

Abstract
The role of the dopamine transporter (DAT) in mediating the neurotoxic effects of methamphetamine (METH) was tested in mice lacking DAT. Dopamine (DA) and serotonin (5-HT) content, glial fibrillary acidic protein (GFAP) expression, and free radical formation were assessed as markers of METH neurotoxicity in the striatum and/or hippocampus of wild-type, heterozygote, and homozygote (DAT −/−) mice. Four injections of METH (15 mg/kg, s.c.), each given 2 hr apart, produced 80 and 30% decreases in striatal DA and 5-HT levels, respectively, in wild-type animals 2 d after administration. In addition, GFAP mRNA and protein expression levels, extracellular DA levels, and free radical formation were increased markedly. Hippocampal 5-HT content was decreased significantly as well (43%). Conversely, no significant changes were observed in total DA content, GFAP expression, extracellular DA levels, or free radical formation in the striatum of DAT −/− mice after METH administration. However, modest decreases were observed in striatal and hippocampal 5-HT levels (10 and 17%, respectively). These observations demonstrate that DAT is required for, and DA is an essential mediator of, METH-induced striatal dopaminergic neurotoxicity, whereas serotonergic deficits are only partially dependent on DAT.
Keywords: dopamine transporter, methamphetamine, microdialysis, serotonin, free radical, GFAP
Methamphetamine (METH) is a highly abused drug worldwide. This phenylethylamine is a powerful psychostimulant that produces both addiction and dependence. It is now commonly accepted that METH causes neurotoxicity in animals (including nonhuman primates) as clearly shown by the degeneration of monoaminergic systems “in vivo,” demonstrating effects on both dopaminergic and serotonergic regions of the CNS. Repeated administration of METH induces long-lasting deficits in the striatal concentrations of dopamine (DA) and its metabolites, tyrosine hydroxylase activity, and dopamine transporter (DAT) binding sites (Gibb and Kogan, 1979; Seiden and Ricaurte, 1987; Seiden and Sabol, 1995). Similarly, reductions in forebrain concentrations of serotonin (5-HT) and its metabolites and the decrease of tryptophan hydroxylase activity after METH administration have been described (Hotchkiss and Gibb, 1980; Ricaurte et al., 1980).
Although several mechanisms have been proposed to explain METH-induced neurotoxicity (Sonsalla et al., 1989; Bowyer et al., 1995; Seiden and Sabol, 1995; Ali et al., 1996; Itzhak and Ali, 1996; Ohmori et al., 1996), two hypotheses are considered primarily. First, the ability of the drug to mobilize DA from intraneuronal stores to the extracellular space via DAT-mediated outward transport results in elevated extracellular DA concentrations. The neurotoxic effects of METH are postulated to occur from subsequent auto-oxidation of DA to highly reactive free radicals (Seiden and Vosmer, 1984; Axt et al., 1990;Marek et al., 1990a,b). An alternative model suggests that redistribution of DA from vesicular storage pools to the cytoplasmic compartment, allowing intraneuronal oxidation, is the primary cause of DA terminal injury (Cubells et al., 1994). These hypotheses suggest that the balance among vesicular, cytoplasmic, and extracellular DA pools plays a key role in the neurotoxic action of METH.
Thus DAT, suggested to be critically involved in inward transport of METH with concomitant outward transport of DA (Liang and Rutledge, 1982b), might be an important determinant of METH neurotoxicity. In fact, it has been shown that DA uptake inhibitors such as amfonelic acid can selectively antagonize the neurochemical response to METH (Axt et al., 1990; Marek et al., 1990a,b). However, pharmacological manipulations with DA uptake inhibitors do not always afford complete protection because they depend highly on the inherent drug properties, route of administration, and dosage. For instance, the DA uptake inhibitor benztropine failed to protect against DA depletion after METH challenge (Marek et al., 1990a). Additionally, it has been suggested that the response of the 5-HT system to METH treatment is mediated by DA (Hotchkiss and Gibb, 1980; Schmidt et al., 1985; Johnson et al., 1987). In this case, amfonelic acid protects 5-HT neurons against METH-induced damage, whereas benztropine does not (Schmidt et al., 1985). These inconsistencies clearly reveal that pharmacological interventions, although helpful, are limited. To assess the role of DAT in METH-induced neurotoxicity directly, we used mice lacking DAT as anin vivo model to determine the effects of METH on dopaminergic and serotonergic neurons.
MATERIALS AND METHODS
Animals. The DAT −/− mice were generated as previously described (Giros et al., 1996). Offspring from heterozygote crossings remained in the same cage until weaning. Mice were genotyped by Southern blot analysis of tail biopsies as described (Giros et al., 1996). They were separated into different cages according to sex and genotype and were maintained under standard housing conditions. Food and water were provided ad libitum. Animal care was in accordance with the Guide for Care and Use of Laboratory Animals (National Institutes of Health publication 865-23, Bethesda, MD) and approved by the Institutional Animal Care and Use Committee.
Drug administration and temperature determination.Three-month-old littermate male wild-type, heterozygote (DAT +/−), and homozygote (DAT −/−) C57BL/129sVj mice were used in these experiments. METH (Sigma, St. Louis, MO) was dissolved in saline (0.9% NaCl). Four subcutaneous injections of METH (15 mg/kg) or vehicle, each given 2 hr apart, or a single dose of METH (30 mg/kg, s.c.) was administered (final injection volume, 10 ml/kg). Rectal body temperatures were determined by a digital thermometer (Physitemp, Clifton, NJ). The probe was inserted into the rectum and maintained until the temperature reading had stabilized. All animal experiments were approved by the Institutional Animal Care and Use Committee.
Measurement of DA, 5-HT, and metabolites. At 48 hr after the first injection of METH the mice were killed, and striata were dissected from the brain. Dissected striata and hippocampi were homogenized in 0.1 m perchloric acid containing 100 ng/ml 3,4-dihydroxybenzylamine (DHBA) as an internal standard. Homogenates were centrifuged for 10 min at 10,000 × g. Supernatants were filtered through 0.22 μm filters and analyzed for levels of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 5-HT, and 5-hydroxy-indoleacetic acid (5-HIAA), using HPLC with electrochemical detection (HPLC-EC). Monoamines and metabolites were separated on a microbore reversed-phase column (C-18; 5 μm, 1 × 150 mm; Unijet, BAS, West Lafayette, IN) by using a mobile phase consisting of 0.03 m citrate-phosphate buffer with 2.1 mm octanesulfonic acid, 0.1 mm EDTA, and 17% methanol, pH 3.6, at a flow rate of 90 μl/min that was detected by a 3 mm glassy carbon Unijet electrode (Unijet, BAS) set at +0.85 V (Gainetdinov et al., 1997). The volume of injection was 5 μl.
Determinations of 5-HT synthesis rate in vivo. To measure the rate of 5-HT synthesis, we injected mice simultaneously with thel-aromatic amino acid decarboxylase inhibitor 3-hydroxybenzylhydrazine (NSD-1015) at 100 mg/kg, intraperitoneally, and saline or METH (30 mg/kg, s.c.) The concentration of 5-hydroxytryptophan (5-HTP) in the striatum 60 min later was determined via HPLC-EC as an in vivo measurement of tryptophan hydroxylase activity (Carlsson et al., 1972). Determinations were performed by using the same column and apparatus as described earlier (Wang et al., 1997) with a mobile phase consisting of (in mm) 50 monobasic sodium phosphate, 0.2 octyl sodium sulfate, 0.1 EDTA, and 10 NaCl plus 10% methanol, pH 2.6. The applied potential was +0.65 V.
RNA preparation and Northern blot analysis. Total RNA from mouse striata was isolated according to Chomczynski and Sacchi (1987). Northern blot analysis was performed according to Fumagalli et al. (1996), using a [32P]cRNA probe for glial fibrillary acidic protein (GFAP) mRNA. Briefly, after prehybridization, membranes were hybridized overnight at 65°C, washed under high stringency (2× SSC/0.1% SDS for 30 min at room temperature, followed by two to three washes in 0.1× SSC/0.1% SDS for 30 min at 68°C), and then exposed to Kodak X-Omat films (Rochester, NY). Blots were quantified with a Molecular Dynamics PhosphorImager (Sunnyvale, CA) and ImageQuant 3.3 software. A 115 base cRNA probe from a pTRIPLEscript vector (Ambion, Austin, TX) complementary to β-actin was used as an internal control to correct for gel-loading efficiencies.
Immunohistochemistry. At 2 d after METH administration the animals were killed, and the brains were dissected and immersion-fixed in 4% paraformaldehyde in PBS, pH 7.2, overnight at room temperature. Then the brains were dehydrated in ethanol, embedded in paraffin, and cut into 6 μm sections. Neuronal cell bodies were visualized by routine hematoxylin and eosin staining. Astrocytes were identified by immunohistochemistry, using anti-GFAP antibodies (Dako, Carpinteria, CA). Briefly, rehydrated sections were treated with 3% H2O2 for 10 min to remove endogenous peroxidase activity, rinsed for 20 min in PBS, and incubated with nonimmune goat serum (in PBS containing 1% BSA) for 20 min before a 60 min incubation with rabbit anti-rat GFAP (1:2000 in 1% PBS/BSA; Dako). Sections then were incubated with a secondary IgG antibody (Vectastain Elite ABC immunohistochemistry kit, Vector Laboratories, Burlingame, CA) for 30 min, washed in PBS, incubated in ABC reagent for 30 min, rinsed with PBS, and stained with a DAB substrate containing cobalt ions.
In vivo microdialysis. In vivo microdialysis in freely moving mice was performed as previously described (Gainetdinov et al., 1997). Mice were anesthetized with chloral hydrate (400 mg/kg., i.p.) and placed in a stereotaxic apparatus. Dialysis probes (2 mm membrane length and 0.24 mm outer diameter, Cuprophane; cutoff 6000 Daltons, CMA-11, CMA/Microdialysis, Solna, Sweden) with CMA-11 guide cannulae were implanted into the right striatum. Because of significant differences in animal size (Giros et al., 1996), the stereotaxic coordinates for implantation of microdialysis probes were anteroposterior (AP) 0.0, dorsoventral (DV) −4.4, lateral (L) 2.5 for wild-type and DAT +/− mice; and AP 0.0, DV −3.2, L 1.8 for DAT −/− mice, relative to bregma (Franklin and Paxinos, 1996). Placement of the probe was verified by subsequent histological examination.
After surgery the animals were returned to their home cages with free access to food and water. At 24 hr after surgery the dialysis probe was connected to a syringe pump (A-99, Razel Scientific Instruments, Stamford, CT) and perfused at 1 μl/min with artificial CSF [composition (in mm): Na+ 150, K+ 3.0, Ca2+ 1.4, Mg2+ 0.8, PO4− 31.0, and Cl− 155, pH 7.3]. After a 1 hr equilibration period the perfusate was collected every 20 min into tubes containing 1 μl of 2 m perchloric acid. At least four control samples were taken before METH was administered. Perfusate samples were assayed for DA, using HPLC-EC by the same chromatographic conditions as those described above. The sensitivity of the method permitted the detection of ∼3 fmol of DA.
Analysis of free radical formation. To evaluate the level of free radical formation, we used the “salicylate” microdialysis technique (Obata and Chiueh, 1992). The microdialysis procedure was performed as above. For trapping hydroxyl free radicals, the striatum was perfused with 5 mm sodium salicylate in Ringer’s solution. Brain dialysate (1 μl/min) was collected every 20 min and assessed immediately for 2,3-dihydroxybenzoic acid (2,3-DHBA) and 2,5-dihydroxybenzoic acid (2,5-DHBA) concentrations by HPLC-EC under the chromatographic conditions described for 5-HTP measurements.
Data analysis. The data are presented as mean ± SEM and were analyzed statistically by ANOVA with Fisher’s Protected Least-Significant Difference test.
RESULTS
Effect of METH on DA and 5-HT content in striatum and hippocampus
METH is known to induce profound deficits in both the dopaminergic and serotonergic systems. In this study HPLC-EC was used to quantitate the effects of METH on the neurochemistry of these systems in wild-type, DA transporter knock-out (DAT −/−), and heterozygous (DAT +/−) mice. Four injections of METH (15 mg/kg, s.c, each given 2 hr apart) produced an 80% decrease in DA levels in the striatum of wild-type animals 2 d after treatment, whereas no diminution was observed in DAT −/− mice (Fig.1A). In addition, DAT +/− mice displayed a much smaller decrease in DA levels as compared with wild-type animals (24%) (Fig. 1A). Similarly, the levels of DOPAC and HVA, the two main DA metabolites, were decreased markedly in the striatum of wild-type mice (80 and 72%, respectively), moderately reduced in DAT +/− mice (35 and 28%, respectively), and unchanged in DAT −/− mice (Fig.1A). These results suggest that DAT plays a determinant role for striatal dopaminergic neurotoxicity.
Fig. 1.
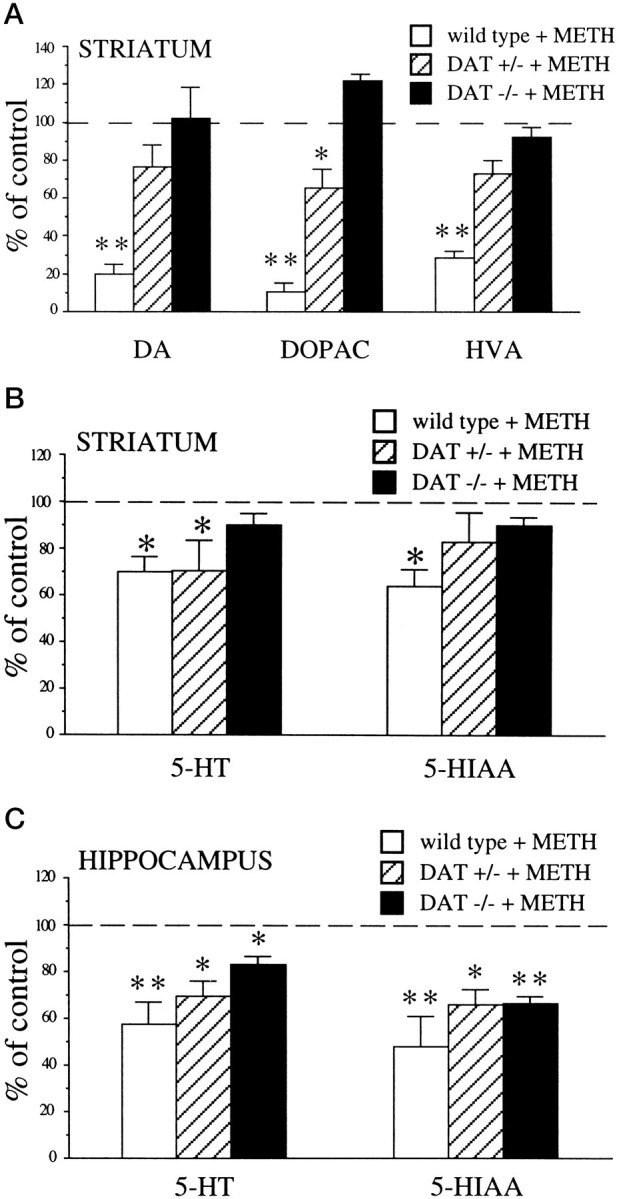
Effect of METH (4 × 15 mg/kg, s.c.) on DA, 5-HT, and metabolite content. A, Striatal levels of DA, DOPAC, and HVA. The data are presented as percentages of striatal monoamine levels in saline-treated controls, which were 18.2 ± 1.6, 1.2 ± 0.1, and 1.5 ± 0.2 ng/mg wet tissue in wild-type mice; 14.2 ± 1.3, 1.0 ± 0.08, and 2.1 ± 0.3 ng/mg wet tissue in DAT +/− mice; and 0.9 ± 0.05, 0.85 ± 0.1, and 3.9 ± 0.3 ng/mg wet tissue in DAT −/− mice for DA, DOPAC, and HVA, respectively. B, Striatal levels of 5-HT and 5-HIAA. The data are presented as percentages of striatal 5-HT and 5-HIAA levels in saline-treated controls, which were 0.6 ± 0.01 and 0.5 ± 0.08 ng/mg wet tissue in wild-type mice; 0.6 ± 0.02 and 0.5 ± 0.02 ng/mg wet tissue in DAT +/− mice; and 0.4 ± 0.03 and 0.6 ± 0.05 ng/mg wet tissue in DAT −/− mice for 5-HT and 5-HIAA, respectively. C, Hippocampal levels of 5-HT and 5-HIAA. The data are presented as percentages of hippocampal 5-HT and 5-HIAA levels in saline-treated controls, which were 0.9 ± 0.07 and 0.9 ± 0.11 ng/mg wet tissue in wild-type mice, 0.9 ± 0.07 and 0.9 ± 0.05 ng/mg wet tissue in DAT +/− mice, and 0.9 ± 0.04 and 1.0 ± 0.1 ng/mg wet tissue in DAT −/− mice for 5-HT and 5-HIAA, respectively. Values represent the mean ± SEM of four to five independent determinations. **p < 0.01 versus controls and *p < 0.05 versus controls.
The serotonergic system also was affected. In striatum, 5-HT levels were decreased by 30% in both wild-type and DAT +/− mice, whereas DAT −/− mice showed only a 10% decrease that was not statistically different from control animals (Fig. 1B). In hippocampus, where 5-HT levels are higher than in striatum, decreases of 43% in wild-type, 31% in DAT +/−, and 17% in DAT −/− mice were observed (Fig. 1C). The main metabolite of serotonin, 5-HIAA, also was affected. In fact, METH decreased 5-HIAA levels by 36% in the striatum of wild-type mice, with no significant changes in either DAT +/− or DAT −/− animals (Fig. 1B). Interestingly, in hippocampus, 5-HIAA levels were diminished not only in wild-type (52%) and DAT +/− (34%) mice but also in DAT −/− mice (33%) (Fig. 1C). In addition, the activity of tryptophan hydroxylase, the rate-limiting enzyme of 5-HT synthesis, was inhibited markedly in both striatum and hippocampus of wild-type (30 and 38%, respectively) and DAT −/− mice (37 and 46%, respectively) (Fig.2). These data suggest that DAT only contributes to, but is not solely responsible for, METH-induced 5-HT changes.
Fig. 2.

Effect of METH on tryptophan hydroxylase activity. 5-HTP accumulation in the striatum and hippocampus of wild-type and DAT −/− mice 60 min after METH treatment (single injection, 30 mg/kg, s.c.) was used as an indicator of tryptophan hydroxylase activity, as described under Materials and Methods. Concentrations of 5-HTP in saline-treated controls were 0.305 ± 0.043 ng/mg tissue in striatum (n = 5) and 0.697 ± 0.026 ng/mg tissue in hippocampus (n = 8). Values represent the mean ± SEM of five to eight animals per group. **p < 0.01 versus controls and *p < 0.05 versus controls.
It should be noted also that the effects described above were of prolonged duration. Because of a high lethality rate in animals repeatedly treated with 15 mg/kg METH, a single injection (30 mg/kg) was used to examine its long-term effects in the two genotypes. Serotonergic and dopaminergic indices of METH neurotoxicity were still apparent in wild-type mice 1 week after treatment with METH (30 mg/kg, s.c.). Dopamine, DOPAC, and HVA levels were reduced in wild-type mice by 80, 45, and 62%, respectively (Fig.3A), whereas 5-HT and 5-HIAA were depressed by 37 and 26%, respectively (Fig. 3B), as compared with the pretreatment levels. By contrast, no significant changes in DAT −/− mice were observed (Fig. 3A,B). These data suggest that the neurotoxic effects were long-lasting and rule out the possibility that DAT elimination only might have delayed, and not completely prevented, METH toxicity.
Fig. 3.

Long-term effect of METH (single injection, 30 mg/kg, s.c.) on striatal content of DA, DOPAC, and HVA (A) or 5-HT and 5-HIAA (B) in mice killed 7 d after the injection. The data are presented as percentages of striatal monoamine levels in saline-treated controls, which were 18.4 ± 1.0, 1.1 ± 0.1, and 1.6 ± 0.08 ng/mg wet tissue in wild-type mice; and 0.9 ± 0.11, 0.9 ± 0.07, and 3.1 ± 0.06 ng/mg wet tissue in DAT −/− mice for DA, DOPAC, and HVA, respectively. The data are presented as percentages of striatal 5-HT and 5-HIAA levels in saline-treated controls, which were 0.7 ± 0.04 and 0.7 ± 0.02 ng/mg wet tissue in wild-type mice and 0.5 ± 0.03 and 0.6 ± 0.05 ng/mg wet tissue in DAT −/− mice for 5-HT and 5-HIAA, respectively. Values represent the mean ± SEM of five independent determinations. *p < 0.05 versus controls. **p < 0.01 versus controls.
Effect of METH on GFAP gene expression, a marker of neuronal damage
The complete lack of toxic effects of METH in the striatum of DAT −/− mice was demonstrated further by determination of the neuroglial response to the neurotoxin. METH administration is known to increase GFAP immunoreactivity in the striatum of mice (Pu and Vorhees, 1993). GFAP is a classical marker of astrogliosis (Chiang et al., 1994). In our study Northern blot experiments showed a dramatic increase (764%) in GFAP mRNA levels in the striatum of wild-type animals 2 d after METH treatment, whereas no effect was observed in the DAT −/− mice (Fig. 4A). GFAP mRNA levels in untreated wild-type and DAT −/− mice were similar, presumably reflecting the resting state of astrocytes. A similar paradigm was used to examine the effects of METH in hippocampus. Interestingly, METH treatment did not increase GFAP mRNA levels in the hippocampus of wild-type or DAT −/− animals (data not shown).
Fig. 4.

METH-induced astrogliosis. A, Northern blot analysis of GFAP mRNA in striatum of saline- and METH-treated (4 × 15 mg/kg, s.c, each given 2 hr apart) wild-type and DAT −/− mice, killed 2 d after the last injection. Experiments were performed as described under Material and Methods. The blot shown represents a 15 hr exposure. Quantitation of GFAP mRNA expression revealed an eightfold increase in METH-treated wild-type mice, with no significant change in DAT −/− mice (wild-type, 764% ± 179; DAT −/−, 87.5% ± 23.9). Results are expressed as a percentage of saline-injected animals of both genotypes. B, Striatal GFAP immunohistochemical staining. Top panel, METH treatment showing METH-induced astrogliosis in the striatum of wild-type mice. Bottom panel, METH treatment showing no astrogliosis after METH challenge in the striatum of DAT −/− mice. Note the increased number and hypertrophied GFAP-positive astrocytes. The right panels are a higher magnification (40×) of the sections shown on the left.
Astrogliosis was tested further by immunohistochemistry. GFAP immunoreactivity was increased markedly in the striatum of wild-type animals (Fig. 4B, top panel), with virtually no staining observed in DAT −/− mice (Fig. 4B, bottom panel). In addition, no increase in GFAP immunoreactivity was observed in the hippocampus of either genotype (data not shown). These results confirm and extend our previous observations and suggest a strong correlation between DAT and METH-induced dopaminergic neurotoxicity in the striatum; moreover, they also indicate that the serotonergic deficits in hippocampus may not be associated with astrogliosis.
Effect of METH on striatal extracellular DA levels
In vivo microdialysis was used to monitor extracellular levels of striatal DA in freely moving wild-type and DAT −/− mice. After the administration of METH (30 mg/kg, s.c.), wild-type animals showed an 18-fold increase in extracellular DA, whereas DAT −/− mice showed no significant change (Fig.5A,). It should be noted that the extracellular basal level of DA in DAT −/− mice is approximately five times higher (Jones et al., 1998a) than that of wild-type mice [predrug concentrations of DA in dialysates were: wild-type, 46.3 ± 18.5 fmol/20 μl (n = 7); DAT −/−, 213.1 ± 68.7 fmol/20 μl (n = 5)]. After METH administration the dialysate levels of DOPAC were decreased by ∼60% in both wild-type and DAT −/− mice (Fig.5B), consistent with METH entering nerve terminals in both genotypes and inhibiting monoamine oxidase, one of the known actions of METH (Seiden et al., 1993).
Fig. 5.
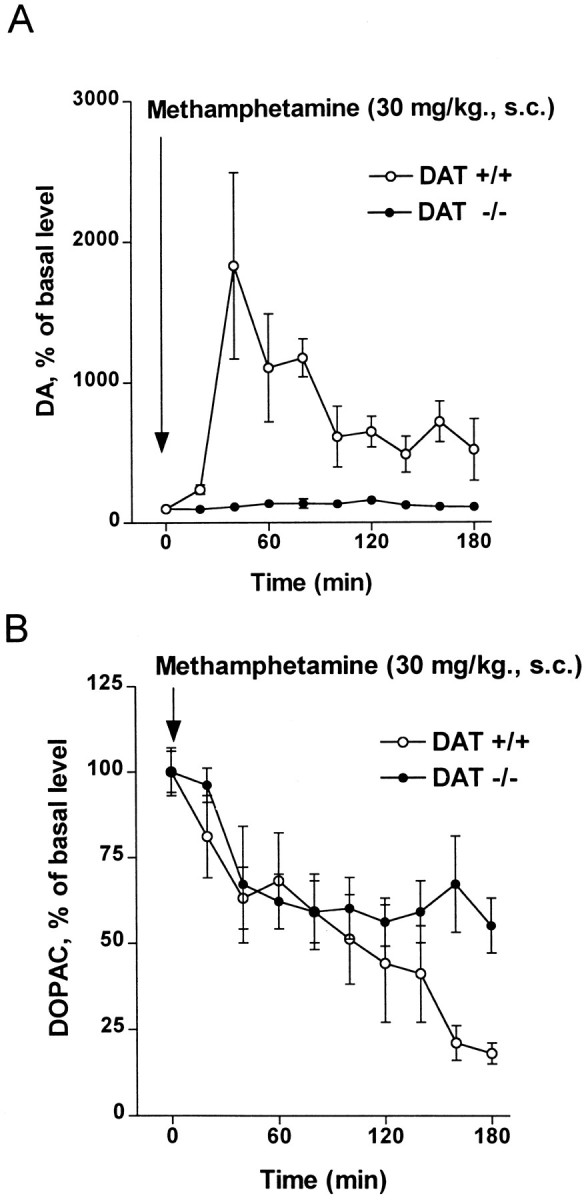
Effect of METH (single injection, 30 mg/kg, s.c.) on extracellular levels of striatal DA (A) and DOPAC (B). Microdialysis was used to measure extracellular monoamine levels in freely moving mice, as described under Materials and Methods. Basal concentrations of DOPAC in dialysates were 11.8 ± 2.3 pmol/20 μl in wild-type and 6.8 ±.2.4 pmol/20 μl in DAT −/− mice. Values represent the mean ± SEM of five to seven independent experiments.
Effect of METH on oxygen radical formation in striatum
It has been suggested that METH neurotoxicity depends on the formation of oxygen radicals (De Vito and Wagner, 1989; Cadet et al., 1994; Cubells et al., 1994; Fleckenstein et al., 1997b). In this case the METH-induced increase in intra- and/or extraneuronal DA represents a readily oxidizable pool. With the use of microdialysis and HPLC-EC, the formation of 2,3-DHBA and 2,5-DHBA products generated by the reaction of oxygen radicals and salicylate (Coudray et al., 1995) were monitored during METH treatment. METH produced a marked increase in DHBA levels in striatum of wild-type mice (three to fourfold), with no significant effect in DAT −/− mice (Fig.6). These data suggest that an intact DAT-mediated transport is required for the formation of potentially dangerous reactive molecules that are presumed to drive METH-induced striatal neurotoxicity.
Fig. 6.
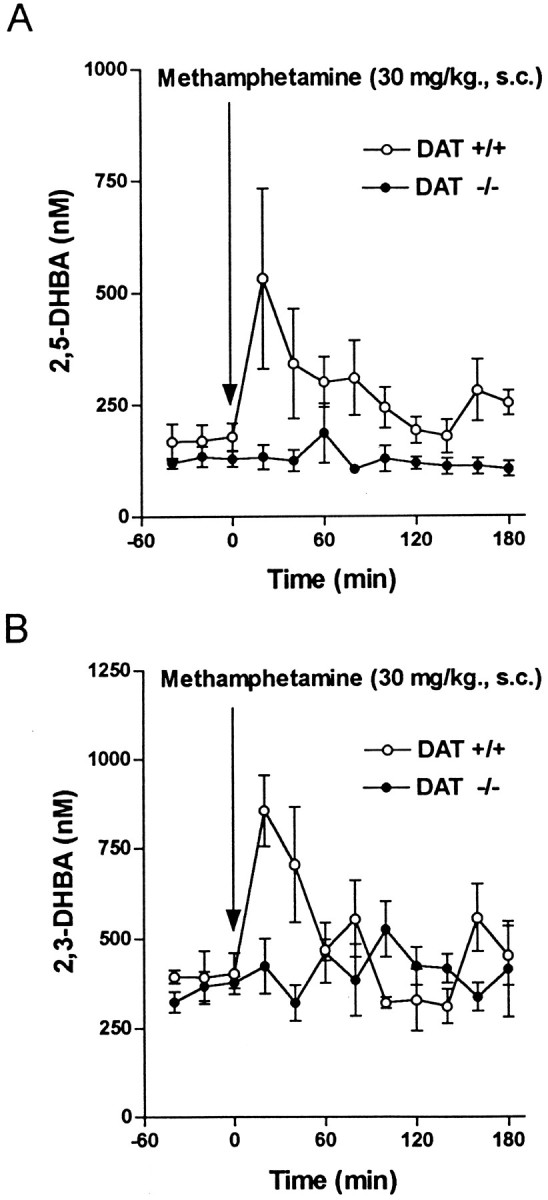
Effect of METH on oxygen radical concentrations in striatum (single injection, 30 mg/kg, s.c.). 2,5-DHBA (A) and 2,3-DHBA (B) dialysate concentrations were measured during the infusion of 5 mm salicylate through microdialysis probe into the striatum of freely moving mice, as described under Materials and Methods. Values represent the mean ± SEM of four to six experiments.
Effect of METH on body temperature
Several reports have suggested a direct association between METH-induced hyperthermia and dopaminergic neurotoxicity in mice (Bowyer et al., 1992, 1994; Albers and Sonsalla, 1995). To test whether body temperature plays a role in the lack of effects observed in DAT −/− mice after METH treatment, we measured rectal temperature in wild-type, DAT +/−, and DAT −/− mice before and after METH treatment (30 mg/kg, s.c.). Interestingly, DAT −/− mice had a lower basal temperature (35.1°C) when compared with DAT +/− (36.3°C) and wild-type animals (37.3°C) (Fig. 7). At 1 hr after the injection of METH, DAT −/− mice showed no increase in body temperature, whereas both wild-type and DAT +/− mice demonstrated a 2°C elevation (Fig. 7).
Fig. 7.

Effect of METH (single injection, 30 mg/kg, s.c.) on body temperature. Temperatures were recorded immediately before and 1 hr after a single injection of METH, as described under Materials and Methods. Values represent the mean ± SEM of eight animals per genotype. **p < 0.001 versus saline-treated wild-type and saline-treated DAT +/− mice;##p < 0.001 versus wild-type saline-treated mice; ∧∧p < 0.001 versus wild-type and DAT +/− saline-treated mice.
DISCUSSION
Repeated administration of METH is thought to produce permanent damage to dopaminergic neurons in the striatum (Wagner et al., 1980;Seiden and Ricaurte, 1987; Ricaurte et al., 1994; Seiden and Sabol, 1995). Although the mechanism of METH neurotoxicity remains controversial, there is strong evidence showing that DA itself acts as a mediator (Gibb and Kogan, 1979; Wagner et al., 1983; Schmidt and Gibb, 1985a; Schmidt et al., 1985; Sonsalla et al., 1986; Marek et al., 1990a; Pu et al., 1994). Thus, the possibility exists that METH, via interaction with DAT, alters the homeostasis of the dopaminergic neuron, directly resulting in neurotoxicity.
DAT −/− mice demonstrate a phenotype that highlights the critical role of DAT in the regulation of DA neuron homeostasis. These mice display an overall drastic reduction in tissue DA levels but maintain markedly elevated extracellular DA levels (Jones et al., 1998a). Immunohistochemical analysis and binding experiments were used to assess the DA neuron integrity of DAT −/− mice and revealed that the nigrostriatal dopaminergic pathways are intact and show no degeneration (Jones et al., 1998a; M. Jaber, unpublished data).
The present study directly demonstrates that DAT is required for the dopaminergic neurotoxicity induced by METH, as evidenced by the marked alteration in parameters of dopaminergic cell integrity in wild-type mice and the virtual absence of any changes in DAT −/− mice. Particularly, maintenance of striatal DA and metabolite content in DAT −/− mice (see Fig. 1A) strongly suggests a tight link between functioning DAT and METH-induced neurotoxicity. In addition, our experiments show that, although METH dramatically increased GFAP mRNA and protein levels in the striatum of wild-type animals, no changes were observed in DAT−/− mice (see Fig.4A,B), indicating that a complete protection of striatal neurons was afforded by DAT deletion.
Several mechanisms may underlie the lack of METH toxicity in DAT −/− mice. First, it is known that amphetamines enter cells through DAT and by diffusion (Mack and Bonisch, 1979; Liang and Rutledge, 1982a; Zaczek et al., 1991a,b; Seiden et al., 1993; Jones et al., 1998b). Our microdialysis experiments demonstrate that DOPAC was decreased in both wild-type and DAT −/− mice (see Fig. 5), suggesting direct monoamine oxidase (MAO) inhibition by METH. Hence, access of METH to the cell interior at this dose (30 mg/kg) occurs even in the absence of DAT. This does not rule out the possibility, however, that a fully active uptake system may be required to produce the concentrations of METH necessary to cause neurotoxicity.
A second mechanism attributed to METH neurotoxicity involves METH displacement of DA from storage vesicles or reversal of the vesicular transporter such that cytoplasmic DA levels are enhanced. Indeed, it has been suggested that METH neurotoxicity is mainly attributable to its ability to increase intracellular DA levels (Cubells et al., 1994). Because DAT −/− mice have much less DA in their vesicular storage pools as compared with wild-type animals (Jones et al., 1998a), the possibility exists that DAT −/− mice lack the METH-induced increase in cytoplasmic DA levels, thus explaining the absence of toxicity. Interestingly, Cappon et al. (1997) recently demonstrated that postnatal day 20 rats have fewer DAT binding sites and lower tissue DA content than adult rats and are resistant to the neurotoxic effects of METH.
A third mechanism may involve the pathway of DA release. For instance, it is known that DA can be released via impulse-dependent vesicular and impulse-independent transporter-mediated release (Raiteri et al., 1979;Hurd and Ungerstedt, 1989). The ability of amphetamine to release DA appears to occur via the second mechanism (Sulzer et al., 1995). We recently provided evidence that reverse transport by DAT, causing the release of DA from the cytoplasm to the extracellular space, is required for the releasing action of amphetamine (Jones et al., 1998b). This METH-induced increase in extracellular DA may be a critical component for METH toxicity (Seiden and Vosmer, 1984; Schmidt et al., 1985; Marek et al., 1990b; O’Dell et al., 1992, 1993). Therefore, in DAT −/− mice the absence of transporter may prevent extracellular DA concentrations from rising to neurotoxic levels. Consistent with this hypothesis, microdialysis experiments clearly showed that extracellular DA levels are elevated dramatically in wild-type animals that are administered METH, but not in DAT −/− mice. However, it should be noted that, although extracellular steady-state DA levels in DAT −/− mice are fivefold over that of wild-type mice, basal levels of free radicals are not increased.
Hyperthermia is suggested to be an important contributor of METH-induced neuropathology (Bowyer et al., 1992, 1994; Albers and Sonsalla, 1995). To this end, drugs protecting against METH toxicity also prevent hyperthermia (Ali et al., 1994; Albers and Sonsalla, 1995;Farfel and Seiden, 1995). In contrast, pretreatment with reserpine, a drug known to produce hypothermia, does not prevent METH-induced neurotoxicity (Ricaurte et al., 1983; Wagner et al., 1983), suggesting that hyperthermia may contribute to, but is not the sole mediator of, METH toxicity. Interestingly, basal core temperatures were 1 and 2°C lower in DAT +/− and DAT −/− mice, respectively, as compared with wild-type animals. Hyperthermia after METH administration was observed only in wild-type and DAT +/− mice. However, in DAT +/− mice, hyperthermia is not paralleled by severe striatal DA depletion as in wild-type mice. Because the severity of DA depletion has been correlated with the degree of hyperthermia (Bowyer et al., 1994), this discrepancy suggests that thermal response does not appear to be a primary prerequisite for METH-induced dopaminergic neurotoxicity; hence, the lack of hyperthermia in DAT −/− mice does not seem to be sufficient to explain the lack of toxicity.
Regardless of the mechanisms proposed, it appears that DA-mediated free radical formation is a major contributor to METH-induced neurotoxicity. Recent evidence demonstrates a correlation between oxygen-based radicals and the severity of DA depletion. In fact, pretreatment with antioxidants and the overexpression of superoxide dismutase in transgenic mice both attenuate METH-induced DA depletion (Wagner et al., 1985; De Vito and Wagner, 1989; Cadet et al., 1994). Our data, showing that METH administration leads to the massive production of free radicals in the striatum of wild-type animals only, corroborate this view and emphasize the pivotal role played by DAT in METH-induced neurotoxicity.
In addition to the dopaminergic system, METH also has profound effects on the serotonergic system. Long-lasting modifications have been observed in several serotonergic markers, including TPH activity, 5-HT content, and 5-HT transporter levels (Gibb et al., 1994). Moreover, DA has been suggested to play a primary role in mediating serotonergic depletion after METH administration. In fact, DA uptake inhibitors greatly improve the deficit in METH-induced TPH activity (Schmidt et al., 1985). Our data show that the striatal serotonergic system is partially protected in DAT −/− mice, whereas METH toxicity is clearly evident in both wild-type and DAT +/− animals, suggesting that striatal serotonergic toxicity may be dependent, at least in part, on free radical formation, given the partial protection afforded by DAT deletion. It should be pointed out, however, that striatal 5-HT synthesis is decreased significantly, indicating a direct METH effect on 5-HT neurons. Our results confirm and extend those of Fleckenstein et al. (1997b), which showed that METH treatment in rats leads to a decrease in striatal TPH activity. Conversely, DAT elimination did not protect hippocampal 5-HT neurons against METH toxicity in which both 5-HT and 5-HIAA content were depressed significantly in both wild-type and DAT −/− mice, suggesting that the action of METH on hippocampal serotonergic neurons is independent of dopaminergic toxicity. Interestingly, the decrease in TPH activity in DAT −/− mice occurs without an elevation in body temperature. These data do not support earlier observations from Fleckenstein et al. (1997b), which showed that hyperthermia contributes to a METH-induced decrease in TPH activity. However, the greater dose used in our experiments (30 vs 15 mg/kg) and the species differences (mice vs rats) potentially could explain this difference.
Another important issue is whether METH-induced DA and 5-HT reductions result from direct nerve terminal toxicity or long-term neurotransmitter depletion. A recent study conducted in humans revealed that METH-induced DA depletion does not necessarily correspond to nerve terminal loss (Wilson et al., 1996). Other authors suggest that DA decreases after METH might be the result of neuronal downregulation in addition to terminal degeneration (Bowyer et al., 1992, 1994). Interestingly, the depletion of DA storage pools detected in untreated DAT −/− mice suggests that impairment of DAT-mediated transport results in decreased intraneuronal accumulation of DA (Jones et al., 1998a). In this case it is important to note that a direct modulatory effect of METH on DAT function has been reported and presumably is mediated by oxygen radicals (Fleckenstein et al., 1997a,c). Thus, it is reasonable to suggest not only that the neurotoxic effect of METH is mediated by DAT but also that direct depletory action of METH on intraneuronal storage of DA may be related to plasma membrane transport. The same considerations may be extended to the serotonergic system. In our study GFAP mRNA levels were not increased in the hippocampus of wild-type mice, where 5-HT and 5-HIAA levels were found to be decreased significantly, implying a dissociation between 5-HT depletion and astrogliosis. Likewise, Cappon et al. (1997) recently showed that postnatal day 20 rats that had been administered METH displayed 5-HT depletion but not concomitant astrogliosis. Thus, because 5-HT depletion persists in DAT −/− mice, METH may affect 5-HT neurons directly without the involvement of dopaminergic mechanisms, and this action presumably occurs via the 5-HT plasma membrane transporter (Schmidt and Gibb, 1985b).
In summary, these results provide direct evidence that an intact and functional DAT is required for the development of METH-induced dopaminergic neurotoxicity; the serotonergic hippocampal deficits after METH appears to be independent of dopamine uptake and to rely, perhaps, on direct action of the drug on 5-HT neurons.
Footnotes
This work was supported in part by National Institute of Health Grants NS-19576 and MH-40159 and unrestricted gifts from Bristol-Myers Squibb and Zeneca Pharmaceuticals. R.R.G. is a recipient of a Tourette Syndrome Association fellowship. We thank Dr. Iain L. Campbell for the generous gift of the GFAP plasmid. We are grateful to Drs. R. M. Wightman and W. C. Wetsel for critical reading of this manuscript. We also thank Dr. A. Bruccoleri and Dr. G. J. Harry for helping with immunohistochemistry and S. Suter and J. A. Holt for excellent technical assistance.
Fabio Fumagalli is a visiting fellow from Center of Neuropharmacology, Institute of Pharmacological Sciences, University of Milan, Via Balzaretti 9, 20133 Milano, Italy.
Raul R. Gainetdinov is a visiting fellow from the Institute of Pharmacology, Russian Academy of Medical Sciences, Baltiyskaya 8, 125315, Moscow, Russia.
Correspondence should be addressed to Dr. Marc G. Caron, Howard Hughes Medical Institute, Box 3287, Duke University Medical Center, Durham, NC 27710.
REFERENCES
- 1.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 2.Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- 3.Ali SF, Newport GD, Slikker W., Jr Methamphetamine-induced dopaminergic toxicity in mice. Role of environmental temperature and pharmacological agents. Ann NY Acad Sci. 1996;801:187–198. doi: 10.1111/j.1749-6632.1996.tb17441.x. [DOI] [PubMed] [Google Scholar]
- 4.Axt KJ, Commins DL, Vosmer G, Seiden LS. α-Methyl-p-tyrosine pretreatment partially prevents methamphetamine-induced endogenous neurotoxin formation. Brain Res. 1990;515:269–276. doi: 10.1016/0006-8993(90)90606-c. [DOI] [PubMed] [Google Scholar]
- 5.Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- 6.Bowyer JF, Davied DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 7.Bowyer JF, Clausing P, Gough B, Slikker W, Jr, Holson RR. Nitric oxide regulation of methamphetamine-induced dopamine release in caudate/putamen. Brain Res. 1995;699:62–70. doi: 10.1016/0006-8993(95)00877-s. [DOI] [PubMed] [Google Scholar]
- 8.Cadet JL, Sheng P, Ali S, Rothman R, Carlson E, Epstein CJ. Attenuation of methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutase transgenic mice. J Neurochem. 1994;62:380–383. doi: 10.1046/j.1471-4159.1994.62010380.x. [DOI] [PubMed] [Google Scholar]
- 9.Cappon GD, Morford LRL, Vorhees CV. Ontogeny of methamphetamine-induced neurotoxicity and associated hyperthermic response. Dev Brain Res. 1997;103:155–162. doi: 10.1016/s0165-3806(97)81791-9. [DOI] [PubMed] [Google Scholar]
- 10.Carlsson A, Davis JN, Kehr W, Lindqvist M, Atack CV. Simultaneous measurement of tyrosine and tryptophan hydroxylase activities in brain in vivo using an inhibitor of the aromatic amino acid decarboxylase. Naunyn Schmeidebergs Arch Pharmacol. 1972;275:153–168. doi: 10.1007/BF00508904. [DOI] [PubMed] [Google Scholar]
- 11.Chiang CS, Stalder A, Samimi A, Campbell IL. Reactive gliosis as a consequence of interleukin-6 expression in the brain: studies in transgenic mice. Dev Neurosci. 1994;16:212–222. doi: 10.1159/000112109. [DOI] [PubMed] [Google Scholar]
- 12.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Coudray C, Talla M, Martin S, Fatome M, Favier A. High-performance liquid-chromatography electrochemical determination of salicylate hydroxylation products as an in vivo marker of oxidative stress. Anal Biochem. 1995;227:101–111. doi: 10.1006/abio.1995.1258. [DOI] [PubMed] [Google Scholar]
- 14.Cubells JF, Rayport S, Rajindron G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular stress. J Neurosci. 1994;14:2260–2771. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 16.Farfel GM, Seiden LS. Role of hyperthermia in the mechanism of protection against serotonergic toxicity. II. Experiments with methamphetamine, p-chloroamphetamine, fenfluramine, dizocilpine, and dextromethorphan. J Pharmacol Exp Ther. 1995;272:868–875. [PubMed] [Google Scholar]
- 17.Fleckenstein AE, Metzger RR, Gibb JW, Hanson GR. A rapid and reversible change in dopamine transporters induced by methamphetamine. Eur J Pharmacol. 1997a;323:R9–R10. doi: 10.1016/s0014-2999(97)00148-9. [DOI] [PubMed] [Google Scholar]
- 18.Fleckenstein AE, Wilkins D, Gibb JW, Hanson GR. Interaction between hyperthermia and oxygen radical formation in the 5-hydroxytryptaminergic response to a single methamphetamine administration. J Pharmacol Exp Ther. 1997b;283:281–285. [PubMed] [Google Scholar]
- 19.Fleckenstein AE, Metzger RR, Beyeler ML, Gibb JW, Hanson GR. Oxygen radicals diminish dopamine transporter function in rat striatum. Eur J Pharmacol. 1997c;334:111–114. doi: 10.1016/s0014-2999(97)01175-8. [DOI] [PubMed] [Google Scholar]
- 20.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1996. [Google Scholar]
- 21.Fumagalli F, Jones SR, Caron MG, Seidler FJ, Slotkin TA. Expression of mRNA coding for the serotonin transporter in aged vs young rat brain: differential effects of glucocorticoids. Brain Res. 1996;719:225–228. doi: 10.1016/0006-8993(96)00119-9. [DOI] [PubMed] [Google Scholar]
- 22.Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- 23.Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- 24.Gibb JW, Hanson GR, Johnson M. Neurochemical mechanisms of toxicity. In: Cho AK, Segal DS, editors. Amphetamine and its analogs: neuropsychopharmacology, toxicology and abuse. Academic; San Diego: 1994. p. 269. [Google Scholar]
- 25.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 26.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophane hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 27.Hurd JL, Ungerstedt U. Ca2+ dependence of the amphetamine, nomifensine, and Lu 19–005 effect on in vivo dopamine transmission. Eur J Pharmacol. 1989;166:261–269. doi: 10.1016/0014-2999(89)90067-8. [DOI] [PubMed] [Google Scholar]
- 28.Itzhak Y, Ali SF. The neuronal nitric oxide synthase inhibitor, 7-nitroindazole, protects against methamphetamine-induced neurotoxicity in vivo. J Neurochem. 1996;67:1770–1773. doi: 10.1046/j.1471-4159.1996.67041770.x. [DOI] [PubMed] [Google Scholar]
- 29.Johnson M, Stone DM, Hanson GR, Gibb JW. Role of the dopaminergic nigrostriatal pathway in methamphetamine-induced depression of the neostriatal serotonergic system. Eur J Pharmacol. 1987;135:231–234. doi: 10.1016/0014-2999(87)90616-9. [DOI] [PubMed] [Google Scholar]
- 30.Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998a;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998b;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang NY, Rutledge CO. Comparison of the release of [3H] dopamine from isolated corpus striatum by amphetamine, fenfluramine, and unlabeled dopamine. Biochem Pharmacol. 1982a;31:983–992. doi: 10.1016/0006-2952(82)90332-x. [DOI] [PubMed] [Google Scholar]
- 33.Liang NY, Rutledge CO. Evidence for carrier-mediated efflux of dopamine from corpus striatum. Biochem Pharmacol. 1982b;31:2479–2484. doi: 10.1016/0006-2952(82)90057-0. [DOI] [PubMed] [Google Scholar]
- 34.Mack F, Bonisch H. Dissociation constants and lipophicility of catecholamines and related compounds. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:1–9. doi: 10.1007/BF00499868. [DOI] [PubMed] [Google Scholar]
- 35.Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res. 1990a;513:274–279. doi: 10.1016/0006-8993(90)90467-p. [DOI] [PubMed] [Google Scholar]
- 36.Marek GJ, Vosmer G, Seiden LS. The effects of monoamine uptake inhibitors and methamphetamine on neostriatal 6-hydroxydopamine (6-OHDA) formation, short-term monoamine depletions, and locomotor activity in the rat. Brain Res. 1990b;516:1–7. doi: 10.1016/0006-8993(90)90889-j. [DOI] [PubMed] [Google Scholar]
- 37.Obata T, Chiueh CC. In vivo trapping of hydroxyl free radicals in the striatum utilizing intracranial microdialysis perfusion of salicylate: effects of MPTP, MPDP+, and MPP+. J Neural Transm. 1992;89:139–145. doi: 10.1007/BF01245361. [DOI] [PubMed] [Google Scholar]
- 38.O’Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1992;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- 39.O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced DA overflow and injury to striatal DA terminals: attenuation by DA D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- 40.Ohmori T, Abekawa T, Koyama T. The role of glutamate in the neurotoxic effects of methamphetamine. Ann NY Acad Sci. 1996;801:315–326. doi: 10.1111/j.1749-6632.1996.tb17452.x. [DOI] [PubMed] [Google Scholar]
- 41.Pu C, Vorhees CV. Developmental dissociation of methamphetamine-induced depletion of dopaminergic terminals and astrocyte reaction in rat striatum. Dev Brain Res. 1993;72:325–328. doi: 10.1016/0165-3806(93)90201-k. [DOI] [PubMed] [Google Scholar]
- 42.Pu C, Fisher JE, Cappon GD, Vorhees CV. The effects of amfonelic acid, a dopamine uptake inhibitor, on methamphetamine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum. Brain Res. 1994;649:217–224. doi: 10.1016/0006-8993(94)91067-7. [DOI] [PubMed] [Google Scholar]
- 43.Raiteri M, Cerrito F, Cervoni AM, Levi G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J Pharmacol Exp Ther. 1979;208:195–202. [PubMed] [Google Scholar]
- 44.Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- 45.Ricaurte GA, Fuller RW, Perry KW, Seiden LS, Schuster CR. Fluoxetine increases long-lasting neostriatal dopamine depletion after administration of d-metamphetamine and d-amphetamine. Neuropharmacology. 1983;22:1165–1169. doi: 10.1016/0028-3908(83)90075-8. [DOI] [PubMed] [Google Scholar]
- 46.Ricaurte GA, Sabol KE, Seiden LS. Functional consequences of neurotoxic amphetamine exposure. In: Cho AK, Segal DS, editors. Amphetamine and its analogs: neuropsychopharmacology, toxicology and abuse. Academic; Orlando, FL: 1994. pp. 297–313. [Google Scholar]
- 47.Schmidt CJ, Gibb JW. Role of the dopamine uptake carrier in the neurochemical response to methamphetamine: effects of amfonelic acid. Eur J Pharmacol. 1985a;109:73–80. doi: 10.1016/0014-2999(85)90541-2. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt CJ, Gibb JW. Role of the serotonin uptake carrier in the neurochemical response to methamphetamine: effects of citalopram and chlorimipramine. Neurochem Res. 1985b;10:637–648. doi: 10.1007/BF00964403. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- 50.Seiden LS, Ricaurte G. Neurotoxicity of methamphetamine and related drugs. In: Meltzer HY, editor. Psychopharmacology: the third generation of progress. Raven; New York: 1987. pp. 359–365. [Google Scholar]
- 51.Seiden LS, Sabol KE. Neurotoxicity of methamphetamine-related drugs and cocaine. In: Chang LW, Dyer RS, editors. Handbook of neurotoxicology. Dekker; New York: 1995. pp. 825–843. [Google Scholar]
- 52.Seiden LS, Vosmer G. Formation of 6-hydroxydopamine in caudate nucleus of the rat brain after a single large dose of methylamphetamine. Pharmacol Biochem Behav. 1984;21:29–31. doi: 10.1016/0091-3057(84)90125-4. [DOI] [PubMed] [Google Scholar]
- 53.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 54.Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- 55.Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- 56.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner GC, Ricaurte G, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 58.Wagner GC, Lucot JB, Schuster CR, Seiden LS. α-Methyltyrosine attenuates and reserpine increases methamphetamine-induced neuronal changes. Brain Res. 1983;270:285–288. doi: 10.1016/0006-8993(83)90602-9. [DOI] [PubMed] [Google Scholar]
- 59.Wagner GC, Carelli RM, Jarvis MF. Pretreatment with ascorbic acid attenuates the neurotoxic effects of methamphetamine in rats. Res Commun Chem Pathol Pharmacol. 1985;47:221–228. [PubMed] [Google Scholar]
- 60.Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, Miller GW, Wightman RM, Caron MG. Knock-out of the vesicular monoamine transporter-2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–1296. doi: 10.1016/s0896-6273(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 61.Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- 62.Zaczek R, Culp S, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. II. Active transport. J Pharmacol Exp Ther. 1991a;257:830–835. [PubMed] [Google Scholar]
- 63.Zaczek R, Culp S, Goldberg H, McCann DJ, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. I. Saturable sequestration. J Pharmacol Exp Ther. 1991b;257:820–829. [PubMed] [Google Scholar]