

Abstract
Nonconservative point mutations of the peripheral myelin protein 22 (PMP22) are associated with Charcot-Marie-Tooth type 1A disease, the most common inherited peripheral neuropathy in humans, and with the Trembler J (TrJ) and Trembler (Tr) alleles in mice. We investigated the intracellular transport of wild-type PMP22 and its TrJ and Tr mutant forms in Schwann cells and in a non-neuronal cell line. In contrast to wild type, mutant proteins were not inserted into the plasma membrane and accumulated in the endoplasmic reticulum and Golgi compartments. Coexpression of each mutant with wild-type PMP22 confirmed the different intracellular distribution of the mutant forms, indicating that neither the TrJ nor Tr protein has a dominant-negative effect on the cellular distribution of wild-type PMP22. Accumulation of PMP22 immunoreactivity in the cell body of myelinating Schwann cells was also observed in nerve biopsies obtained from CMT1A patients carrying the TrJ point mutation. We propose that impaired trafficking of mutated PMP22 affects Schwann cell physiology leading to myelin instability and loss.
Keywords: Schwann cell, PMP22, Trembler J, Trembler, Charcot-Marie-Tooth disease, transfection, chimeric protein, cellular sorting, myelin
Peripheral neuropathies comprise a large and heterogeneous group of disorders, and the molecular mechanisms involved in these pathologies are not yet clearly understood. A subset of these neuropathies is hereditary, and the most common of these is Charcot-Marie-Tooth disease type 1 (CMT1), a motor and sensory neuropathy with an estimated prevalence of 1:2500. The first symptoms of CMT1 diseases appear mostly during the second or third decade of life and are characterized by progressive weakness of the distal muscles, mild sensory impairment, and reduction of peripheral nerve conduction velocities. Nerve biopsies of CMT1 patients show demyelination, remyelination, onion bulb formation, and loss of myelinated axons. CMT1A, the most frequent form, appears to be caused by different mechanisms, such as altered gene dosage or point mutations of the peripheral myelin protein 22 (PMP22). In particular, the majority of CMT1A patients presents a duplication of a 1.5 megabase region of chromosome 17 containing the PMP22 gene, and several distinct nonconservative point mutations of PMP22 (L16P, H12Q, M69K, S72L, S72W, S76I, S79C, S79P, L80P, G100R, G107V, T118M, L147R, G150D) (De Jonghe et al., 1997) have been found in families affected by different subtypes of the disease, displaying phenotypes with varying degrees of disease severity.
The PMP22 gene encodes a transmembrane glycoprotein that contains four transmembrane hydrophobic regions and two extracellular domains, and the amino and C termini are exposed to the cytosol (D’Urso and Müller, 1997). It is highly expressed by myelinating Schwann cells and at lower levels also in several non-neural tissues (Spreyer et al., 1991; Welcher et al., 1991; Baechner et al., 1995). PMP22 is localized mainly in the compact portion of myelin, and it is involved in regulating myelin stability and cell growth (Suter and Snipes, 1995;Zoidl et al., 1995; D’Urso et al., 1997). Point mutations in the PMP22 gene have been found in the spontaneous mouse mutants Trembler J (TrJ) (Suter et al., 1992a) and Trembler (Tr) (Suter et al., 1992b). Both mutants carry nonconservative amino acid changes, leucine to proline (L16P) in TrJ and glycine to aspartic acid (G150D) in Tr, that introduce a helix-breaking amino acid or a charged residue into the first and fourth transmembrane domains of the protein, respectively. Such mutations could modify the tertiary structure of the molecule and interfere with its membrane interactions. TrJ and Tr mice display a disease phenotype very similar to CMT1, with progressive impairment of the hind limbs, severe hypomyelination in the peripheral nervous system (PNS) associated with aberrant Schwann cell growth, and a slow progression of the disease. PNS myelin alterations are more severe in homozygous TrJ and Tr animals compared with heterozygous, suggesting a gene dosage effect. Furthermore, the identical TrJ single mutation has been found in a family affected by a severe form of CMT1A disease (Valentijn et al., 1992), whereas the Tr amino acid substitution was detected in two patients suffering from Dejerine-Sottas demyelinating neuropathy (Ionasescu et al., 1997). Thus, Tr and TrJ mice are regarded as animal models for CMT1A neuropathy.
In vivo studies have shown that PMP22-deficient mice, or mice with only one functional copy of the PMP22 gene, are affected by delayed onset of myelination, tomacula formation at young age, and subsequent severe demyelination (Adlkofer et al., 1995). Transgenic rats (Sereda et al., 1996) or mice (Magyar et al., 1996) that carry additional copies of the PMP22 gene present peripheral hypomyelination and a phenotype similar to that described in CMT1A patients. Furthermore, it has been reported that rat Schwann cells engineered to overexpress PMP22 (Zoidl et al., 1995) or Schwann cells isolated from nerve biopsies of CMT1A patients (Hanemann et al., 1997) are delayed in their growth rates compared with controls.
Taken together, it seems that PMP22 is involved in PNS myelin formation and maintenance and in regulating Schwann cell proliferation and differentiation (for review, see Müller et al., 1997).
We set out to investigate how single point mutations may affect the cellular fate of the PMP22 protein and whether the mutated molecules interfere with the function of the wild-type form. In this study we provide experimental evidence that Schwann cells are not able to target the TrJ and Tr proteins to the plasma membrane. TrJ and Tr PMP22 are abundantly present in the endoplasmic reticulum (ER) and the Golgi apparatus. Coexpression of each mutant with the wild-type protein showed that native and mutated forms display a different cellular distribution. Only the wild-type PMP22 is present in the cytoplasm and the plasma membrane. Similar results were obtained both in Schwann cells and in HeLa cells, a non-neural cell line. Immunostaining and confocal microscopy of sural nerve sections obtained from CMT1A patients carrying the TrJ point mutation are consistent with thein vitro studies, showing accumulation of PMP22 immunoreactivity in the cell body of myelinating Schwann cells. Our observations raise the intriguing possibility that an intracellular accumulation of PMP22 could activate a cascade of events that lead to functional impairment of Schwann cells and destabilize the myelin structure.
MATERIALS AND METHODS
cDNA constructs of PMP22 wild type and mutants. The PMP22 open reading frame sequence, obtained from the pGDSV7-PMP22 vector (Fabbretti et al., 1995), was used to generate the wild-type and mutant constructs subcloned into the pRc-CMV vector (Invitrogen, San Diego, CA). The cDNAs encoding the PMP22 mutants were constructed using PCR site-directed mutagenesis (Ho et al., 1989; Fabbretti et al., 1995). We used two pairs of complementary inverse primers containing the mutated codons 5′-TCGCGGTGCCGGTGCTGCT-3′ and 5′-AGCAGCACCGGCACCGCGA-3′ for the L16P mutation and 5′-TTCTCAGCGATGTCATCTA-3′, 5′-TAGATGACATCGCTGAGAA-3′ for the G150D mutation. We engineered an eight-amino acid Flag epitope in frame at the 3′ end of the wild-type and mutant PMP22 sequence by PCR. To generate the C terminus Flag constructs, the 5′ primer contained an initiation consensus sequence and 27 nucleotides of the PMP22 cDNA starting at the initiation codon (5′-TGCCGCCAGAATGCTCCTCCTGTTGCTGAGTATCA TCGTC-3′), and the 3′ primer included the Flag sequence (underlined) and 12 nucleotides of the 3′ end PMP22 coding region (5′-AGATCTTCA CTTGTCATCGTCGTCCTTGTAGTCTTCGCGTTTCCG-3′). Tagged constructs were also subcloned into the pRc-CMV vector. All constructs were sequenced in both directions using an ABI automated sequencer.
Cell cultures and transfections. HeLa cells were routinely cultured in DMEM supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin and streptomycin, and 2 mml-glutamine.
Schwann cells were isolated from sciatic nerves of newborn rats according to the method of Brockes et al. (1979). To eliminate contamination of fibroblasts, cultures were maintained in DMEM, 10% FCS, and 10 mm cytosin arabinoside for 1 week and treated twice with fibroblast-specific anti-Thy1.1 antibody (Serotec, Oxford, UK) and baby rabbit complement (Cedarlane, Hornby, Ontario, Canada). Cells were then expanded and cultivated in DMEM supplemented with 10% FCS, 2 mm forskolin (Sigma, St. Louis, MO), and 100 mg/ml bovine glial growth factor (GGF) (Upstate Biotechnology, Lake Placid, NY). Before transfection, HeLa cells or Schwann cells were plated on poly-l-lysine-coated coverslips in 24-multiwell dishes and grown in the appropriate medium to 60–70% confluence. For transfection, cells were washed three times with serum-free medium (OPTI MEM I, Life Technologies, Gaithersburg, MD) and incubated at 37°C for 6 hr in 0.5 ml OPTI MEM I containing 2.5 μg plasmid DNA and 4 mg liposomes (LipofectAMINE, Life Technologies). Cells were cultured further in complete DMEM until they were processed for immunostaining. To select stable transformants of HeLa PMP22 expressors, cells were split once the day after transfection and maintained in complete DMEM containing 400 μg/ml of Geneticin G418 (Life Technologies).
Cotransfections were performed following the same procedure described above using equal amounts of each plasmid DNA.
Northern blot analysis. Total RNA was prepared from confluent monolayers of controls and PMP22-expressing HeLa cells using the guanidinium isothiocyanate acid–phenol method (Chomczynski and Sacchi, 1987). Ten micrograms of total RNA for each sample were loaded per lane and electrophoresed on a formaldehyde agarose gel. RNA was transferred to a nylon membrane (Schleicher & Schuell, Dassel, Germany) and UV-fixed. Northern blots were analyzed with a random primed (32P)dCTP-labeled PMP22 cDNA or GAPDH cDNA probes. Blots were prehybridized with 0.5% sodium phosphate, pH 7.0, and 7% SDS at 60°C for 30 min, and hybridized with labeled probes in the same buffer at 60°C overnight. Filters were washed twice in 2× SSC/1% SDS at 62°C for 10 min and in 0.1× SSC/1% SDS at 62°C for 20 min, and then exposed to x-ray film (Agfa).
Immunofluorescence and confocal microscopy. For single and double immunostaining of cultures grown on glass coverslips, cells were washed with PBS, fixed in Bouin’s solution or 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.05% Triton X-100 in PBS for 5 min, and incubated for 30 min in PBS containing 3% normal goat serum to block nonspecific binding. Incubation with primary antibodies was performed for 1 hr at room temperature followed by extensive washes in PBS and incubation with the appropriate fluorescein- or Cy3-conjugated secondary antibodies. To detect extracellular immunoreactivity, live cultures were incubated with the primary antibody diluted in medium before fixation, and no permeabilization was performed. Finally, samples were rinsed in PBS, and coverslips were mounted on slides in a solution of 2.5% Dabco (Sigma) in PBS-buffered glycerol. PMP22 expression was detected using rabbit polyclonal PMP22 antibodies that recognize the extracellular domains of the molecule (1:200) (prepared as described by D’Urso et al., 1997), or a mouse monoclonal anti-Flag-M2 (1:100; IBI, Kodak).
For subcellular localization we used mouse monoclonal anti-BiP (1:300; StressGen), mouse monoclonal anti-58K Golgi protein (1:50; Sigma), and mouse monoclonal anti-LAMP (1:25; H4B4, Developmental Studies Hybridoma Bank, Iowa) antibodies. The secondary antibodies used to detect primary monoclonals were an affinity-purified goat anti-mouse coupled to fluorescein (1:25; Southern Biotechnology, Birmingham, AL) and a Cy3-conjugated goat anti-mouse IgG (1:500; Dianova, Hamburg, Germany). PMP22 polyclonal antibodies were visualized with Cy3-conjugated goat anti-rabbit (1:300; Dianova) or with a biotin-conjugated goat anti-rabbit (1:100) combined with a avidin-FITC (1:400) (both Vector Laboratories, Burlingame, CA). Controls treated with secondary antibodies alone showed no staining.
Human sural nerve biopsies were fixed in 4% paraformaldehyde and embedded in paraffin. Tissue sections (5–10 μm) were treated with xylol and ethanol solutions (100, 70, and 50%) before incubation with primary and secondary antibodies according to the procedure described above. For immunostaining of neurofilaments and S-100 we used mouse monoclonal antibodies [1:800 (Affinity) and 1:100 (Boehringer Mannheim, Mannheim, Germany), respectively] (data not shown).
All labeled probes were analyzed using a confocal laser scanning microscope system (MRC 1024, Bio-Rad, Munich, Germany) supplied with an argon-krypton laser. For double-immunostained samples, data from two channels were collected simultaneously. After collection, data from each channel were analyzed in merged images, where colocalization of the green (fluorescein) and the red (Cy3) fluorochromes appear yellow, or individually to show the localization of each antigen.
RESULTS
Expression and cellular sorting of wild-type, TrJ, and Tr PMP22 in transfected HeLa cells
To investigate the effect of TrJ and Tr mutation on the cellular sorting of the proteins we used an in vitro model system. Sibling HeLa cell cultures were transiently transfected with plasmids coding the wild-type (wt), the TrJ, or the Tr forms of PMP22. Twenty-four or thirty-six hours after transfection, PMP22 expression was analyzed by immunofluorescence using antibodies directed against the extracellular domains of the protein (D’Urso and Müller, 1997). In permeabilized cells, wt-PMP22 was found at the plasma membrane and widely distributed in the cytoplasm (Fig.1A), as expected for an integral membrane glycoprotein. In contrast, both mutant proteins were highly concentrated in the perinuclear regions (Fig. 1C,E). To look exclusively for PMP22 inserted into the plasma membrane, immunofluorescence was performed on nonpermeabilized cells. PMP22 immunoreactivity was detected only in the plasma membrane of transfectants expressing wt-PMP22 (Fig. 1B). In contrast, PMP22 immunoreactivity was absent from the plasma membrane surfaces of cells expressing either TrJ or Tr PMP22. The intracellular pools of the different forms of PMP22 proteins could be localized in subcellular compartments along their cellular sorting pathway. To identify the intracellular structures with which the mutated proteins were associated, we used specific antibodies directed against BiP, a resident luminal protein of the ER, or a 58 kDa protein of the Golgi membranes. Double-immunostaining experiments revealed the presence of all three PMP22 forms within the ER compartments (Fig.2A,D,G) and also a consistent amount of the proteins concentrated in the Golgi (Fig.2B,E,H). Again, only the wt-PMP22 reached the plasma membrane (Fig. 2A). Moreover, merged confocal images showed that a portion of the intracellular pool of all PMP22 forms was degraded through a lysosomal pathway, as indicated by colocalization with the lysosomal-associated membrane protein LAMP2 (Fig. 2C,F,I).
Fig. 1.

Expression of wild-type, TrJ-, and Tr-PMP22 in transfected HeLa cells. Mutants are not inserted in the plasma membrane and accumulate in the perinuclear regions. PMP22 immunostaining of permeabilized cultures expressing wild type (A), TrJ (C), and Tr (E) PMP22. In nonpermeabilized transfectants (B, D, F), a PMP22 antibody directed against an extracellular domain of the protein recognizes only the wild-type form on the membrane (B). No signal was obtained in cultures transfected with TrJ-PMP22 (D) or Tr-PMP22 (F). Scale bar: 10 μm in A,B, C, E; 20 μm inD, F.
Fig. 2.

Cellular sorting of wild-type, TrJ-, and Tr-MP22 proteins in transfected HeLa cells. Merged confocal images of transfected cultures immunostained for PMP22 (green; red only ina) and the ER protein BiP (a, d, g, red;green only in a), the 58 kDa Golgi protein (b, e, h, red), and the lysosomal associated protein LAMP2 (c, f, i, red). Colocalization of the antigens appears yellow. Scale bar, 50 μm.
For both mutants no membrane staining was obtained when transfected cultures were analyzed at different time points after transfection.
Coexpression of wt-PMP22 and PMP22 mutants in HeLa cells
Because the PNS myelin of TrJ/+ and Tr/+ animals as well as CMT1A patients carrying PMP22 mutations is severely affected, one hypothesis could be that the mutant proteins can interfere with the membrane targeting of the wild-type form. Clonal lines of PMP22 expressors were selected from HeLa cells transfected with a plasmid containing the wt-PMP22 cDNA and the neomycin resistance gene. PMP22 expression was monitored by Northern blotting (Fig.3A) and immunofluorescence (Fig. 3B). PMP22 mRNA and protein were detected only in cells transfected with a PMP22 expression plasmid; no signals were obtained from nontransfected cells (Fig. 3A, lane 1) or from cultures transfected with the plasmid containing only the neo-gene (Fig. 3A, lane 2). Confocal microscopy of PMP22 expressors showed PMP22 immunofluorescence localized at the cell–cell interfaces and in the cytoplasm (Fig.3B). In nonpermeabilized cultures, staining was found exclusively at the plasma membranes (data not shown). We examined the sorting of mutants when coexpressed with the wt-PMP22. We inserted a Flag octapeptide epitope tag at the C terminus of the TrJ- and Tr-PMP22. This epitope does not contain any targeting sequence that might participate in specific cellular or membrane translocation. In addition, we have recently shown that the Flag peptide does not interfere with the cellular sorting and membrane insertion of the wild-type PMP22 protein in HeLa cells (D’Urso and Müller, 1997). Tagged TrJ- and Tr-PMP22 were expressed in stable wt-PMP22 expressors, and their localization was analyzed using a monoclonal antibody that specifically recognizes the Flag epitope. Confocal images of cotransfected cultures double-immunostained using anti-Flag and anti-PMP22 antibodies showed a clear difference in the subcellular localization of the antigens. Both TrJ-PMP22 (Fig.4B) and Tr-PMP22 (Fig.4D) were present only in the cytoplasmic compartments, in contrast to wild-type protein, which was mostly targeted to the plasma membranes along the cell–cell borders of the PMP22 expressors (Fig. 4A,C). In perinuclear areas of double-expressors, colocalization of Flag and PMP22 staining was observed, indicating that the two PMP22 forms coexisted in the same subcellular compartments (data not shown). In these experiments no evidence for induced apoptosis was observed. When stable PMP22 expressors were transfected with another transmembrane protein of peripheral myelin, protein zero (P0), both proteins were sorted correctly and inserted into the plasma membrane (data not shown). Taken together, the results obtained in the HeLa cells demonstrate that the TrJ and Tr mutant proteins are not inserted into the plasma membrane, indicating an impairment in their cellular sorting. We further investigated the cellular distribution of the wt and mutated proteins in the natural cellular environment for PMP22, the Schwann cells.
Fig. 3.

Stable transfection of PMP22 in HeLa cells. wt-PMP22 expression in clonal lines was analyzed by (A) Northern blotting. PMP22 mRNA was present only in cells transfected with the expression vector (lane 3). No signal was detectable in the untransfected HeLa cells (lane 1) or cells transfected with the neomycin resistance gene (lane 2). B, In PMP22 expressors the protein was localized in the plasma membrane and in the perinuclear regions, as detected by immunofluorescence and confocal microscopy. Scale bar, 10 μm.
Fig. 4.

Membrane targeting of wt-PMP22 is not impaired by coexpression of mutant PMP22. A clonal line of HeLa cells that stably expressed wt-PMP22 were transiently transfected with tagged Flag-TrJ and Flag-Tr PMP22, and the localization of PMP22 was determined by immunofluorescence using PMP22 and anti-Flag antibodies. PMP22 mutants were identified by their C-terminal Flag-epitope (B, D). The membrane targeting of the wt-PMP22 (A, C) is not affected in cells that coexpress TrJ (B) or Tr (D) mutants. Examples of coexpressing cells are marked by arrows.
TrJ- and Tr-PMP22 are not inserted into the plasma membrane of Schwann cells but accumulate in the ER–Golgi compartments
In vivo, PMP22 protein is expressed by myelinating Schwann cells and once synthesized is readily inserted into compact myelin. In myelinating cocultures of Schwann cells and dorsal root ganglia neurons, PMP22 is targeted to the myelin membrane where it colocalizes with other myelin proteins, such as P0 and myelin basic protein (MBP), and is also distributed within the cytoplasm of myelinating Schwann cells (D’Urso et al., 1997). Normally, PMP22 protein is not detectable by immunohistochemistry in nonmyelinating Schwann cells or in pure Schwann cell cultures, and this is consistent with its axonally regulated expression. We wanted to determine whether the TrJ and Tr proteins were impaired in their transport to the plasma membrane of Schwann cells. First we performed a transient transfection of all PMP22 forms into cultured rat Schwann cells. PMP22 immunostaining of permeabilized transfectants showed that the wild-type protein was distributed within the entire cytoplasm of expressors (Fig.5A), whereas both mutants appear to be concentrated exclusively in the perinuclear areas (Fig.5C,E). Confocal optical sections of live stained transfected cells showed that a portion of wt-PMP22 was inserted into the plasma membrane (Fig. 5B). The signal was quite weak compared with that obtained in myelinating cultures (D’Urso et al., 1997), confirming that the presence of axons enhances the amount of protein targeted to the cell surface of Schwann cells. In nonpermeabilized cells, no TrJ or Tr proteins were detected at the plasma membrane at any time point after transfection (Fig. 5D,F).
Fig. 5.

TrJ and Tr proteins concentrate in the perinuclear regions of transfected Schwann cells and do not reach the plasma membrane. PMP22 immunostaining of permeabilized Schwann cells transfected with wt- (a), TrJ- (c), Tr-PMP22 (e). When cells were not permeabilized, a weak signal was obtained for PMP22 immunoreactivity on the surface of cells expressing wild-type protein (b, arrows). No membrane staining was visible in TrJ (d) and Tr (f) transfectants. Scale bar, 10 μm.
To identify the subcellular compartments where the mutant proteins were trapped in Schwann cells, we performed double immunostaining combining PMP22 antibody together with antibodies against BiP or the 58 kDa Golgi protein. Merged confocal images showed that most of the mutant proteins were retained within the endoplasmic reticulum (Fig.6C,E). However, a reproducible fraction of the proteins was further transported into the Golgi apparatus (Fig. 6D,F). In cells expressing the wild-type form, the protein was transported through the ER (Fig.6A) and Golgi (Fig. 6B) compartments and localized along the cellular processes (Fig.6A,B), clearly showing a wider distribution than the mutants. A minimal amount of the proteins colocalized with the lysosomal marker LAMP2 (data not shown), indicating that in the case of Schwann cells lysosomal degradation is not a major route of PMP22 metabolism.
Fig. 6.

TrJ and Tr proteins accumulate in the ER and Golgi compartments of transfected Schwann cells and do not show a dominant-negative effect on the cellular distribution of wt-PMP22. Confocal optical sections of transfected Schwann cells double-immunostained for PMP22 (red) and the ER marker BiP (a, c, d, green), or the 58 kDa Golgi protein (b, d, f, green). In merged images, the yellow signals show colocalization of the antigens. gand h are superimposed confocal micrographs of Schwann cells cotransfected with wt-PMP22 (red) and Flag-TrJ (g, green) or Flag-Tr (h, green). Anti-flag and anti-PMP22 immunostaining colocalize (yellow) only in the perinuclear regions. Mutants do not interfere with the sorting of wt-PMP22.
TrJ and Tr proteins do not impair the transport of wt-PMP22 when coexpressed in Schwann cells
When tissue sections obtained from mutant animals or from patients’ sural nerve biopsies are used to analyze the distribution of PMP22, it is not possible to study the cellular localization of wt-PMP22 versus that of its mutant forms. This is so because antibodies able to distinguish between mutant and wt-PMP22 are not available, andin vivo it is not feasible to follow the post-transcriptional pathways of the newly synthesized molecules. To overcome this problem, we generated an in vitro system by coexpressing a Flag-tagged TrJ- or Tr-PMP22 with the wild-type protein in cultured Schwann cells, an approach already tested successfully on HeLa cell transfectants. Cotransfected Schwann cells were immunostained with anti-PMP22 and anti-Flag antibodies and analyzed by simultaneous confocal microscopy. The distribution of the two antigens clearly demonstrated that neither the TrJ nor the Tr protein has a dominant-negative effect on the cellular distribution of wt-PMP22. The latter was widely present throughout the cytoplasm of double-positive cells (Fig. 6G,H), in contrast to TrJ (Fig.6G) and Tr (Fig. 6H) proteins, which did not leave the cytoplasm surrounding the nucleus. This area was the only region in which the Flag and PMP22 staining colocalized.
Schwann cell bodies overloaded with PMP22 immunoreactivity are present in CMT1A nerve
In the majority of cases, CMT1A disease is associated with a 1.5 Mb DNA duplication on chromosome 17p11.2-p12, which contains the PMP22 gene. Valentijn and colleagues (1992) have described a family affected by CMT1A, which showed a complete segregation of a point mutation in the PMP22 coding region. This mutation, L16P, is identical to the mutation found in TremblerJ mice (Suter et al., 1992a). We analyzed the distribution of PMP22 immunoreactivity in a sural nerve biopsy obtained from a patient displaying clinical and electrophysiological symptoms of CMT1A and carrying the L16P point mutation as detected by DNA analysis. Serial paraffin sections (5–10 μm) were double-stained with PMP22 polyclonal antibody combined with anti-neurofilament or anti-S100 monoclonal antibodies and analyzed by confocal microscopy. Although PMP22 immunostaining was present in myelin, it was mostly localized in the cytoplasm of myelinating Schwann cells (Fig.7A–C), as confirmed by colocalization with S-100 (data not shown). Enlarged cell bodies of Schwann cells that wrapped axons were a frequent feature observed in the CMT1A sections analyzed. These observations are consistent with recent similar studies performed on CMT1A patients carrying a PMP22 duplication (Nishimura et al., 1996) and on Trembler mice (Naef et al., 1997). In control nerves, PMP22 immunoreactivity was confined mostly to the myelin membranes surrounding axons of different sizes (Fig.7D) and did not exhibit enlarged Schwann cell bodies. Similar results were obtained in tissue sections from patients with an acquired inflammatory demyelinating neuropathy (data not shown). These observations, together with our in vitro data, suggest that also in CMT1A nerves the mutated PMP22 protein accumulates in the cytoplasmic compartments, whereas the native form accounts for the immunoreactivity found in compact myelin. So far, however, it is impossible to distinguish between the two forms of the protein in vivo.
Fig. 7.
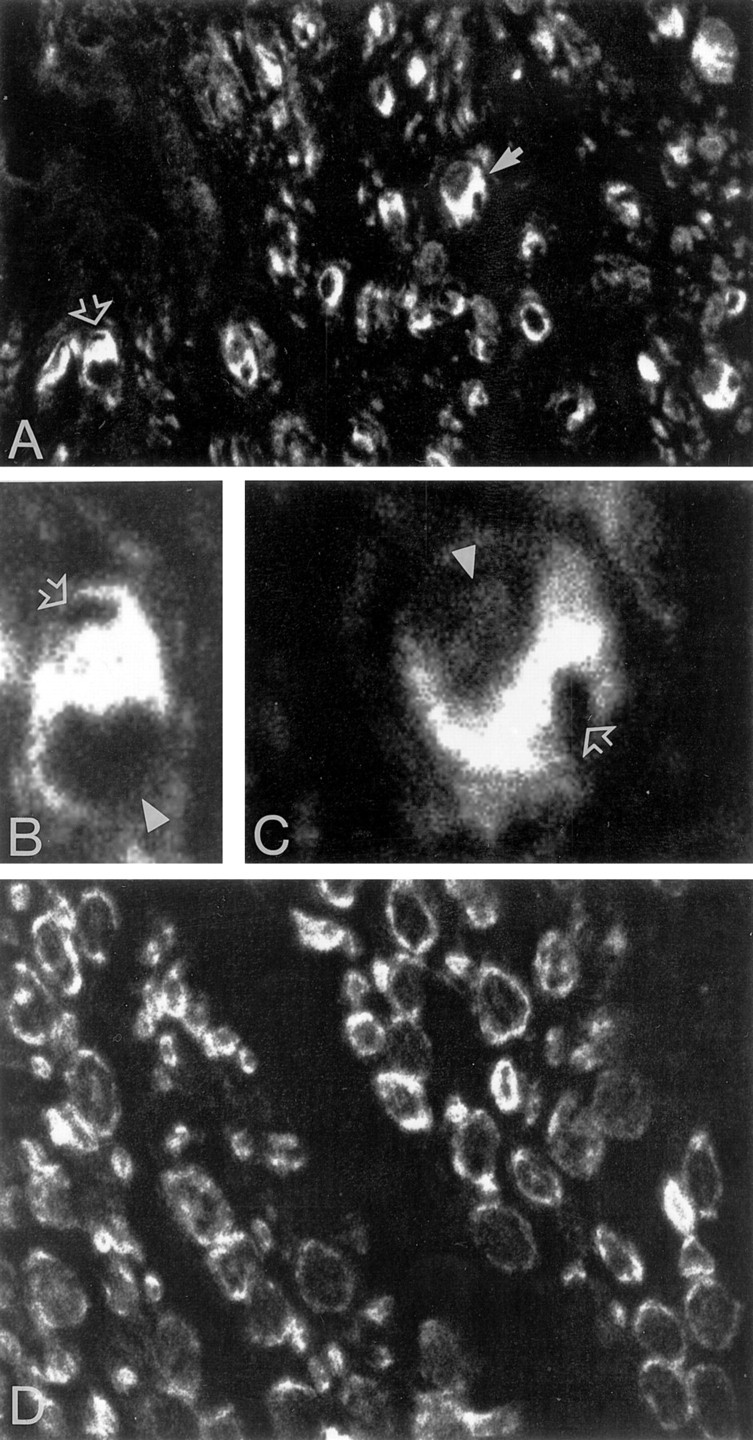
PMP22 is abundant in the cytoplasm of myelinating Schwann cells in CMT1A sural nerve. A, PMP22 immunostaining in CMT1A tissue section highlights cell bodies of Schwann cells. Two examples are enlarged in B(open arrow in A) and C(full arrow in A). InB and C, open arrowsindicate the position of dislocated axons, andarrowheads the position of the nuclei. D, PMP22 immunoreactivity in control sural nerve. Magnification 60× inA and D; 100× in B andC.
DISCUSSION
The mouse mutants Trembler J and Trembler carry point mutations identical to those found in a CMT1A family (Valentjin et al., 1992) and in two Dejerine Sottas Syndrome patients (Ionasescu et al., 1997), respectively, and they are considered to be animal models of CMT1A disease. Indeed, CMT1A is the most frequent form of hereditary neuropathies in humans, and perturbed peripheral myelin is a common feature of the pathology of these diseases. In our in vitromodel we show that both the TrJ- and Tr-PMP22 forms, when expressed in Schwann cells, accumulate in the ER and are also present in the Golgi apparatus. The combination of live immunostaining and confocal analyses clearly demonstrates that the mutant proteins are not targeted to the plasma membrane, in contrast to the wt-PMP22, which is always detectable on the cell surface of expressing cells. To investigate the functional consequence of the expression of the mutant PMP22 on wt-PMP22 we have tagged PMP22 at the C terminus with the Flag epitope. In this way we were able to distinguish between the wild-type and the mutant forms in the same cell. We have reported previously that the C- or N-terminal Flag epitope does not interfere with the intracellular sorting of PMP22, and that the fusion protein is glycosylated and correctly transported to the plasma membrane (D’Urso and Müller, 1997). We have shown here that wt-PMP22 reaches the plasma membrane even when PMP22 mutants are coexpressed. Interestingly, the impaired transport of the mutated proteins does not influence the correct subcellular distribution of the native form, and it does not exhibit a dominant-negative effect on the cellular distribution of wt-PMP22. Furthermore, this shows that overloading of the ER and the presence in the Golgi of the PMP22 mutants do not impair the function of these organelles, as indicated by the correct sorting of the native protein. wt-PMP22 obviously colocalizes to some extend with BiP and Golgi staining during its transport to the plasma membrane. In parallel, the similar results obtained in transfected HeLa cultures demonstrate that the inability of the mutant proteins to be transported to the cell surface is a general bona fide phenomenon that is independent from the cell type. In CMT1A nerve, PMP22 immunostaining was mostly but not exclusively concentrated in the cell body of myelinating Schwann cells. Thus, it is likely that also in vivo the mutated and wild-type proteins are segregated differently, and that the PMP22 immunoreactivity associated with myelin sheaths is caused by the native protein expressed in these cells. Accumulation of PMP22 in the perinuclear regions of Schwann cells has been reported also in Trembler J (Notterpek et al., 1997) and Trembler mice (Naef et al., 1997). Thus, this subcellular gathering of PMP22 appears to be a common feature observed in Schwann cells expressing the mutated PMP22. Interestingly, both TrJ and Tr mutations seem to have the same defect in intracellular trafficking of the protein. However, it remains unclear whether the underlying mechanism is the same. Moreover, both mutations are located in transmembrane domains of the protein, and because these are the most conserved regions and probably relevant for a proper function of the protein, it is not surprising that such mutations affect PMP22 severely.
During the preparation of this manuscript, a paper was published on the intracellular trafficking of Tr-PMP22 in transfected COS-7 cells (Naef et al., 1997). In part, our results are in close agreement with these data, because they show that the mutated PMP22 protein localizes mainly in the ER. However, in contrast to our results in COS-7 cells it is reported that Tr protein impairs the correct transport of the wild-type form to the plasma membrane. Our data have been generated in Schwann cells, the “natural host” for this protein, as well as in HeLa cells. It might be that the dominant-negative effect shown in COS-7 cultures can be observed under certain conditions but that it is not a general phenomenon. Furthermore, these results are also in contrast to the in vivo observations showing PMP22 immunoreactivity in the myelin sheath of Tr/+ mice.
We propose that the disease phenotype associated with the TrJ- and Tr-PMP22 mutations involves the combination of different mechanisms. With a heterozygous genetic background, only the product of the wild-type allele is correctly targeted and inserted into the myelin membrane, whereas the mutated PMP22 is retained in the cytoplasmic compartments. A decreased amount of the functional protein present in myelin may cause a gene dosage effect. Additionally, there may be a minimum amount of PMP22 needed to maintain compact myelin. If this hypothesized critical amount is not realized, there may also be a consequent abnormal interaction of PMP22 and another protein, or complex of proteins, in the lipid bilayer. Thus a reduced level of myelin-associated PMP22 may lead to an unstable myelin structure. At the same time, most of the mutated PMP22 proteins accumulate in the ER. Overloading of the ER with membrane proteins may have pronounced effects on cell signaling, which can induce a cell response involving different second messenger pathways and transcription factors (Thomas et al., 1995; Pahl and Baeuerle, 1997a,b). An ER-overloaded response has been associated with different genetic diseases caused by naturally occurring mutations. In cystic fibrosis the mutated cystic fibrosis transmembrane conductance regulator (CFTR) is entirely retained in the ER together with a portion of the wild type (Cheng et al., 1990; Yang et al., 1993; Lukacs et al., 1994). In Alzheimer’s disease, wild-type and mutant forms of presenilins, which are responsible for the majority of early onset familial forms of the disease, are localized in the ER (Kovacs et al., 1996). In both cases upregulation of specific transcription factors has been described (Pahl and Baeuerle, 1997a). It is likely that distinct sets of genes are activated, and this may change the phenotype of the affected cell in different ways. Our experimental evidence raises the interesting hypothesis that the disease phenotype associated with PMP22 mutations is attributable not only to an impaired membrane targeting but also to secondary intracellular changes effected by ER–Golgi overloading. Transfection of primary Schwann cells with epitope-tagged protein offer a useful model to further substantiate this hypothesis.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft (Mu 630/5-3). We are grateful to Dr. C. Schneider for kindly providing the pGDSV7-hgas3/PMP22 plasmid used in preliminary studies.
Correspondence should be addressed to Dr. Donatella D’Urso, Molecular Neurobiology Laboratory, Department of Neurology, Heinrich-Heine-University, Moorenstrasse 5, 40225 Düsseldorf, Germany.
REFERENCES
- 1.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in PMP22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 2.Baechner D, Liehr T, Hameister H, Altenberger H, Grehel H, Suter U, Rautenstrauss B. Widespread expression of the peripheral myelin protein-22 gene (PMP22) in neural and non-neural tissues during murine development. J Neurosci Res. 1995;42:733–741. doi: 10.1002/jnr.490420602. [DOI] [PubMed] [Google Scholar]
- 3.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 4.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 5.De Jonghe P, Timmerman V, Nelis E, Martin J-J, Van Broeckhoven C. Charcot-Marie-Tooth disease and related peripheral neuropathies. J Periph Nerve System. 1997;2:370–387. [PubMed] [Google Scholar]
- 6.D’Urso D, Müller HW. Ins and outs of peripheral myelin protein-22: mapping transmembrane topology and intracellular sorting. J Neurosci Res. 1997;49:551–562. doi: 10.1002/(SICI)1097-4547(19970901)49:5<551::AID-JNR5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 7.D’Urso D, Schmalenbach C, Zoidl G, Prior R, Müller HW. Studies on the effects of altered PMP22 expression during myelination in vitro. J Neurosci Res. 1997;48:31–42. doi: 10.1002/(sici)1097-4547(19970401)48:1<31::aid-jnr3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Fabbretti E, Edomi P, Brancolini C, Schneider C. Apoptotic phenotype induced by overexpression of wild-type gas3/PMP22: its relation to the demyelinating peripheral neuropathy CMT1A. Genes Dev. 1995;9:1846–1856. doi: 10.1101/gad.9.15.1846. [DOI] [PubMed] [Google Scholar]
- 9.Hanemann CO, Rosenbaum C, Kupfer S, Wosch S, Stoegbauer F, Müller HW (1997) Improved culture methods to expand Schwann cells with altered growth behaviour from CMT1A patients. Glia, in press. [DOI] [PubMed]
- 10.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 11.Ionasescu VV, Searby CC, Ionasescu R, Chatkupt S, Patel N, Koenigisberger R. Dejerine-Sottes neuropathy in mother and son with same point mutation of PMP22 gene. Muscle Nerve. 1997;20:97–99. doi: 10.1002/(sici)1097-4598(199701)20:1<97::aid-mus13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 12.Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 13.Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J. 1994;13:6076–6086. doi: 10.1002/j.1460-2075.1994.tb06954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, Zielasek J, Toyka KV, Suter U. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 dosage. J Neurosci. 1996;16:5351–5380. doi: 10.1523/JNEUROSCI.16-17-05351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller HW, Suter U, van Broeckhover C, Hanemann O, Nelis E, Timmerman V, Sancho S, Barrio L, Bolhuis P, Dermietzel R, Frank M, Gabreël-Festen A, Gillen C, Haites N, Levi G, Marinari E, Martini R, Nave K, Bautenstrauss B, Schachner M, Schenone A, Schneider C, Schröder M, Willecke K. Neurobiology of disease, Vol 4, pp 206–211. Academic; London: 1997. Advances in Charcot-Marie-Tooth disease research: cellular function of CMT-related proteins, transgenic animal models and pathomechanisms. [DOI] [PubMed] [Google Scholar]
- 16.Naef R, Adlkofer K, Lescher B, Suter U. Aberrant protein trafficking in Trembler suggests a disease mechanism for hereditary human peripheral neuropathies. Mol Cell Neurosci. 1997;9:13–25. doi: 10.1006/mcne.1997.0604. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura T, Yoshikawa H, Fujimura H, Sakoda S, Yanagihara T. Accumulation of peripheral myelin protein 22 in onion bulbs and Schwann cells of biopsied nerves from patients with Charcot-Marie-Tooth disease type 1A. Acta Neuropathol. 1996;92:454–460. doi: 10.1007/s004010050546. [DOI] [PubMed] [Google Scholar]
- 18.Notterpek L, Shooter EM, Snipes GJ. Upregulation of the endosomal-lysosomal pathway in the Trembler-J neuropathy. J Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pahl HL, Baeuerle PA. The ER-overload response: activation of NF-kB. Trends Biochem. 1997a;22:63–67. doi: 10.1016/s0968-0004(96)10073-6. [DOI] [PubMed] [Google Scholar]
- 20.Pahl HL, Baeuerle PA. Endoplasmic reticulum-induced signal transduction and gene expression. Trends Cell Biol. 1997b;7:50–55. doi: 10.1016/S0962-8924(96)10050-7. [DOI] [PubMed] [Google Scholar]
- 21.Sereda M, Griffiths I, Pühlhofer A, Steward H, Rossner MJ, Zimmermann F, Magyar JP, Schneider A, Hund E, Meinck HM, Suter U, Nave KA. A transgenic rat model for Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/s0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 22.Spreyer P, Kuhn G, Hanemann CO, Gillen G, Schaal H, Kuhn R, Lemke G, Müller HW. Axon-regulated expression of a Schwann cell transcript that is homologous to a “growth arrest-specific” gene. EMBO J. 1991;10:3661–3668. doi: 10.1002/j.1460-2075.1991.tb04933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suter U, Snipes GJ. Biology and genetics of hereditary motor and sensory neuropathies. Annu Rev Neurosci. 1995;18:45–75. doi: 10.1146/annurev.ne.18.030195.000401. [DOI] [PubMed] [Google Scholar]
- 24.Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22 kDa peripheral myelin protein in trembler-J mouse. Proc Natl Acad Sci USA. 1992a;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992b;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- 26.Thomas PJ, Bao-He Q, Pedersen PL. Defective protein folding as a basis of human disease. Trends Biochem. 1995;20:456–459. doi: 10.1016/s0968-0004(00)89100-8. [DOI] [PubMed] [Google Scholar]
- 27.Valentijn LJ, Baas F, Wolterman RA, Hoogendijk JE, van den Bosch NHA, Zorn I, Gabreëls-Festen AAWM, de Visser M, Bolhuis PA. Identical point mutations of PMP-22 in Trembler-J mouse and Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;2:288–291. doi: 10.1038/ng1292-288. [DOI] [PubMed] [Google Scholar]
- 28.Welcher AA, Suter U, De Leon M, Snipes JG, Shooter EM. A myelin protein is encoded by the homologue of a growth arrest-specific gene. Proc Natl Acad Sci USA. 1991;88:7195–7199. doi: 10.1073/pnas.88.16.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi non-lysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoidl G, Blass-Kampmann S, D’Urso D, Schmalenbach C, Müller HW. Retroviral-mediated gene transfer of the peripheral myelin protein PMP22 in Schwann cells: modulation of cell growth. EMBO J. 1995;14:1122–1128. doi: 10.1002/j.1460-2075.1995.tb07095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]