

Abstract
Depolarization-induced suppression of inhibition (DSI) is a transient reduction of GABAA receptor-mediated IPSCs that is mediated by a retrograde signal from principal cells to interneurons. Using whole-cell recordings, we tested the hypothesis that mGluRs are involved in the DSI process in hippocampal CA1, as has been proposed for cerebellar DSI. Group II mGluR agonists failed to affect either evoked monosynaptic IPSCs or DSI, and forskolin, which blocks cerebellar DSI, did not affect CA1 DSI. Group I and group III mGluR agonists reduced IPSCs, but only group I agonists occluded DSI. (S)-MCPG blocked (1S,3R)-ACPD-induced IPSC suppression and markedly reduced DSI, whereas group III antagonists had no effect on DSI. Many other similarities between DSI and the (1S,3R)-ACPD-induced suppression of IPSCs also were found. Our data suggest that a glutamate-like substance released from pyramidal cells could mediate CA1 DSI by reducing GABA release from interneurons via the activation of group I mGluRs.
Keywords: IPSP, GABA, retrograde signal, voltage clamp, mGluR, synaptic inhibition
Depolarization-induced suppression of inhibition (DSI) is a phenomenon seen in both hippocampal CA1 pyramidal cells (Pitler and Alger, 1992, 1994; Alger et al., 1996;Morishita and Alger, 1997a) and cerebellar Purkinje cells (Llano et al., 1991; Vincent et al., 1992; Vincent and Marty, 1993). It involves the transient (∼1 min) suppression of GABAAreceptor-mediated (GABAARmediated) IPSCs impinging on these cells after depolarization of their membranes that is sufficient to open voltage-gated Ca2+ channels. Despite the clearly postsynaptic locus of induction of DSI, the actual suppression of inhibition occurs via a presynaptic mechanism. Many experiments have led to the conclusion that the quantal content of GABAAR-mediated IPSCs is reduced, with no evidence of a decrease in postsynaptic GABAAR responsiveness—neither iontophoretic GABAAR-mediated responses nor quantal size is reduced—during DSI. Taken together, the postsynaptic site of induction plus the presynaptic site of expression strongly imply that a retrograde signal must pass between the principal cells and their interneurons (Alger and Pitler, 1995).
It has been suggested recently that glutamate, released from Purkinje cells and acting on a presynaptic group II mGluR on the GABA-releasing basket cells, is the retrograde messenger for DSI in the cerebellum (Glitsch et al., 1996). Supporting evidence includes the findings that the specific mGluR agonist DCG-IV mimics and occludes DSI and that the mGluR antagonist l-AP3 reduces DSI [for review of mGluR pharmacology, see Pin and Duvoisin (1995)]. Group II mGluRs (mGluR2 and mGluR3) decrease cAMP production (Conn et al., 1994), which can reduce GABA release (Capogna et al., 1995). Glitsch et al. (1996) found that in cerebellum forskolin, an activator of adenylate cyclase, also reduced DSI, which was consistent with their hypothesis.
Despite many similarities, hippocampal DSI and cerebellar DSI differ in some ways (Alger and Pitler, 1995). For example, whereas cerebellar DSI reduces TTX-insensitive mIPSC frequency, in CA1 pyramidal cells mIPSCs are unaffected by DSI. That and other data have suggested that there are two mechanisms for DSI expression in cerebellum, but only one is significantly present in CA1. In view of these differences it was of particular interest to test the hypothesis that the retrograde process in hippocampus might be mediated by glutamate acting on presynaptic group II mGluRs.
We made whole-cell voltage-clamp recordings of monosynaptic, evoked IPSCs in CA1 pyramidal cells in the rat hippocampal slice and used a battery of mGluR agonists and antagonists to test the mGluR hypothesis of DSI. Antagonism of DSI by (S)-MCPG and similarities between the actions of mGluR agonists and DSI support the hypothesis that glutamate, or a glutamate-like compound, could be the retrograde messenger of DSI in CA1 pyramidal cells, although the mechanism does not involve the group II mGluR subtype. This hypothesis has interesting implications for understanding both the mechanism by which the DSI process mediates DSI and the apparent differences between hippocampal and cerebellar DSI.
Some of the data in this report have appeared in abstract form (Alger et al., 1997).
MATERIALS AND METHODS
Preparation of hippocampal slices. Adult male Sprague Dawley rats (125–250 gm) were anesthetized deeply with halothane and decapitated; the brain was removed, and the hippocampi were dissected free. The hippocampi were mounted on an agar block in a slicing chamber containing ice-cold saline. Transverse slices (400 μm) were cut with a Vibratome (Technical Products International, Chicago, IL) and allowed to recover in a holding chamber at the interface of a physiological saline and humidified 95% O2/5% CO2atmosphere at room temperature. After a minimum 1 hr incubation, a single slice was transferred to a submersion-type recording chamber (Nicoll and Alger, 1981), where it was perfused with oxygenated saline (29–31°C) at a flow rate of 0.5–1 ml/min.
Solutions. Patch electrodes with resistances 3–6 MΩ usually were filled with (in mm): CsCH3SO3 100, CsCl 50 or 60, BAPTA 2, CaCl2 0.2, MgCl2 1, MgATP 2 or 4, HEPES 10, Tris-GTP 0.3, and 2-(triethylamino)-N-(2,6-dimethylphenyl) acetamide (QX-314) 5, pH adjusted to 7.3 with KOH, osmolarity 310–320 mOsm. In some experiments 145 mm KCl was used in place of the Cs salts; other constituents were the same. The results using these two electrode solutions did not differ. Physiological saline contained (in mm): NaCl 120, KCl 3.5, NaH2PO41.25, NaHCO3 25, CaCl2 2, MgSO4 2, and glucose 10, equilibrated with a 95% O2/5% CO2 mixture, pH 7.3. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX; 20 μm) andd,l-2-amino-5-phosphonovaleric acid (APV; 50 μm) were used in all experiments in the extracellular saline to block ionotropic glutamate responses. TTX, 0.5 μm, was present in the bath solution for experiments on miniature IPSCs (mIPSCs) to block action potential-dependent transmitter release. Recording of mIPSCs was initiated only after high-intensity stimulation elicited no IPSC.
The metabotropic glutamate receptor (mGluR) agonistsl-quisqualic acid (Quis), (S)-3,5-dihydroxyphenylglycine (DHPG), (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid [(1S,3R)-ACPD], (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV), (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (l-CCG-I), l(+)-2-amino-4-phosphonobutyric acid (l-AP4), as well as the mGluR antagonistsl(+)-2-amino-3-phosphonopropionic acid (l-AP3), (S)-4-carboxyphenylglycine (4CPG), (RS)-α-methylserine-O-phosphate (MSOP), (S)-2-amino-2-methyl-4-phosphonobutanoic acid (M-AP4), and (S)-α-methyl-4-carboxyphenylglycine [(S)-MCPG] were purchased from Tocris Cookson (Bristol, UK). CNQX was acquired from Research Biochemicals (Natick, MA), BAPTA from Molecular Probes (Eugene, OR), and TTX from Calbiochem (La Jolla, CA). QX-314 was generously donated by Astra (Sodertalje, Sweden) or was purchased from Alomone Labs (Jerusalem, Israel). All other drugs and chemicals were from Sigma (St. Louis, MO). Drugs were either iontophoretically- or bath-applied. All agonists and antagonists of mGluRs were prepared as concentrated stock solutions: (1S,3R)-ACPD, l-CCG-I, quisqualate,l-AP4, and DHPG were dissolved at 1000× final concentration in 1 equivalent (eq) of NaOH (except for DHPG, which was prepared in distilled water); l-AP3 and (S)-MCPG were solubilized at 100× final concentration in 1 eq or 1.1 eq of NaOH. All other bath-applied drugs were prepared as 1:1000 concentrated stock solutions.
Electrophysiology and data analysis. Tight-seal whole-cell recordings were obtained from CA1 pyramidal cells via the “blind” technique (Blanton et al., 1989). The cells were voltage-clamped at −70 mV immediately after break-in, using either an Axopatch 200A or Axoclamp 2B amplifier (Axon Instruments, Foster City, CA). Acceptable cells had resting potentials more than or equal to −55 mV and input resistances >40 MΩ. Series resistance and input resistance were monitored continuously (every 2, 16, or 120 sec) by observing changes in the amplitude characteristics of the capacitive current elicited by a 5 mV, 50 msec hyperpolarizing rectangular voltage step. Experiments with unstable series resistances or series resistances >30 MΩ were discarded. Series resistance compensation was between 50 and 70%. Liquid junction potentials were small and were not corrected for. Evoked inhibitory postsynaptic currents were recorded by stimulating either stratum oriens or stratum radiatum at 0.33 or 0.5 Hz with a bipolar concentric stimulating electrode (Rhodes Electronics). Extracellular field EPSPs were recorded with electrodes (resistances 2–5 MΩ) filled with buffered salt solution having the same composition as the physiological saline. Mossy-fiber field EPSPs recorded in s. lucidum were evoked by a stimulating electrode placed in s. granulosum of the dentate gyrus. CA1 field EPSPs were recorded in s. radiatum. To record CA1 field EPSPs, we removed the CA3 region and placed a stimulating electrode in s. radiatum near the cut edge. Field EPSPs were evoked at 0.1 Hz. Signals were filtered at 2 kHz with an eight-pole Bessel filter (Frequency Devices, Haverhill, MA), digitized at 10 kHz with a DigiData 1200 interface board (Axon Instruments), and analyzed with pCLAMP 6 software (Axon Instruments). Data also were stored on VHS videotape after being acquired at 22 kHz with a 14-bit PCM digitizer (Neuro-Corder DR-484, Neuro Data Instruments).
Iontophoresis of (1S,3R)-ACPD was performed in some experiments. (1S,3R)-ACPD was dissolved at 25 mm in 1 eq of NaOH and was present at full strength in the iontophoretic pipettes. The drug was ejected from glass pipettes with resistances of 1–2 MΩ positioned in the vicinity of the recording patch electrode. Iontophoretic currents of −155 to −600 nA lasting from 2 to 4 sec were used.
Positive voltage step commands to ∼0 mV for a duration of 1–2 sec were used to elicit DSI every 90–120 sec. With this protocol an unclamped Ca2+ spike current and K+ currents were present in the current trace. DSI was expressed as the percent reduction of the control response by calculating the mean amplitude of 7 or 10 IPSCs in the control (pre-DSI) period and the mean amplitude of the same number of IPSCs after the DSI step. Because DSI often is not maximal immediately after the step and takes ∼1–3 sec to develop (Pitler and Alger, 1994;Alger et al., 1996), we typically omitted the first two IPSCs during the DSI period from the calculations.
To quantify drug effects on DSI, we compared the mean of two to four complete DSI trials in control (predrug period) with the mean of the equal number of DSI trials at the time of maximum drug effect. The percent reduction of IPSC amplitude by the given drug was calculated by comparing the mean amplitudes in the pre-DSI period in control and during the drug effect. The percent reduction of IPSCs by iontophoretic (1S,3R)-ACPD application was computed in the same manner, comparing the mean amplitude of 7–10 IPSCs before and after drug ejection at the time of maximum effect. Unless otherwise stated, a Student’s paired t test was used to determine statistical significance of effects (p < 0.05), and all data are reported as the mean ± SEM.
The significance of (1S,3R)-ACPD effects on mIPSCs was assessed by Kolmogorov–Smirnov (K–S) statistics with a significance level of p < 0.005. Cumulative frequency amplitude distributions of TTX-insensitive mIPSCs were obtained over 1 min in the control period and during the subsequent application of (1S,3R)-ACPD. Averaged cumulative frequency amplitude distributions were constructed by normalizing individual distributions to the median amplitude of the corresponding control distributions. Data from individual cells were averaged by calculating the normalized amplitudes at fixed cumulative frequency intervals.
RESULTS
(1S,3R)-ACPD reduces IPSCs and DSI
The results presented in this report are based on whole-cell voltage-clamp experiments done on 134 cells recorded in the CA1 pyramidal layer of the rat hippocampal slice. The ionotropic glutamate receptor antagonists CNQX (20 μm) and APV (50 μm) were present in all experiments except for those on field potential EPSPs.
Confirming previous reports (Desai et al., 1994; Jouvenceau et al., 1995), we found that the mGluR agonist (1S,3R)-ACPD bath-applied at 50 (n = 15) or 100 μm (n = 4) substantially suppressed the evoked monosynaptic GABAAR-mediated IPSC (55.6 ± 5.3%; Fig.1A,D, n= 19). In 18 of these cells DSI was present; the mean suppression of the IPSC during DSI was 51 ± 3.4% of the control amplitude. (1S,3R)-ACPD reduced the IPSC by 53.9 ± 5.3% and reduced DSI in these cells to 17 ± 4% suppression. In 10 cells we tested for recovery and found that DSI recovered after 50 μm (1S,3R)-ACPD was washed out (control DSI, 54 ± 4.3%; wash DSI, 50 ± 5.6%) (Fig.1C).
Fig. 1.

Activation of mGluRs by (1S,3R)-ACPD reduces DSI of evoked monosynaptic GABAAR-mediated IPSCs recorded from hippocampal CA1 pyramidal cells. A, Illustrated is a control DSI trial on a series of evoked IPSCs (these inward currents are shown as downward deflections). DSI was elicited by a depolarizing voltage step [depolarizing voltage steps (see Materials and Methods) are indicated by filled arrows in all figures] and is represented by the transient reduction of the IPSCs. The center trace is from a DSI trial on the same cell during the fifth minute of bath application of (1S,3R)-ACPD. The IPSCs are reduced in amplitude, and DSI is occluded. The right trace is from a DSI trial recorded 22 min after washout of (1S,3R)-ACPD. B, Recorded from another cell, the first current trace (left) shows the control DSI trial. The center trace shows the effects of (1S,3R)-ACPD (recorded during the fourth minute of bath application). The right traceis from a DSI trial after the stimulation intensity had been increased (ADJ. STIM. INTENSITY) to evoke IPSCs comparable in amplitude to those recorded in control. Note that DSI remains reduced after this manipulation. C, Combined data summarizing the action of (1S,3R)-ACPD on DSI from 10 experiments as in A. In the same graph (separated by the break in the ordinate) are data recorded from a different set of cells summarizing experiments performed as in B. Data in control and recovery are labeled CON and REC, respectively.D, Summarized is the suppression of the IPSC amplitude produced by bath-applied (1S,3R)-ACPD. Data in graphs C and D were obtained from the number of cells shown in parentheses above the bars. Asterisks in the figures indicate significant differences from control values (Student’s paired t test; p < 0.05). In this and other figures stimulus artifacts were blanked for clarity in the display.
The block of DSI induced by (1S,3R)-ACPD could not be explained simply by the reduction in IPSC size because when, still in (1S,3R)-ACPD, the stimulus intensity was increased to produce an IPSC similar to the control IPSC, DSI did not increase (Fig. 1B,C). DSI recovered when (1S,3R)-ACPD was washed from the bath, however (n = 5). Thus it appears that activation of mGluRs mimics and occludes hippocampal CA1 DSI as it does cerebellar DSI (Glitsch et al., 1996).
Both DSI and (1S,3R)-ACPD decrease the frequency of TTX-insensitive mIPSCs in cerebellum (Llano et al., 1991; Llano and Marty, 1995). In hippocampus, DSI does not block mIPSCs (Pitler and Alger, 1994; Alger et al., 1996). We recorded mIPSCs during 1 min intervals from six cells in TTX and then applied (1S,3R)-ACPD (50 μm) and compared the mIPSCs in (1S,3R)-ACPD with those in control. There was no change in mIPSC frequency (control, 4.8 ± 1 Hz; (1S,3R)-ACPD + TTX, 5.29 ± 1 Hz) or amplitude (Fig. 2). That bath-applied (1S,3R)-ACPD was active in these cells was evident by the small inward currents, 30–60 pA (see, for example, Fig.2), that it induced. Note that in experiments performed with Cs-based electrode filling solutions and iontophoretic application of (1S,3R)-ACPD, direct postsynaptic membrane effects, such as these inward currents, were undetectably small (see Figs. 4,6,7) and cannot account for the effects we report.
Fig. 2.

(1S,3R)-ACPD does not affect the amplitude or frequency of TTX-insensitive mIPSCs. Thetop trace illustrates the actions of bath-applied (1S,3R)-ACPD (duration of application is indicated by the solid bar) on a continuous record of spontaneous mIPSCs in the presence of 0.5 μm TTX. (1S,3R)-ACPD produced an inward current of ∼30 pA. A1, Traces of mIPSCs on an expanded time scale recorded during the control period before the application of (1S,3R)-ACPD. A2, Shown are mIPSCs during the sixth minute of (1S,3R)-ACPD perfusion.B1, Corresponding amplitude histograms of the mIPSCs before (filled bars) and in the presence of (1S,3R)-ACPD (open bars). Measurements were taken for 1 min in each condition.B2, Cumulative amplitude distributions obtained in B1 for mIPSCs recorded in control (solid line) and in the presence of (1S,3R)-ACPD (dashed line) for the experiment shown in A. C, Averaged cumulative amplitude distributions of mIPSCs obtained from six cells in control and then in (1S,3R)-ACPD. D, Bar graph shows mean mIPSC frequencies in control and in the presence of (1S,3R)-ACPD (n = 6). (1S,3R)-ACPD had no significant effect on the frequency of mIPSCs. Individual amplitude distributions of events in B2 and C in control and in (1S,3R)-ACPD are not statistically significant, as determined by Kolmogorov–Smirnov tests (p < 0.005).
Fig. 4.

Group III mGluRs are not involved in CA1 DSI.A1, The first current trace (left) illustrates two control DSI trials. Thecenter trace shows two DSI trials recorded from the same cell during the fifth minute of application of the group III mGluR agonist, l-AP4. The trials in the right trace also were recorded in l-AP4 after the stimulation intensity had been increased (ADJ. STIM. INTENSITY) to elicit IPSCs comparable in amplitude to those recorded in control. A2, The bar graph summarizes results from 11 experiments similar to those inA1. DSI was not affected significantly byl-AP4. B, An experiment showing that the suppression of CA1 field EPSPs by l-AP4 can be blocked by M-AP4, under our conditions, and hence that M-AP4 is an effective group III antagonist. EPSPs displayed above the graph are averages of six consecutive responses recorded at the indicated time points from one slice. Results from five experiments are summarized in the bar graph to the right. The continuous trace inC shows that M-AP4 (the duration of application is indicated by the solid bar) blocks neither DSI (filled arrows) nor the suppression of IPSCs induced by iontophoresis of (1S,3R)-ACPD (open arrows). Below the trace are IPSCs recorded at the indicated time points before (Pre-DSI) and during DSI (DSI) as well as before (Pre-ACPD) and after (ACPD) iontophoresis of (1S,3R)-ACPD. Individual IPSCs are the averages of five consecutive responses. The bar graph to theright summarizes results from four experiments. (1S,3R)-ACPD was iontophoresed by a −155 nA, 2 sec current. Asterisks denote significant differences from control values.
Fig. 6.

Selective block of DSI and the (1S,3R)-ACPD-induced suppression of IPSCs by (S)-MCPG, but not 4CPG. The current trace (CONTROL) in A illustrates the transient suppression of IPSCs during DSI (filled arrows) and after iontophoresis of (1S,3R)-ACPD (open arrows). The trace to the right of control, (MCPG) shows that both forms of IPSC suppression are antagonized during the 12th min of application of (S)-MCPG. The recovery trace shown to the far right was taken 40 min after we started to wash (S)-MCPG from the recording chamber. All current traces in A were recorded from the same cell. To theleft, in the bottom part of A, are IPSCs recorded at the indicated time points before (Pre-DSI) and during (DSI) as well as before (Pre-ACPD) and after (ACPD) iontophoretic application of (1S,3R)-ACPD. The bar graph in thecenter summarizes the results from five cells. The bar graph to the extreme right shows the dose dependence of (S)-MCPG effects on DSI. B, Shown is a continuous record in which the evoked IPSCs were subjected to the same stimulating protocol as in A. Belowthe record are IPSCs recorded at the indicated time points. Note that 4CPG (duration of application is indicated by the solid bar) does not antagonize DSI or the (1S,3R)-ACPD-induced suppression of IPSCs. The bar graph illustrates the results from five cells. IPSCs inA and B are averaged traces from five consecutive responses. (1S,3R)-ACPD was iontophoresed by a −155 nA, 2 sec current. Asterisksindicate significant differences from the control values.
Fig. 7.
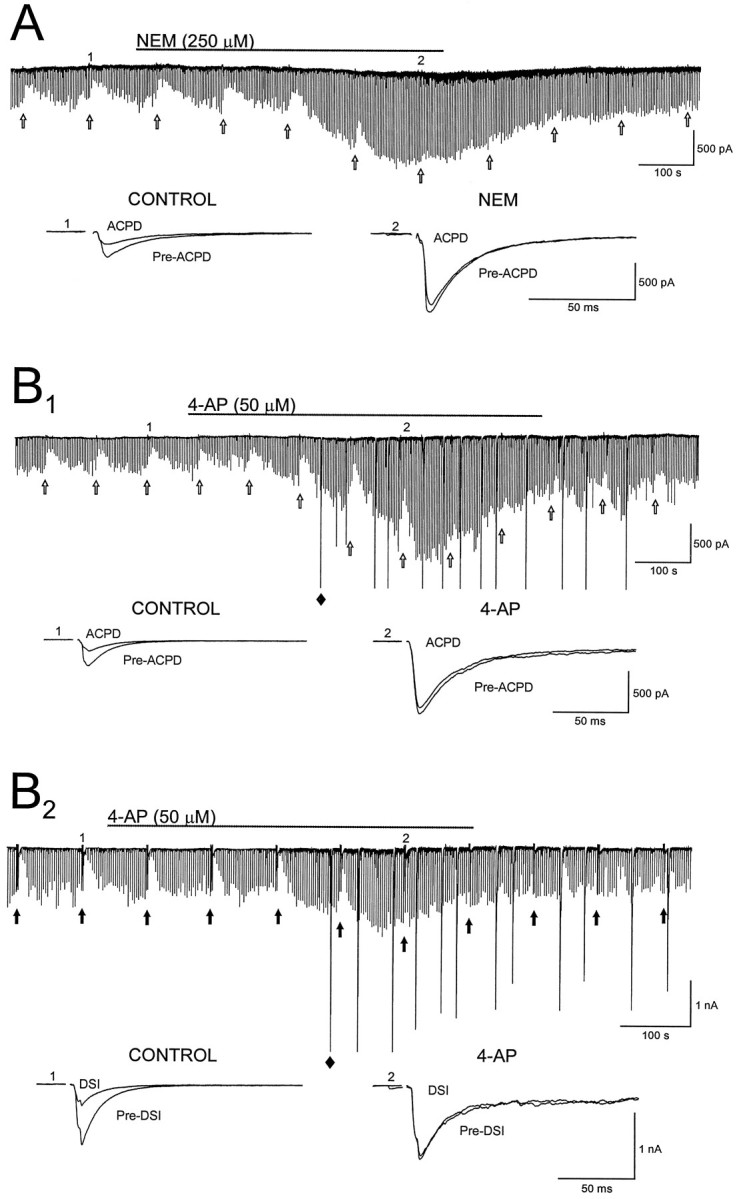
Agents that block DSI also block (1S,3R)-ACPD-induced depression of evoked monosynaptic IPSCs. A, N-ethylmaleimide (NEM; duration of application is indicated by thesolid line) blocks IPSC suppression induced by iontophoretic application of (1S,3R)-ACPD (open arrows). Below the current trace are IPSCs recorded before (Pre-ACPD) and after (ACPD) iontophoresis of (1S,3R)-ACPD at the indicated time points before (CONTROL) and during (NEM) application of NEM. B1, (1S,3R)-ACPD-induced depression of IPSCs is blocked by 4-aminopyridine (4-AP; duration of application is denoted by the solid line). IPSCs shownbelow the continuous trace were taken at the indicated time points. 4-AP induced large spontaneous inward currents (e.g.,filled diamond), presumed to be GABADresponses (Perrault and Avoli, 1992). B2, 4-AP blocks DSI of evoked IPSCs. IPSCs below the continuous trace were taken before (Pre-DSI) and after (DSI) the depolarizing steps at the indicated time points. Individual IPSCs in A and Bare averages of five consecutive IPSCs. Iontophoresis of (1S,3R)-ACPD (25 mm) inA and B1 was accomplished by a −155 nA, 2 sec current. Large spontaneous inward currents inB2 are truncated to fit the figure.
Evidence against group II or group III mGluR involvement in DSI
(1S,3R)-ACPD is an agonist at group I and group II mGluRs; hence it was not clear which class was responsible for IPSC suppression in CA1. In cerebellum the highly selective group II agonist DCG-IV (0.5–5 μm) potently blocked IPSCs and occluded DSI (Glitsch et al., 1996). In CA1 we found that bath application of 10 μm DCG-IV to five pyramidal cells, in which robust DSI of evoked IPSCs was present, affected neither the IPSC amplitudes nor DSI (control DSI, 43 ± 4.7%; DCG-IV DSI, 46 ± 6.1%) (Fig. 3A1). We confirmed (Kamiya et al., 1996) that 1 μm DCG-IV was effective in CA3, however, reducing evoked field potentials by 79 ± 4.2% (n = 7) (Fig. 3A2), and in a separate study (Morishita and Alger, 1997b) we verified that 10 μm DCG-IV reduces CA3 IPSCs (see Poncer et al., 1995). The EC50 of l-CCG-I for phosphoinositide (PI) hydrolysis caused by expressed mGluR1 is ∼50 μm (Suzdak et al., 1994). The EC50 of l-CCG-I for the inhibition of forskolin-stimulated cAMP production by expressed mGluR4 is ∼50 μm, whereas its EC50 in the same assay when mediated by expressed mGluR2 is 0.3 μm. Thus at low concentrations l-CCG-I is relatively selective for mGluR2. We found that l-CCG-I, at 3 μm, had no effect on either evoked IPSCs or DSI (n = 6; Fig.3B). In one cell we then increased the dose ofl-CCG-I to 100 μm and found that both IPSCs and DSI were reduced dramatically. The lack of effect of group II mGluR agonists on IPSCs means that these receptors probably are not involved in DSI.
Fig. 3.

Group II mGluRs are not involved in CA1 DSI.A1, Continuous trace of IPSCs showing several DSI trials. Below the trace are averages of five consecutive IPSCs recorded at the indicated time points before (Pre-DSI) and after the voltage step (DSI) in control and in the presence of the specific group II mGluR agonist, DCG-IV, applied at 10 μm (duration of application is indicated by thesolid bar above the continuous trace). Next to the averaged traces is a bar graph summarizing the data from five cells. DSI is not significantly altered by DCG-IV. A2, The graph shows that 1 μm DCG-IV suppressed mossy fiber–CA3 field EPSPs (n = 7 slices) and hence is active under our conditions. EPSPs above the graph are averages of six consecutive responses recorded at the indicated time points from one slice. B, The first trace (left) shows a control DSI trial, and the trial to theright is from the same cell during the eighth minute of application of the group II mGluR agonist, l-CCG-I. Results from six such experiments are shown in the bar graph located to theright of the current traces. l-CCG-I had no effect on DSI.
The mGluR agonist, l-AP4, is selective for group III mGluRs (mGluR4, mGluR6, mGluR7, and mGluR8). Although mGluR6 does not appear to be present in hippocampus and the levels of mGluR4 and mGluR8 are very low in CA1 (Testa et al., 1994; Shigemoto et al., 1997), mGluR7 is present (Okamoto et al., 1994; Saugstad et al., 1994; Shigemoto et al., 1997). l-AP4 bath-applied at 200 μm to six cells reduced the IPSCs by a mean of 39 ± 4.9%. Gereau and Conn (1995b) found that l-AP4 did not block IPSCs when glutamatergic transmission was blocked by APV and CNQX, implying thatl-AP4 acted at another site, probably the excitatory nerve terminals onto the interneurons. Our experiments were done in CNQX and APV; hence this explanation could not account for our data. Nevertheless, glutamate still was released in the presence of CNQX and APV, so we considered whether or not synaptically released glutamate could affect IPSCs by activation of mGluRs on interneurons. Adenosine inhibits glutamate release without affecting GABA release (Lambert and Teyler, 1991). We found that 50 μm adenosine did not alter the ability of l-AP4 to reduce IPSCs (47 ± 14.3% reduction in control vs 46 ± 15.9% reduction in adenosine; n = 4), a result that is explained most easily as a direct inhibitory effect of group III mGluRs on GABAergic interneurons, rather than as an indirect effect.
Despite its suppression of monosynaptic IPSCs, l-AP4 did not reduce DSI significantly (control DSI, 42 ± 4.7%;l-AP4 DSI, 37 ± 4.0%) (Fig.4A1,A2, p = 0.2; n = 11), even when stimulus intensity was increased to restore IPSC amplitudes to control levels (Fig. 4A). Moreover, neither the group III mGluR antagonist MSOP, 200 μm(control DSI, 48 ± 7.8%; MSOP DSI, 50 ± 7.8%;n = 6), nor the antagonist M-AP4, 2.5 mm, affected DSI (Fig. 4C, n = 4). As also shown in Figure 4C, M-AP4 had no effect on iontophoretically applied (1S,3R)-ACPD-induced IPSC suppression, although M-AP4, 1 mm, completely and reversibly blocked the effects of 50 μml-AP4 on the CA1 field EPSP (Fig. 4B). These data argue against a role for group III mGluRs in DSI.
Group II and group III mGluRs produce their effects mainly by inhibiting adenylate cyclase (Conn et al., 1994). We found (data not shown) that bath application of 50 μm forskolin enhanced monosynaptic IPSC amplitude (by 60 ± 14%; n = 8), as previously reported (Capogna et al., 1995). Forskolin had no effect on DSI, however, which in these cells amounted to a depression of 54 ± 3.6% in control and 45 ± 5.9% in forskolin (p = 0.09). Because forskolin did occlude DSI and the effects of the group II agonists in cerebellum (Glitsch et al., 1996), our results are consistent with a lack of participation of group II or group III mGluRs in hippocampal CA1 DSI, assuming that the depression of synaptic transmission mediated by these receptors is caused by cAMP reduction.
Group I mGluR agonists mimic and occlude DSI
The efficacy of (1S,3R)-ACPD and a high concentration of l-CCG-I in reducing IPSCs and DSI suggested that group I mGluRs (mGluR1 and mGluR5) could be responsible. At low concentrations (≤10 μm) quisqualate is fairly selective for group I (Suzdak et al., 1994). We tested the effects of quisqualate at 2 (n = 3), 5 (n = 3) and 10 μm (n = 2) and found that each was highly effective in reducing IPSC amplitudes (pooling results from these experiments gave a mean suppression of IPSC amplitudes of 70 ± 5.6% from control) (Fig.5D, n = 8) and DSI from 48 ± 3.6% to 19 ± 3.7% (Fig. 5A,C,n = 7). DHPG, 100 μm, is quite specific for group I mGluRs (Ito et al., 1992; Schoepp, 1994; Brabet et al., 1995; Gereau and Conn, 1995b). DHPG reduced IPSCs (to 52.9 ± 4.4% of control, Fig. 5D) and DSI (from 51 ± 5.3% in control to 17 ± 4.2% in DHPG) (Fig. 5B,C,n = 6). Thus the activation of group I mGluRs can mimic and occlude DSI.
Fig. 5.

Group I mGluR agonists, l-quisqualate and DHPG, reduce the amplitude of evoked monosynaptic IPSCs and occlude DSI. A, The first trace (left) shows the DSI of IPSCs recorded in the control saline. The center trace shows IPSCs recorded during the 10th min of bath application of l-quisqualate (QUIS). Theright trace shows a DSI trial still inl-quisqualate after the stimulation intensity had been increased (ADJ. STIM. INTENSITY) to elicit IPSCs comparable to those in the control condition. B, Illustrated are the effects of DHPG on IPSCs and DSI; trace sequences are as in A. Both l-quisqualate and DHPG suppressed IPSCs and occluded DSI, and the effects persisted even after the stimulation intensity had been increased. C, A graph summarizes the effects of quisqualate and DHPG on DSI.D, A graph shows the effect of these agonists on IPSC amplitudes. Asterisks indicate significant differences from control values.
(S)-MCPG reduces (1S,3R)-ACPD-induced suppression of IPSCs and DSI
The mGluR antagonist, l-AP3, blocks cerebellar DSI; however, when bath-applied at 1 mm, l-AP3 had no consistent effect on CA1 IPSCs or DSI. Of four cells it had no apparent effect on DSI in three, and in one cell both the IPSC and DSI were diminished. When tested on DSI and iontophoretic (1S,3R)-ACPD-induced IPSC suppression in another group of cells, l-AP3 had no significant effect on either (n = 3; data not shown). Thus l-AP3 appeared to be an ineffective mGluR antagonist in CA1 in our hands.
We examined the effect of (S)-MCPG on (1S,3R)-ACPD-induced IPSC suppression and DSI by bath-applying it at concentrations from 0.5 to 5 mm. (1S,3R)-ACPD was applied iontophoretically from a pipette containing 25 mm (1S,3R)-ACPD (see Materials and Methods). Evoked IPSCs were suppressed on alternate trials by DSI or by iontophoretic (1S,3R)-ACPD. (S)-MCPG caused a dose-dependent reduction in DSI, decreasing it, for example, by 10 ± 6.9% at 0.5 mmand by 57 ± 1.3% at 5 mm (Fig.6A). (S)-MCPG was more effective in blocking (1S,3R)-ACPD suppression of IPSCs than DSI, causing reductions of 42 ± 12.7% and 82 ± 16.8% at 0.5 and 5 mm, respectively, in the same cells. (S)-MCPG had no significant effect on IPSC amplitudes even at 5 mm (control IPSC, −1269 ± 172 pA; (S)-MCPG IPSC, −1039 ± 138 pA;p = 0.1; n = 5). We also tested the antagonist 4-carboxyphenylglycine (4CPG) because this has been proposed to distinguish between mGluR1 and mGluR5 effects (Brabet et al., 1995). At 200 μm (KB = 14.9 ± 7.7 μm for blocking mGluR1 effects in LLC-PK1 cells) (Brabet et al., 1995), 4CPG did not affect DSI (control DSI, 54 ± 9.2%; 4CPG DSI 62 ± 7.4%) (see Fig. 6B) and also did not affect (1S,3R)-ACPD-induced IPSC depression (control (1S,3R)-ACPD, 35 ± 8.3%; 4CPG (1S,3R)-ACPD, 40 ± 8.3%). These results suggest that mGluR5 rather than mGluR1 might be involved in IPSC suppression (see Discussion).
(1S,3R)-ACPD does not block DSI by reducing voltage-dependent Ca2+ currents in postsynaptic CA1 pyramidal cells
Activation of mGluRs can cause a modest reduction of voltage-dependent Ca2+ current (VDCC) in CA1 cells (10–30%) (Lester and Jahr, 1990; Swartz and Bean, 1992; Trombley and Westbrook, 1992). Inasmuch as DSI induction depends on VDCCs in CA1 (Pitler and Alger, 1992; Lenz et al., 1997), it was conceivable that the reduction of DSI by mGluRs was dependent on this effect. However, (1) mGluR agonists had no consistent effect on clamp current during the DSI-inducing voltage steps, i.e., in an arbitrary group of 17 cells, the net current did not change in 10, decreased in the outward direction in four, and increased in the outward direction in three; (2) adenosine, which causes a greater reduction in VDCC in CA1 cells than does (1S,3R)-ACPD (Kavalali et al., 1997), had no effect on DSI; and, finally, (3) reported mGluR effects on VDCCs are strictly dependent on the presence of GTP, or GTPγS, in the recording electrode. Omitting GTP from our pipette solution decreased the magnitude of G-protein-dependent responses (Pitler and Alger, 1994). Nevertheless, the (1S,3R)-ACPD reduction of DSI in the absence of pipette GTP was not altered (n = 4; data not shown). Thus the effect of (1S,3R)-ACPD cannot be ascribed to any apparent action on postsynaptic VDCCs in CA1 pyramidal cells. In view of its strong suppressant effect on IPSCs, it seems most likely that (1S,3R)-ACPD occludes DSI by reducing IPSCs by a presynaptic inhibition of GABA release, although there are other possibilities.
Other similarities between (1S,3R)-ACPD-induced IPSC suppression and DSI
The previous data are consistent with a role for glutamate and mGluR in DSI. To test the hypothesis further, we examined other properties of mGluR-induced IPSC suppression. DSI can be reduced by 250–300 μm NEM (Morishita et al., 1997) and by bath application of the K+ channel blocker 4-AP, 50 μm (Alger et al., 1996). If an mGluR mediates the signal for DSI, then NEM and 4-AP should reduce the (1S,3R)-ACPD-induced IPSC suppression. From the actions of mGluR1 agonists and the lack of effect of DCG-IV in CA1, we infer that the actions of (1S,3R)-ACPD are on group I mGluRs in CA1. A limited-duration application (10 min) of NEM, 250 μm, blocked DSI at a time when IPSC amplitudes were increased over control values, as previously reported. NEM also consistently antagonized the effects of (1S,3R)-ACPD on IPSCs (control IPSC suppression by (1S,3R)-ACPD, 56 ± 10.6%; IPSC suppression by (1S,3R)-ACPD in NEM, 6 ± 3.3%; n = 6; p < 0.01) (Figure7A). NEM effects were not reversible over the time course of our experiments (Morishita et al., 1997). The (1S,3R)-ACPD-induced suppression of IPSCs and DSI (Fig. 7B1) was reversed by 4-AP, and 4-AP reduced DSI of evoked IPSCs (Fig. 7B2) (control DSI, 38 ± 4%; 4-AP DSI, 11 ± 2.9%;n = 6), as previously reported (Alger et al., 1996).
DSI does not affect the normal paired-pulse depression (PPD) of IPSCs that is present at a 200 msec interstimulus interval (Alger et al., 1996; Morishita and Alger, 1997a). (1S,3R)-ACPD, 50 μm, reduced IPSCs by 56 ± 4.7%, and yet PPD, which was 77 ± 2.8% in control, was 81 ± 6.9% during (1S,3R)-ACPD application (Fig.8, p = 0.5;n = 7). Thus (1S,3R)-ACPD, like DSI, reduced IPSC amplitude, but it did not alter PPD.
Fig. 8.

(1S,3R)-ACPD does not significantly alter paired-pulse depression of evoked monosynaptic IPSCs. A1, Continuous trace illustrating the effects of bath-applied (1S,3R)-ACPD (duration of application is indicated by the solid bar) on paired-pulse depression (PPD) of IPSCs. Pairs of IPSCs were elicited every 5 sec with an interstimulus interval of 200 msec.Below the continuous trace are IPSCs elicited by the first (filled circle) and second (open circle) stimulus of the paired-pulse stimulation recorded at the indicated time points. In the right records the traces at 1 and 2 are overlapped (offset for ease of comparison) after the first IPSC in 2 was scaled up to match the amplitude of the first IPSC in 1. Note that the ratio of second to first IPSCs does not change in (1S,3R)-ACPD. A2, Plots generated from the experiment in A1illustrating the amplitudes of IPSCs arising from the first (filled circles) and second (open circles) stimulus and corresponding PPD ratio (IPSC2/IPSC1,filled triangles). B, Average PPD ratio obtained from seven cells recorded in control and then in the presence of (1S,3R)-ACPD. The IPSCs illustrated inA1 are averaged from five consecutive IPSC pairs.
DISCUSSION
We examined the general hypothesis that glutamate, or an analog, might, via activation of an mGluR, act as a retrograde signal to suppress IPSCs in hippocampal DSI. Our data do not support the hypothesis that group II mGluRs mediate CA1 DSI (cf. Glitsch et al., 1996). This was not surprising, because there is little evidence that mGluR2 exists in CA1 (Shigemoto et al., 1997), and mGluR3 mRNA expression is low (Testa et al., 1994). We confirmed that, although active in CA3 (cf. Poncer et al., 1995; Morishita and Alger, 1997b), DCG-IV does not depress monosynaptic evoked IPSCs or mIPSCs in CA1 (Gereau and Conn, 1995a), and we found that neither DCG-IV nor low concentrations of l-CCG-I affected DSI. Forskolin, which blocks cerebellar DSI (Glitsch et al., 1996), had no effect on CA1 DSI.
Both group I and group III mGluR agonists suppressed IPSCs; however, only group I agonists occluded DSI, suggesting that only they mimic the DSI mechanism. MSOP and M-AP4, effective group III antagonists, did not block DSI. Moreover, because group III mGluRs reduce cAMP, as do group II receptors, the results of Glitsch et al. (1996) also suggest that forskolin should have reduced CA1 DSI if l-AP4-sensitive receptors had been involved.
Gereau and Conn (1995a) reported that l-AP4 blocked only polysynaptic, but not monosynaptic, IPSCs in CA1. In our handsl-AP4 consistently reduced IPSCs in CNQX and APV, even when adenosine was added to suppress glutamate release and further prevent the activation of polysynaptic IPSCs. The differences in results probably are explained by the activation of different interneurons in the two studies.
(S)-MCPG, a weak competitive mGluR antagonist, reduced DSI in a dose-dependent way. The efficacy of (S)-MCPG in blocking PI hydrolysis either produced by expressed group I mGluRs (Brabet et al., 1995) or measured in brain tissue (Littman and Robinson, 1994) depends on the agonist and is greater when (1S,3R)-ACPD, rather than glutamate, is used. We found that (S)-MCPG was more potent in reversing (1S,3R)-ACPD-induced IPSC reduction than in reducing DSI. Indeed, Littman and Robinson (1994) show that 3 mm (S)-MCPG reducedl-glutamate-induced PI hydrolysis in hippocampal tissue suspensions by only ∼20%, in good agreement with our data on DSI. Although (S)-MCPG antagonizes both group I and group III mGluRs (Manzoni et al., 1995) the evidence against group III mGluR involvement makes a role for group I mGluRs in DSI more likely. Alternatively, an undefined mGluR subtype could mediate the inhibition of interneurons. The signal could be a glutamate analog, and not glutamate itself. In any case, the block of DSI by (S)-MCPG suggests that a glutamate-like substance plays a role.
Additional tests of the mGluR hypothesis were based on previously established properties of DSI that should be duplicated by any putative DSI signal in CA1. The candidate mechanism should (1) not affect mIPSCs (Pitler and Alger, 1994; Alger et al., 1996), (2) be blocked by agents that block G-proteins, such as pertussis toxin (Pitler and Alger, 1994) or NEM (Morishita et al., 1997), (3) be reduced by bath application of 50 μm 4-AP, and (4) not alter PPD (Alger et al., 1996;Morishita and Alger, 1997a). As assessed by these criteria, mGluR activation is a candidate mechanism for DSI induction. The K+ channel blocker 4-AP reversed the depressant action of (1S,3R)-ACPD on CA1 IPSCs and decreased DSI. [4-AP prevents the trans-ACPD-induced depression of EPSCs in cortical neurons (Sladeczek et al., 1993), suggesting that 4-AP sensitivity may be a general property of mGluR-mediated synaptic depression.] The sulfhydryl alkylating agent, NEM, enhances IPSCs and abolishes DSI (Morishita et al., 1997), and NEM abolishes the depressant action of (1S,3R)-ACPD on IPSCs, implying that G-proteins are involved in both responses (cf. Shapiro et al., 1994).
(1S,3R)-ACPD, like DSI, reduced IPSCs without affecting PPD of IPSCs, whereas many mechanisms that reduce transmitter release do affect PPD (Davies et al., 1990; Misgeld et al., 1995; Alger et al., 1996) by altering the probability of transmitter release by the first pulse according to the inverse relationship between probability of release by the first and second pulses of a pair (Martin, 1977). When evoked GABA release is decreased by substituting Sr2+ for extracellular Ca2+, PPD changes to paired- pulse facilitation (PPF) (Morishita and Alger, 1997a). Neither PPD nor Sr2+-induced PPF changed during DSI; thus the DSI process does not lower the probability of release at presynaptic terminals as other agents do. Baskys and Malenka (1991) found that (1S,3R)-ACPD enhanced paired-pulse EPSC facilitation while depressing EPSCs, raising the possibility that (1S,3R)-ACPD may suppress glutamate and GABA release via different mechanisms, although the effect on EPSCs was present only in young animals (<30 d).Barnes-Davies and Forsythe (1995) reported that (1S,3R)-ACPD reduced EPSCs at the calyx of Held in rat auditory brainstem slices without affecting PPF or the presynaptic action potential. Subsequently, Takahashi et al. (1996)showed that (1S,3R)-ACPD reduced Ca2+ entry into the calyx. It recently has been reported that a DSI-like process occurs in a dissociated hippocampal cell culture (Ohno-Shosaku et al., 1998). Despite having many similarities to DSI in the hippocampal slice, DSI in tissue culture altered the paired-pulse ratio. Why some forms of presynaptic inhibition alter paired-pulse release and others do not is unknown. In CA1, mGluR activation and DSI are similar in this regard, however.
Activation of mGluR can reduce voltage-dependent Ca2+ currents (Lester and Jahr, 1990; Sahara and Westbrook, 1993; Takahashi et al., 1996). DSI is dependent on Ca2+ influx into the pyramidal cell through voltage-dependent Ca2+ channels (Lenz et al., 1997), and thus mGluR agonists could reduce DSI by decreasing postsynaptic Ca2+ influx. On the other hand, the mGluR-induced inhibition of Ca2+ currents is dependent on postsynaptic G-proteins (Lester and Jahr, 1990; Sahara and Westbrook, 1993), and neither the (1S,3R)-ACPD-induced IPSC reduction nor occlusion of DSI requires GTP in the recording electrode. Thus mGluR effects on DSI probably are not caused by a reduction in Ca2+ influx, although we cannot rule out a contribution of this mechanism.
If a group I mGluR is involved in DSI and (1S,3R)-ACPD-induced IPSC suppression, it is more likely to be mGluR5 than mGluR1. Expression of mGluR1 mRNA in CA1 is low (Testa et al., 1994) and antibody staining for mGluR1 is confined to a discrete group of interneurons near the border of s. oriens and the CA1 alveus, whereas expression of mGluR5 mRNA is very high and antibody staining is dense and widely distributed in CA1. (S)-MCPG was somewhat less potent in blocking mGluR5a-induced than mGluR1a-induced PI hydrolysis in LLC-PKI cells when glutamate was the agonist (Brabet et al., 1995); when (1S,3R)-ACPD was the agonist, (S)-MCPG was very effective at both receptors. In contrast, 4CPG was a potent antagonist of (1S,3R)-ACPD actions only on mGluR1a-, not mGluR5a-, mediated effects; thus the combination of (1S,3R)-ACPD and 4CPG can distinguish between mGluR1a and mGluR5. Because IPSC suppression by (1S,3R)-ACPD was blocked by (S)-MCPG, but not by 4CPG, mGluR5 receptors may inhibit GABA release in CA1.
Results of ultrastructural labeling studies have been inconsistent regarding the localization of group I mGluRs, with one study finding evidence for axonal localization of group I mGluRs (Romano et al., 1995) and the other not (Shigemoto et al., 1997). If group I mGluRs are not present on or near inhibitory nerve terminals, the group I mGluRs known to exist on the somata and dendrites of the interneurons could be responsible for the effects we report.
Implications of mGluR involvement in DSI
The hypothesis that glutamate acting via mGluRs is the retrograde signal in hippocampal CA1 DSI can explain some puzzles. In cerebellum there are two types of DSI (Alger and Pitler, 1995). One acts on TTX-insensitive mIPSCs; the other is blocked when TTX is applied. Because TTX blocks CA1 DSI, we infer that DSI in CA1 and the second form of cerebellar DSI are identical. We suggest that glutamate, or an analog, could be the universal DSI messenger. The different types of DSI would be determined by the classes of mGluR on the interneurons and the effector mechanisms to which they are coupled. The group II mGluRs would be linked to the cAMP-dependent mechanism, which can block even TTX-insensitive mIPSCs. The mGluRs hypothetically responsible for CA1 DSI instead would be linked to other mechanisms such that only TTX-sensitive release processes are affected.
A simple model for our results is that glutamate, released from pyramidal cell somatic–dendritic regions, acts directly on interneurons. Lledo et al. (1998) report that a vesicle-fusion-dependent process presumably in pyramidal cell somatic– dendritic regions is required for LTP. If glutamate were packaged in vesicles in the dendrites, then this process also could be involved in DSI. Alternatively, glutamate also can be released from glial cells (Barres, 1991; Parpura et al., 1994) and affect neuronal neurotransmitter release via mGluRs (Arague and Haydon, 1997). An unknown signal from pyramidal cells could induce glutamate release from glial cells or the numerous glutamate-containing terminals of nearby excitatory fibers and mediate DSI indirectly. It will be important to test these implications of the mGluR hypothesis for DSI.
Footnotes
This work was supported by National Institutes of Health Grants NS30219 and NS22010 to B.E.A. We thank E. Elizabeth for expert typing and editorial assistance. We also thank F. Le Beau, R. Lenz, S. Mason, L. Martin, and N. Varma for their comments on a draft of this manuscript.
W.M. and S.A.K. contributed equally to this work.
Correspondence should be addressed to Dr. B. E. Alger, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201.
Dr. Kirov’s present address: Children’s Hospital, Department of Neurology, Harvard Medical School, 300 Longwood Avenue, Room 211, Boston, MA 02115.
REFERENCES
- 1.Alger BE, Pitler TA. Retrograde signaling at GABAA receptor synapses in the mammalian CNS. Trends Neurosci. 1995;18:333–340. doi: 10.1016/0166-2236(95)93923-l. [DOI] [PubMed] [Google Scholar]
- 2.Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA. Retrograde signaling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol (Lond) 1996;496:197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alger BE, Morishita W, Kirov SA. Evaluation of glutamate as the retrograde signal in hippocampal CA1 DSI. Soc Neurosci Abstr. 1997;23:10. [Google Scholar]
- 4.Arague A, Haydon PG. Astrocytes modulate evoked synaptic transmission between cultured hippocampal neurons. Soc Neurosci Abstr. 1997;23:359. [Google Scholar]
- 5.Barnes-Davies M, Forsythe ID. Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brainstem slices. J Physiol (Lond) 1995;488:387–406. doi: 10.1113/jphysiol.1995.sp020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barres BA. New roles for glia. J Neurosci. 1991;11:3685–3694. doi: 10.1523/JNEUROSCI.11-12-03685.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol (Lond) 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole-cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 9.Brabet I, Mary S, Bockaert J, Pin J-P. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacology. 1995;34:895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- 10.Capogna M, Gähwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J Neurosci. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conn PJ, Boss V, Chung DS. Second-messenger systems coupled to metabotropic glutamate receptors. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 59–98. [Google Scholar]
- 12.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai MA, McBain CJ, Kauer JA, Conn PJ. Metabotropic glutamate receptor-induced disinhibition is mediated by reduced transmission at excitatory synapses onto interneurons and inhibitory synapses onto pyramidal cells. Neurosci Lett. 1994;181:78–82. doi: 10.1016/0304-3940(94)90564-9. [DOI] [PubMed] [Google Scholar]
- 14.Gereau RW, IV, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995a;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gereau RW, IV, Conn PJ. Roles of specific metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995b;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 16.Glitsch M, Llano I, Marty A. Glutamate as a candidate retrograde messenger at interneurone–Purkinje cell synapses of rat cerebellum. J Physiol (Lond) 1996;497:531–537. doi: 10.1113/jphysiol.1996.sp021786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H. 3,5-Dihydroxyphenyl-glycine: a potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- 18.Jouvenceau A, Dutar P, Billard J-M. Presynaptic depression of inhibitory postsynaptic potentials by metabotropic glutamate receptors in rat hippocampal CA1 pyramidal. Eur J Pharmacol. 1995;281:131–139. doi: 10.1016/0014-2999(95)00223-8. [DOI] [PubMed] [Google Scholar]
- 19.Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J Physiol (Lond) 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- 21.Lambert NA, Teyler TJ. Adenosine depresses excitatory, but not fast inhibitory, synaptic transmission in area CA1 of the rat hippocampus. Neurosci Lett. 1991;122:50–52. doi: 10.1016/0304-3940(91)90190-5. [DOI] [PubMed] [Google Scholar]
- 22.Lenz RA, Wagner JJ, Alger BE. Involvement of N- and L-type Ca2+ channels in DSI of GABAAergic IPSCs in rat hippocampal CA1 neurons. Soc Neurosci Abstr. 1997;23:2276. [Google Scholar]
- 23.Lester RAJ, Jahr CE. Quisqualate receptor-mediated depression of calcium currents in hippocampal neurons. Neuron. 1990;4:741–749. doi: 10.1016/0896-6273(90)90200-y. [DOI] [PubMed] [Google Scholar]
- 24.Littman L, Robinson MB. The effects of l-glutamate and trans-(+)-1-amino-1,3-cyclopentanedicarboxylate on phosphoinositide hydrolysis can be pharmacologically differentiated. J Neurochem. 1994;63:1291–1302. doi: 10.1046/j.1471-4159.1994.63041291.x. [DOI] [PubMed] [Google Scholar]
- 25.Llano I, Marty A. Presynaptic metabotropic glutamatergic regulation of inhibitory synapses in rat cerebellar slices. J Physiol (Lond) 1995;486:163–176. doi: 10.1113/jphysiol.1995.sp020800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 27.Lledo P-M, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- 28.Manzoni OJ, Castillo PE, Nicoll RA. Pharmacology of metabotropic glutamate receptors at the mossy fiber synapses of the guinea pig hippocampus. Neuropharmacology. 1995;34:865–871. doi: 10.1016/0028-3908(95)00060-j. [DOI] [PubMed] [Google Scholar]
- 29.Martin AR. Junctional transmission. II. Presynaptic mechanisms. In: Kandel ER, editor. Handbook of physiology, Sec 1, The nervous system, Vol I, Pt 1. American Physiological Society; Bethesda, MD: 1977. pp. 329–355. [Google Scholar]
- 30.Misgeld U, Bijak M, Jarolimek W. A physiological role for GABAB receptors and the effects of baclofen in the mammalian central nervous system. Prog Neurobiol. 1995;46:423–462. doi: 10.1016/0301-0082(95)00012-k. [DOI] [PubMed] [Google Scholar]
- 31.Morishita W, Alger BE. Sr2+ supports DSI and provides new evidence for a presynaptic expression mechanism in rat hippocampal slices. J Physiol (Lond) 1997a;505:307–317. doi: 10.1111/j.1469-7793.1997.307bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morishita W, Alger BE. Differential effects of type II mGluR agonist, DCG-IV, on DSI of GABAAergic IPSCs in the CA1 region of rat hippocampus in vitro. Soc Neurosci Abstr. 1997b;23:356. [Google Scholar]
- 33.Morishita W, Kirov SA, Pitler TA, Martin LA, Lenz RA, Alger BE. N-ethylmaleimide blocks depolarization-induced suppression of inhibition and enhances GABA release in the rat hippocampal slice in vitro. J Neurosci. 1997;17:941–950. doi: 10.1523/JNEUROSCI.17-03-00941.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicoll RA, Alger BE. A simple chamber for recording from submerged brain slices. J Neurosci Methods. 1981;4:153–156. doi: 10.1016/0165-0270(81)90049-2. [DOI] [PubMed] [Google Scholar]
- 35.Ohno-Shosaku T, Sawada S, Yamamoto C. Properties of depolarization-induced suppression of inhibitory transmission in cultured rat hippocampal neurons. Pflügers Arch. 1998;435:273–279. doi: 10.1007/s004240050512. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem. 1994;269:1231–1236. [PubMed] [Google Scholar]
- 37.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte–neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 38.Perrault P, Avoli M. 4-Aminopyridine-induced epileptiform activity and a GABA-mediated long-lasting depolarization in the rat hippocampus. J Neurosci. 1992;12:104–115. doi: 10.1523/JNEUROSCI.12-01-00104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pin J-P, Duvoisin R. The metabotrophic glutamate receptors: structure and function. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 40.Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G-protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 42.Poncer J-C, Shinozaki H, Miles R. Dual modulation of synaptic inhibition by distinct metabotropic glutamate receptors in the rat hippocampus. J Physiol (Lond) 1995;485:121–134. doi: 10.1113/jphysiol.1995.sp020717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romano C, Sesma MA, McDonald CT, O’Malley K, van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 44.Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saugstad JA, Kinzie JM, Mulvihill ER, Segerson TP, Westbrook GL. Cloning and expression of a new member of the l-2–4-phosphonobutyric acid-sensitive class of metabotropic glutamate receptors. Mol Pharmacol. 1994;45:367–372. [PubMed] [Google Scholar]
- 46.Schoepp DD. 3,5-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada N, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sladeczek F, Momiyama A, Takahashi T. Presynaptic inhibitory action of a metabotropic glutamate receptor agonist on excitatory transmission in visual cortical neurons. Proc R Soc Lond [Biol] 1993;253:297–303. doi: 10.1098/rspb.1993.0117. [DOI] [PubMed] [Google Scholar]
- 50.Suzdak PD, Thomsen C, Mulvihill E, Kristensen P. Molecular cloning, expression, and characterization of metabotropic glutamate receptor subtypes. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 1–30. [Google Scholar]
- 51.Swartz KJ, Bean BP. Inhibition of calcium channels in rat CA3 pyramidal neurons by a metabotropic glutamate receptor. J Neurosci. 1992;12:4358–4371. doi: 10.1523/JNEUROSCI.12-11-04358.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- 53.Testa CM, Catania MV, Young AB. Anatomical distribution of metabotropic glutamate receptors in mammalian brain. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 99–123. [Google Scholar]
- 54.Trombley PQ, Westbrook GL. l-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J Neurosci. 1992;12:2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vincent P, Marty A. Neighboring cerebellar Purkinje cells communicate via retrograde inhibition of common presynaptic interneurons. Neuron. 1993;11:885–893. doi: 10.1016/0896-6273(93)90118-b. [DOI] [PubMed] [Google Scholar]
- 56.Vincent P, Armstrong CM, Marty A. Inhibitory synaptic currents in rat cerebellar Purkinje cells: modulation by postsynaptic depolarization. J Physiol (Lond) 1992;456:453–471. doi: 10.1113/jphysiol.1992.sp019346. [DOI] [PMC free article] [PubMed] [Google Scholar]