuang G, Yang Z, Chen Z. Detection of promoter hypermethylation of the CpG island of E‐cadherin in gastric cardiac adenocarcinoma. Eur J Med Res. 2009;14:453‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wang X, Kang GH, Campan M, et al. Epigenetic subgroups of esophageal and gastric adenocarcinoma with differential GATA5 DNA methylation associated with clinical and lifestyle factors. PLoS ONE. 2011;6:e25985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hanai J‐I, Cao P, Tanksale P, et al. The muscle‐specific ubiquitin ligase atrogin‐1/MAFbx mediates statin‐induced muscle toxicity. J Clin Investig. 2007;117:3940‐3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guo W, Zhang M, Guo Y, Shen S, Guo X, Dong Z. FBXO32, a new TGF‐β/Smad signaling pathway target gene, is epigenetically inactivated in gastric cardia adenocarcinoma. Neoplasma. 2015;62:646‐657. [DOI] [PubMed] [Google Scholar]
- 86. Guo W, Dong Z, Guo Y, et al. Aberrant methylation and loss expression of RKIP is associated with tumor progression and poor prognosis in gastric cardia adenocarcinoma. Clin Exp Metas. 2013;30:265‐275. [DOI] [PubMed] [Google Scholar]
- 87. Espinosa‐Parrilla Y, Muñoz X, Bonet C, et al. Genetic association of gastric cancer with miRNA clusters including the cancer‐related genes MIR29, MIR25, MIR93 and MIR106: results from the EPIC‐EURGAST study. Int J Cancer. 2014;135:2065‐2076. [DOI] [PubMed] [Google Scholar]
- 88. Gu J, Li Y, Fan L, et al. Identification of aberrantly expressed long non‐coding RNAs in stomach adenocarcinoma. Oncotarget. 2017;8:49201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Guo W, Dong Z, Shi Y, et al. Methylation‐mediated downregulation of long noncoding RNA LOC100130476 in gastric cardia adenocarcinoma. Clin Exp Metas. 2016;33:497‐508. [DOI] [PubMed] [Google Scholar]
- 90. Wang Y, Feng X, Jia R, et al. Microarray expression profile analysis of long non‐coding RNAs of advanced stage human gastric cardia adenocarcinoma. Mol Genet Genom. 2014b;289:291‐302. [DOI] [PubMed] [Google Scholar]
- 91. Li S, Zhu J, Li J, Li S, Li B. MicroRNA‐141 inhibits proliferation of gastric cardia adenocarcinoma by targeting MACC1. Archiv Med Sci. 2018;14:588‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu W, Dong Z, Liang J, et al. Downregulation of potential tumor suppressor miR‐203a by promoter methylation contributes to the invasiveness of gastric cardia adenocarcinoma. Cancer Invest. 2016;34:506‐516. [DOI] [PubMed] [Google Scholar]
- 93. Wang S, Lv C, Jin H, et al. A common genetic variation in the promoter of miR‐107 is associated with gastric adenocarcinoma susceptibility and survival. Mutat Res. 2014;769:35‐41. [DOI] [PubMed] [Google Scholar]
- 94. Gao S, Zhou F, Zhao C, et al. Gastric cardia adenocarcinoma microRNA profiling in Chinese patients. Tumor Biol. 2016;37:9411‐9422. [DOI] [PubMed] [Google Scholar]
- 95. Sun LL, Li WD, Lei FR, Li XQ. The regulatory role of microRNAs in angiogenesis‐related diseases. J Cell Mol Med. 2018;22(10):4568‐4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Conti DV, Wang K, Sheng X, et al. Two novel susceptibility loci for prostate cancer in men of African ancestry. J Natl Cancer Inst. 2017;109:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature. 2016;538:161‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]