

Abstract
Many nervous systems contain rhythmically active subnetworks that interact despite oscillating at widely different frequencies. The stomatogastric nervous system of the crab Cancer borealis produces a rapid pyloric rhythm and a considerably slower gastric mill rhythm. We construct and analyze a conductance-based compartmental model to explore the activation of the gastric mill rhythm by the modulatory commissural neuron 1 (MCN1). This model demonstrates that the period of the MCN1-activated gastric mill rhythm, which was thought to be determined entirely by the interaction of neurons in the gastric mill network, can be strongly influenced by inhibitory synaptic input from the pacemaker neuron of the fast pyloric rhythm, the anterior burster (AB) neuron. Surprisingly, the change of the gastric mill period produced by the pyloric input to the gastric mill system can be many times larger than the period of the pyloric rhythm itself. This model illustrates several mechanisms by which a fast oscillatory neuron may control the frequency of a much slower oscillatory network. These findings suggest that it is possible to modify the slow rhythm either by direct modulation or indirectly by modulating the faster rhythm.
Keywords: neural oscillators, central pattern generators, crustaceans, coupled oscillators, neuromodulation, stomatogastric ganglion, stomatogastric nervous system, compartmental model
Neurons and networks that produce oscillatory behavior are ubiquitous in the nervous system (Meda et al., 1984; Llinás and Yarom, 1986; Alonso and Llinás, 1989; Bal and McCormick, 1993; Steriade et al., 1993; Calabrese, 1995; Gray, 1995; Welsh et al., 1995; Marder and Calabrese, 1996). The neural networks that control rhythmic movements often involve the coupling of network oscillators with substantially different periods. For example, the period of the respiratory pattern generator that controls breathing in mammals is influenced by the activity in the locomotory and swallowing motor patterns (Kawahara et al., 1989; Corio et al., 1993;McFarland and Lund, 1993; Lafortuna et al., 1996). As the presence of oscillatory neurons and networks in the brain becomes more apparent, it is important to develop an understanding of the multitude of mechanisms that come into play as oscillatory networks of different intrinsic periods interact.
There is extensive theoretical and experimental work on networks of coupled oscillators when the individual oscillators are relatively close in period (Pinsker, 1977a,b; Ayers and Selverston, 1979; Kopell, 1988; Kopell and Ermentrout, 1988; Rand et al., 1988; Somers and Kopell, 1993), but less theory on the interactions of networks of oscillators whose intrinsic frequencies are significantly different. The best understood case is that of an entraining oscillator, in which, if the periods of the two oscillators are not well matched, 2:1, 3:1, or 3:2 patterns of coupling can result (Kopell, 1988).
The stomatogastric nervous system of decapod crustaceans generates four different rhythmic motor patterns, the periods of all of which differ significantly. Because of the small number of neurons within the stomatogastric ganglion (STG), the synaptic circuitry among these neurons has been established, and much is known about their properties and responses to modulatory inputs. The modulatory commissural neuron 1 (MCN1) is a modulatory projection neuron that can activate the gastric mill rhythm (Coleman et al., 1995). In the process of developing a compartmental model of the mechanisms by which MCN1 activates the gastric mill rhythm in the crab Cancer borealis, we discovered that the period of the model gastric mill rhythm is sensitive to the strength and frequency of a periodic inhibitory synaptic input it receives from the faster pyloric rhythm. Interestingly, input from the fast pyloric oscillator can influence the period of the slower gastric mill rhythm over a range much larger than the period of the pyloric rhythm.
The model of the interaction of the MCN1 neuron with its gastric mill circuit targets allows the exploration of a number of interesting features concerning the slow antiphase oscillation of a reciprocally inhibitory pair of neurons [the lateral gastric (LG) neuron and interneuron 1 (Int1)] and its regulation by a combination of slow modulatory excitation and fast periodic inhibition. We find that a pivotal transition of the gastric mill rhythm, namely activation of the LG neuron burst, arises from the interaction between a fast, pyloric-timed disinhibition of the LG neuron and a slow depolarization that the LG neuron receives from MCN1. This transition occurs with a fixed latency between the LG neuron burst onset and the pyloric pacemaker neuron burst just preceding it, at a time determined by the interaction between the strength and time course of the slow excitatory drive from MCN1 and the strength and period of the fast synaptic input. This work reveals a novel mechanism describing the regulation of a slow circuit oscillator by a synaptic interaction with a fast oscillator.
Some of this work has been published previously in abstract form (Manor et al., 1996).
MATERIALS AND METHODS
Experiments. Animals (Cancer borealis) and electrophysiological procedures were as described in Bartos and Nusbaum (1997). An AT-MIO-16E2 board was used for data acquisition with LabWindows/CVI software (National Instruments) on a PC. Data were analyzed using Unix shell scripts.
Model. Int1, MCN1, and the LG neuron were modeled as conductance-based Hodgkin and Huxley (1952)-type neurons. These three neurons were modeled with three compartments (Fig.1A). In MCN1, the three compartments represented the soma, axon, and axonal terminals. These compartments were used to separate the site of the electrical coupling between MCN1 and the LG neuron (axon) from the site of MCN1 synaptic release (axonal terminals). The soma compartment was passive, whereas the axon and the axonal terminals included voltage-dependent conductances. In the cases of Int1 and the LG neuron, the three segments represented the soma, axon, and neurite. This spatial arrangement was chosen to isolate the spike-generation zone (axon) and the site of synaptic inputs (neurite) from the soma so that the membrane potentials recorded from the soma were within the biological range. The AB neuron inhibition to Int1 was modeled as a periodic synaptic current with a conductance given by the α function:
where τ is the time constant of the α function (in msec),EAB→Int1 is the reversal potential (set to −70 mV), and gmax is the maximal (peak) synaptic conductance (see Table 2).
Fig. 1.

The MCN1-elicited gastric mill rhythm in the crabCancer borealis. A, Schematic representation showing the compartmental model used to model the MCN1-elicited gastric mill rhythm. Int1, MCN1, and the LG neuron were modeled with three compartments each. The AB neuron input to Int1 was modeled as a periodic injection of an inhibitory synaptic conductance into Int1 (see Results). B, Biological intracellular recordings of Int1 and the LG neuron in the absence (left) and presence (right) of MCN1 stimulation. The most hyperpolarized membrane potential in the traces was −58 mV for the LG neuron and −46 mV for Int 1. Adapted from Coleman et al. (1995). C, Model traces of Int1 and the LG neuron membrane potentials in the absence (left) and presence (right) of MCN1 stimulation. The most hyperpolarized membrane potential in the traces was −56 mV for the LG model neuron and −71 mV for the model Int1.
Table 2.
Parameters of synaptic currents for canonical model
| Synapse | Presynaptic site | Postsynaptic site | syn (nS/cm2) | Esyn (mV) | κ (mV−1) | Vκ (mV) | λ (mV−1) | Vλ (mV) | τ3 (msec) | τ4 (msec) |
|---|---|---|---|---|---|---|---|---|---|---|
| MCN1 → LG (Chemical) | Axonal terminals | Neurite | 0.3 | 45 | −0.5 | −50 | 4000 | 4000 | ||
| MCN1 → LG (Electrical) | Axon | Axon | 0.09 | |||||||
| MCN1 → Int1 | Axonal terminals | Neurite | 0.0015 | 45 | −1 | −50 | 30 | 30 | ||
| Int1 → LG | Neurite | Neurite | 1.3 | −80 | −0.5 | −49 | 50 | 50 | ||
| LG → Int1 | Neurite | Neurite | 1.3 | −80 | −1 | −25 | 1 | −25 | 3 | 97 |
| Soma | Soma | 1.3 | −80 | −1 | −25 | 1 | −25 | 3 | 97 | |
| Axon | Axon | 1.3 | −80 | −1 | −25 | 1 | −25 | 3 | 97 | |
| LG → MCN1 | Axon | Axonal terminals | 100 | −80 | −2 | −30 | 30 | 30 | ||
| AB → Int1 | Neurite | 1.8 | −70 | 80 |
All simulations were done with NEURON (Hines, 1993). The soma segments were spheres with diameter of 125 μm. All other segments were cylinders with a length of 1000 μm and a diameter of 2.5 μm. The axial resistance Ra was 200 Ωcm. NEURON automatically divided the segments into isopotential compartments (of length λ/10, where λ is the passive space constant).
Equations. In each segment of the model neurons, the membrane potential V was obtained by numerical integration of the differential equation:
where C is the specific capacitance (1 μF/cm2) and Itotal is the sum of all currents flowing in that segment:
Each ionic current was modeled with:
where q is 0 or 1 and m and hare governed by the following equations:
The parameter values for the ionic and leak currents in each neuron are given in Table 1.
Table 1.
Parameters of ionic currents for canonical model
| Cell | Current | Site | ion (mS/cm2) | Eion (mV) | State | k (mV−1) | Vk (mV) | l (mV−1) | V1 (mV) | τ1 (msec) | τ2 (msec) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MCN1 | Na+ | Axon, soma | 3.5 | 45 | m3 | −0.08 | −21 | 0 | 0 | ||
| h | 0.13 | −33 | −0.12 | −62 | 0 | 5 | |||||
| K+ | Axon, soma | 2.5 | −80 | m4 | −0.045 | −33 | 0.065 | −5 | 4 | 100 | |
| Leak | Axon, soma | 0.0073 | −60 | ||||||||
| Terminals | 0.1 | −40 | |||||||||
| LG | Na+ | Axon | 3.5 | 45 | m3 | −0.08 | −21 | 0 | 0 | ||
| h | 0.13 | −33 | −0.12 | −62 | 0 | 5 | |||||
| K+ | Axon | 4 | −80 | m4 | −0.045 | −33 | 0.065 | −5 | 4 | 100 | |
| Leak | Axon | 0.0073 | −60 | ||||||||
| Soma | 0.1 | −40 | |||||||||
| Neurite | 0.1 | −40 | |||||||||
| Int1 | Na+ | Axon | 3.5 | 45 | m3 | −0.08 | −26 | 0 | 0 | ||
| h | 0.13 | −38 | −0.12 | −67 | 0 | 5 | |||||
| K+ | Axon | 6 | −80 | m4 | −0.045 | −25 | −0.065 | −30 | 4 | 150 | |
| h | Axon | 2 | 10 | m | 2 | −65 | 2 | −65 | 200 | 2500 | |
| Leak | Axon | 0.0073 | −30 | ||||||||
| Soma | 0.1 | −40 | |||||||||
| Neurite | 0.1 | −40 |
The synaptic currents were computed using:
Synaptic currents were modeled as graded functions of the presynaptic voltage Vpre:
with parameter values provided by Table2.
In the MCN1 axon and the LG neuron neurite, a current describing the electrical coupling between the two segments was added to the sum of currents:
where Vneighbor is the voltage of the coupled segment.
Tuning the model parameters. We tuned the parameters of the biophysical model so that it reproduced the known output of the biological network as closely as possible (Coleman et al., 1995).
Because there is little information on the intrinsic ionic currents of individual neurons in this network, each neuron was modeled using only leak, fast Na+, and delayed-rectifier K+ currents. The Int1 model cell was also modeled with a hyperpolarization-activated inward current (Ih) to help it escape from LG neuron inhibition. In the absence of MCN1 stimulation, Int1 and the LG neuron constitute an asymmetric pair of reciprocally inhibitory neurons, where the LG neuron is quiescent and Int1 is active (Coleman et al., 1995). This asymmetry was introduced into the model by shifting the steady-state activation curve of the Int1 Na+current by +7 mV, the steady-state inactivation curve of the Int1 Na+ current by −5 mV, and the steady-state activation curve of the Int1 K+ current by −4 mV relative to those of the LG neuron. The steady-state Na+ and K+ activation curves and time constants were adjusted so that the spike frequencies of the model LG neuron and Int1 were comparable to those of the biological cells.
Most synapses within the STG have a large graded component (Graubard, 1978; Graubard et al., 1980, 1983), and therefore we modeled most of the synaptic connections as graded. A graded synaptic connection from Int1 to the LG neuron is important and consistent with the known biological data. For example, in response to the AB neuron’s inhibition of Int1, Int1 hyperpolarizes and stops firing action potentials, and the LG neuron depolarizes but does not necessarily fire (Fig. 1B). It thus appears that during the AB neuron’s inhibition of Int1, Int1 still inhibits the LG neuron and prevents it from firing action potentials.
In contrast with other synapses in the network under study, the synaptic connection from the LG neuron to Int1 has a large spike-mediated component, apparent from the large IPSPs observed in the Int1 trajectory during its interburst phase (Fig.1B). The presynaptic threshold of the model LG neuron to Int1 synapse was adjusted so that individual action potentials in the LG neuron produced fast, large IPSPs in Int1. To model the fast rise of the synaptic current in response to the presynaptic action potential, we combined the fast rise-time of the synaptic current and its slow decay into τS(Vpre), the synaptic time constant of the LG neuron to Int1 synapse: fast (3 msec) at values more depolarized than −25 mV, corresponding to the rise of the IPSPs, and slow (100 msec) at more hyperpolarized membrane potentials, corresponding to the decay rate of the IPSPs. The model synapse from the LG neuron to Int1 is purely spike-mediated, and this is reflected in the voltage dependence of the synaptic time constant.
The synaptic connection from the LG neuron to MCN1 (the presynaptic inhibition) was modeled using a steep sigmoidal input/output curve (Table 2). When the LG neuron was in its burst phase, it strongly inhibited the compartment representing the axonal terminals of MCN1 and completely eliminated the chemical MCN1 excitation of Int1 and the LG neuron. When the LG neuron was not bursting, it had no effect on the MCN1 chemical excitation of Int1 and the LG neuron.
The biological MCN1 was stimulated with a 15 Hz extracellular stimulus. The model MCN1 was stimulated with a constant current of 20 nA/cm2 in the soma compartment, producing an average firing rate of 15 Hz.
The electrical coupling between MCN1 and the LG neuron contributes significantly to LG neuron activity (Coleman et al., 1995). It is also clear that the electrical coupling alone, without the chemical excitation from MCN1, is not sufficient to maintain LG neuron bursting (Coleman et al., 1995). We therefore adjusted the strength of the model MCN1 to LG neuron electrical coupling such that the LG neuron (1) when switching to a burst produced action potentials with the electrical coupling but not without it and (2) could not maintain firing indefinitely and stopped producing action potentials when the effect of the chemical excitation waned.
As a result of the above process, we obtained a set of parameters, termed the canonical model, that produced activity resembling that of the biological system (Fig. 1C). Despite the fact that the model does not exactly reproduce the biological voltage traces, the results reported are general and robust. The parameters of the canonical model are listed in Tables 1 and 2.
Figure 2 shows expanded time base recordings of the canonical model in the absence of MCN1 activity (Fig.2A) and at the transitions (dotted lines) between Int1 and LG neuron bursts (Fig. 2B). The transition associated with the termination of LG neuron activity occurs because the last spike in the LG neuron burst fails, as a consequence of the decreasing excitation from MCN1, allowing Int1 to depolarize. These recordings also illustrate both the spike-mediated IPSPs evoked in Int1 by the LG neuron, and the small depolarizations in the LG neuron caused by the electrical coupling between it and the active MCN1. The spiking frequency of MCN1 was 16 Hz when Int1 was bursting and 14 Hz when the LG neuron was bursting (and inhibiting MCN1).
Fig. 2.

Expanded time-base recordings of the canonical model Int1 (top traces), LG neuron (middle traces), and MCN1 soma (bottom traces) in the absence (A) and presence (B1,B2) of MCN1 stimulation. Dotted linesshow the transitions between Int1 and LG neuron bursts.
RESULTS
The gastric mill rhythm in the STG of the crab Cancer borealis, exhibits a network-generated oscillation with a period of 7–15 sec. This rhythm can be elicited by tonic MCN1 stimulation (Fig. 1B). When MCN1 is silent, the LG neuron shows subthreshold depolarizations that are time-locked with the fast pyloric rhythm, and Int1 fires action potentials except when inhibited in pyloric time. MCN1 action potentials elicit EPSPs in Int1 and the LG neuron. When MCN1 is stimulated sufficiently, these EPSPs result in a rhythm in which the LG neuron bursts in antiphase with Int1 (Fig.1B). At the heart of the circuit is the reciprocally inhibitory Int1 and LG neuron pair (Fig. 1A). The LG neuron also presynaptically inhibits the terminals of MCN1 in the STG and is electrically coupled to MCN1.
On the basis of the anatomical and physiological data, Coleman et al. (1995) suggested the following verbal model for the MCN1-activated gastric mill rhythm. When MCN1 is activated, it excites Int1 rapidly and produces a slower excitation of the LG neuron. The rapid excitation of Int1 enhances its pyloric rhythm-timed bursts, causing a stronger inhibition of the LG neuron. Here Int1 is active, and the LG neuron is inhibited. Because the LG neuron continuously receives slow excitation from MCN1, it slowly depolarizes. Eventually, the LG neuron escapes from Int1 inhibition and starts to fire. The LG neuron burst inhibits Int1, which stops firing and hyperpolarizes. At the same time, the LG neuron presynaptically inhibits the terminals of MCN1, thereby shutting off the transmitter-mediated excitation to itself and to Int1. The LG neuron continues to fire, partially because it continues to receive electrical excitation from MCN1. The electrical coupling by itself is not sufficient to sustain LG neuron firing, and as the effect of the chemical excitation from MCN1 wanes, the LG neuron burst terminates and the cycle repeats. The hypothesis resulting from this verbal model (Coleman et al., 1995; Marder, 1996) was that the period of the gastric mill rhythm would be determined by the time course and strength of the slow EPSP evoked in the LG neuron by MCN1.
The voltage traces of both the biological rhythm (Fig.1B) and the canonical model (Fig. 1C) suggest that the verbal model described above contains only part of the story. In the absence of MCN1 stimulation, Int1 receives pyloric-timed inhibition from the AB neuron. When Int1 is hyperpolarized, it releases less inhibitory transmitter, producing a pyloric-timed disinhibition in the LG neuron. When MCN1 is stimulated to produce a gastric mill rhythm, note that each transition of the LG neuron from inactive to active coincides with the peak of a pyloric-timed disinhibition. This suggests that the gastric mill period might be determined by the interaction between the strength and time course of the MCN1-produced slow depolarization of the LG neuron and the strength and period of the pyloric-timed disinhibitions of the LG neuron. In this paper we examine systematically these four parameters and their interactions with the other parameters in the model.
The effect of the slow MCN1 excitation of the LG neuron
We examined the effect of the time constant of the MCN1 to LG neuron slow chemical excitation (τMCN1→LG; both τ3 and τ4 in Table 2) on the model gastric mill period. Figure 3 shows the model gastric mill period as a function of τMCN1→LG for a pyloric period of 1 sec. The dotted lines indicate the values for the canonical model. The overall trend was a linear increase of gastric mill periods with τMCN1→LG. As the time constant was varied 13-fold, the period varied almost 10-fold, indicating a strong dependence of the model gastric mill rhythm on the slow MCN1 to LG neuron excitation, as predicted by Coleman et al. (1995). At most values of τMCN1→LG, however, the model gastric mill rhythm alternated between two and three discrete periods. As τMCN1→LG increased, these discrete periods were locally constant and increased in a stepwise manner, with the step size determined by the pyloric period. The overall linear dependence of gastric mill period on τMCN1→LG was not affected by the pyloric period (data not shown). The staircase-like graph in Figure 3implies that factors other than τMCN1→LG might also influence the gastric mill period.
Fig. 3.

Dependence of the model gastric mill period on the time constant of the MCN1 excitation of the LG neuron (τMCN1→LG). A 200 sec simulation was run with a fixed value of τMCN1→LG, and the gastric mill periods (times from the onset of an LG neuron burst to the onset of the subsequent LG neuron burst) were measured. The procedure was repeated for different values of τMCN1→LG, with increments of 250 msec. The gastric mill periods are plotted versus τMCN1→LG (from 1 to 13 sec). Dotted lines indicate the values corresponding to the canonical model.
The effect of the pyloric period on the gastric mill period
We tested the dependence of the gastric mill period on the pyloric period. Figure 4A shows the behavior of the canonical model (when the pyloric period was 1 sec). The period of the gastric mill oscillation alternated between two values, equal to 9 and 10 times the pyloric period. This discrete variation in the gastric mill period is consistent with the staircase effect seen in Figure 3. To assess fully the effect of different pyloric periods on the gastric mill period, a 200 sec simulation was run at a fixed pyloric period, and the gastric mill periods were measured. This procedure was repeated for a range of naturally occurring pyloric periods.
Fig. 4.
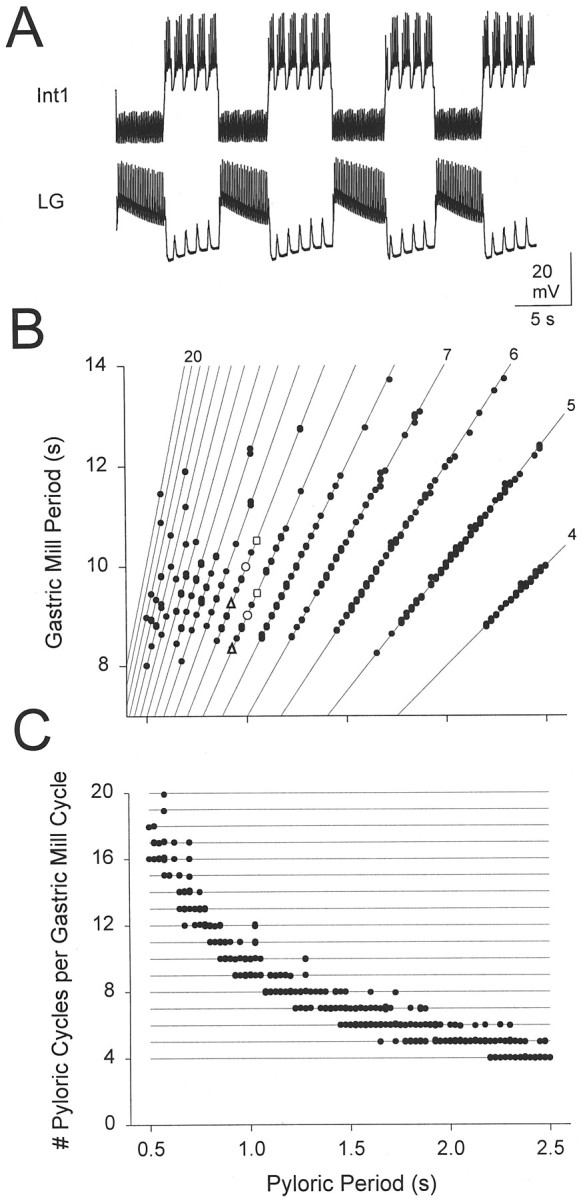
The effect of the pyloric period on the gastric mill period. A, Voltage traces of Int1 and the LG neuron for a pyloric period of 1 sec. B, Gastric mill period as function of pyloric period. A 200 sec simulation was run at a fixed pyloric period, and the gastric mill periods were measured. The procedure was repeated for different pyloric periods, with increments of 25 msec. Open triangles, open circles, and open squares show the gastric mill periods for pyloric periods of 0.925 sec, 1 sec (shown inA), and 1.05 sec, respectively. Solid lines have integer slopes ranging from y = 4x to y = 20x. C, Number of pyloric cycles per gastric mill cycle as function of pyloric period.
Figure 4B shows these gastric mill periods plotted against the pyloric period. The solid lines are given by PGastric =kPpyl, where Ppylis the pyloric period (the horizontal axis),PGastric is the gastric mill period (the vertical axis), and k is an integer between 4 and 20. All values of the gastric mill period fell on these lines, indicating thatPGastric was always an integer multiple ofPpyl. At some values ofPpyl, PGastric had a unique value. For most Ppyl values however, the model produced oscillations at two or three different periods. In general, small increments in Ppyl resulted in a linear increase of PGastric. For example, compare PGastric at Ppyl = 0.925 sec (▵),Ppyl = 1 sec (○) andPpyl = 1.05 sec (□). This, however, was alocal trend: PGastric did not increase beyond 14 sec. As PGastric approached this upper limit, a further increase in Ppylresulted in a decrement of n (the number of pyloric cycles per gastric mill cycle) by 1. These sharp transitions restrictedPGastric to values between 8 and 14 sec. Onaverage, PGastric increased modestly with Ppyl (slope = 0.73, with a linear fit).
In Figure 4C, the data in Figure 4B are replotted with the number of pyloric cycles per gastric mill cycle as a function of pyloric period. This plot highlights the fact that in all cases the gastric mill period was an integer function of the pyloric period and demonstrates that as the pyloric period increased there was less variability in the number of pyloric cycles in each gastric mill period.
The onset of the LG burst is locked to the pyloric time
In the model gastric mill rhythm, cycle periods are integer multiples of the pyloric period. This caused us to ask whether the same relationship holds in data taken from biological preparations. Therefore, we analyzed sections of data from MCN1-stimulated gastric mill rhythms. We examined the onset and termination times of the LG neuron bursts, relative to the pyloric rhythm. Because the AB and pyloric dilator (PD) neurons are electrically coupled and fire together, the units on the pyloric dilator nerve (Fig.5A, pdn), which are extracellularly recorded spikes of the two PD neurons, coincide with the AB neuron burst. The pyloric latency of the LG neuron burst (recorded as the large unit on the lateral gastric nerve) onset is the time between the onset of the burst on the pyloric dilator nerve just previous to the onset of the LG neuron burst, and the onset of the LG neuron burst (Fig. 5A). The pyloric latency of the LG neuron burst termination is the time between the onset of the pyloric dilator nerve burst just previous to the termination of the LG neuron burst and the termination of the LG neuron burst (Fig. 5A).
Fig. 5.
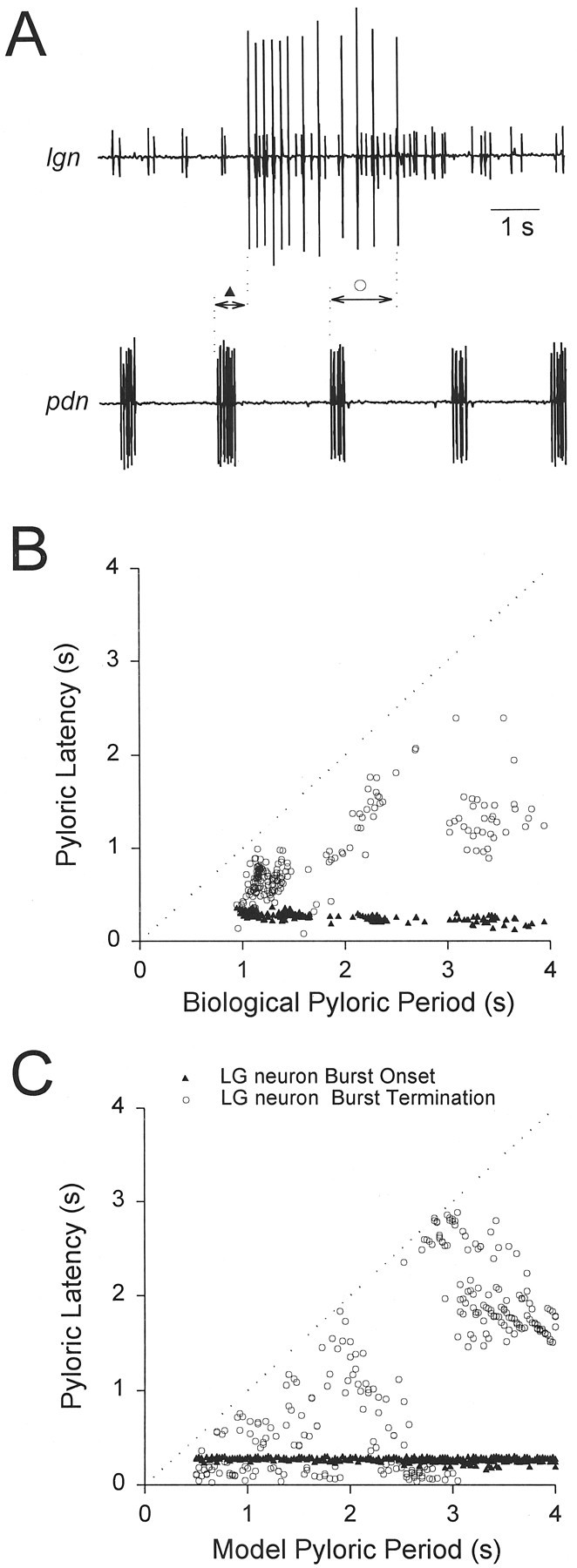
Pyloric latencies of LG neuron burst onset (▴) and termination (○). A, Extracellular recordings from the lateral gastric nerve (lgn) and the pyloric dilator nerve (pdn). Pyloric latencies (experimental) were calculated as the time delay from the onset of the previouspdn burst to the onset/termination of thelgn burst (see Results). B, Experimental pyloric latencies of LG neuron burst onset and termination are plotted against pyloric period. The pyloric period was changed by DC current injection in the AB neuron. The dotted linerepresents the pyloric latency of the next pdn burst.C, Model pyloric latencies of LG neuron burst onset and termination are plotted against pyloric period.
To obtain a range of pyloric periods, we injected DC current into the biological AB neuron. Figure 5B shows the pyloric latencies of the LG neuron burst onset and termination as a function of pyloric period, in one biological preparation. The dotted line (x = y) represents the time of the next AB neuron burst. In this experiment, over the whole range of pyloric periods, the pyloric latencies of the LG neuron burst onset and termination were 265 ± 48 and 883 ± 409 msec, respectively (mean ± SD). The variability of the pyloric latency of the LG neuron burst onset was much smaller than that of the LG neuron burst termination. In five preparations, the range of the standard deviations for the LG neuron burst onset was between 21 and 60 msec, whereas the range of the standard deviations for the LG neuron burst termination was between 181 and 427 msec. These results suggest that the biological LG neuron burst onset, but not its termination, is time-locked to the pyloric rhythm.
Figure 5C shows the pyloric latency of the model LG neuron burst onset and termination as a function of the pyloric period. For a range of pyloric periods between 500 and 4000 msec, the pyloric latencies of the LG neuron burst onsets and terminations were 261 ± 29 and 1201 ± 890 msec (mean ± SD), respectively. As in the experiments, the model LG neuron burst onset was time-locked to the pyloric rhythm. However, this figure also emphasizes that the LG neuron burst termination was not entirely independent of the pyloric rhythm. Indeed, at pyloric periods above 3.1 sec, the pyloric latency of the LG neuron burst termination was always larger than 1.4 sec. These large pyloric latencies occurred because the LG neuron burst duration had an upper and lower limit (as described later in Results). Therefore, for these pyloric periods, the LG neuron burst duration was considerably larger (at least 1.4 sec) than one pyloric period but smaller than two pyloric periods.
Sensitivity of the gastric mill period to model parameters
To examine the dependence of the gastric mill period on model parameters we used two procedures. First, we did a sensitivity analysis of the rhythm period to small changes in parameters around those of the canonical model to determine which small changes in parameters altered the model behavior the most. Second, we varied each parameter over a large range to determine the overall dependence of model behavior on each parameter. We start with the results of the sensitivity analysis.
We varied each parameter by ±10% of its canonical value, measured the model gastric mill periods, and compared them with the period of the canonical model (Fig. 6). We define the sensitivity of period as:
where period refers to the average of all gastric mill periods in a 200 sec run.
Fig. 6.

Sensitivity test of model maximal synaptic conductances. Speriod is the ratio of the variation of period to the variation in parameter. Each parameter is varied from its canonical value by −10% (gray bars) and +10% (black bars), and the periods in a 200 sec run are measured. Values shown are mean ± SD.
Negative values of Speriod indicate a decrease in period when the parameter value increases. We found that the gastric mill period was most sensitive to the synaptic conductance of the AB neuron inhibition of Int1. The model gastric mill oscillations were also sensitive to the strength of the reciprocally inhibitory synapses between Int1 and the LG neuron, especially whensyn was decreased.Speriod did not depend on the MCN1 to Int1 excitation, but was somewhat dependent on the MCN1 to LG neuron excitation. Speriod was relatively independent of the electrical coupling conductance between MCN1 and the LG neuron.
Speriod did not have a large dependence on other model parameters, including the pyloric period, the maximal conductance of Ih in Int1, and the time constants of the MCN1 to LG neuron, MCN1 to Int1, AB neuron to Int1, Int1 to LG neuron, and LG neuron to Int1 synapses (these Speriodvalues were <1.25 in absolute value; data not shown).
Sensitivity of the gastric mill period to the AB neuron input
We have shown that the MCN1 excitation of the LG neuron is an important factor in determining the gastric mill period (Fig. 3). However, the sensitivity analysis in Figure 6 indicated that the gastric mill period was also extremely sensitive to the strength of the AB neuron inhibition of Int1. The interplay between the MCN1 excitation and the AB neuron input is explored in Figure7. Figure 7A shows the MCN1 to LG conductance and the membrane potentials of Int1 and the LG neuron for a ±10% change in AB→Int1 from its canonical value. The decrease resulted in a 42% longer gastric mill period (Fig. 7A, left traces), and the increase resulted in a 17% shorter gastric mill period (Fig. 7A,right traces).
Fig. 7.

The effect of the strength of the pyloric input.A, The maximal conductance of the AB neuron to Int1 inhibition was changed by −10% (left panel) and +10% (right panel) of the canonical value (middle panel). Each panel shows voltage traces of Int1, the LG neuron, and the conductance of the MCN1 to LG neuron chemical synapse (gMCN1→LG).B, gMCN1→LG is shown for the canonical model (solid trace), +10% of the maximal conductance of the AB neuron to Int1 inhibition (dashed trace), and −10% of the maximal conductance of the AB neuron to Int1 inhibition (dotted trace). The valuesgmin and gmaxdenote, respectively, the minimum attained and the maximum possible values of gMCN1→LG for the current level of MCN1 stimulation. C, Voltage traces of Int1, the LG neuron (−52 mV), and the gMCN1→LG (280 pS/cm2) conductance, when inhibition from the AB neuron to Int1 was removed (−100% of the canonical model).
The effect of AB→Int1 on the gastric mill period can be examined by asking howAB→Int1 determines the timing of the LG burst onset and termination. The LG neuron burst onset occurs when an AB neuron-evoked disinhibition, riding on the slow MCN1-evoked excitation, is sufficient to trigger a burst. The strength of this disinhibition is given by AB→Int1. WhenAB→Int1 is small, the rising phase of the slow MCN1-evoked excitation builds up for a longer time before a later AB neuron-evoked disinhibition triggers a burst in the LG neuron, and the rising phase terminates at a larger value ofgMCN1→LG. Likewise, whenAB→Int1 is large, the AB neuron-evoked disinhibition triggers a burst in the LG neuron at an earlier time on the rising phase of the slow MCN1-evoked depolarization, and the rising phase terminates at a smaller value ofgMCN1→LG (Fig. 7A).
Figure 7B is a superposition of thegMCN1→LG traces from Figure 7A. Notice that because gMCN1→LG exponentially approaches its maximal value, the slope ofgMCN1→LG is steeper early in its rise than later. This explains why the duration of the rising phase of the slow MCN1-evoked depolarization is more sensitive to a decrease inAB→Int1 than to an increase inAB→Int1. Also, with smallerAB→Int1, the falling phase of the slow MCN1-evoked excitation starts at a largergMCN1→LG value. However, for all values ofAB→Int1 the falling phase of the slow MCN1-evoked depolarization terminates at approximately the samegMCN1→LG = gmin (note the minima of the traces in Fig. 7B). It follows that whenAB→Int1 is small, the LG neuron burst duration expands, because it takes longer for the MCN1 excitation to decay back to its minimum.
It is important to note that the effect of changingAB→Int1 was less prominent for the falling phase of the MCN1-evoked excitation than for its rising phase, because of the exponential nature of thegMCN1→LG decay. It is a property of exponential decay that the rate of decay is faster at larger values. When AB→Int1 was small, the falling phase of the slow MCN1-evoked excitation started at a largergMCN1→LG value and therefore the initial decay rate of gMCN1→LG was faster. The duration of the LG burst was expanded because of a largergMCN1→LG value at the start of the falling phase of the MCN1-evoked slow depolarization, but this effect on burst duration was partially damped because of the faster decay rate.
When AB→Int1 was set to 0 (Fig.7C), the model gastric mill rhythm was completely disrupted. In this case, despite reaching its saturation level (gMCN1→LG =gmax), the MCN1 chemical excitation of LG could not overcome the uninterrupted Int1 inhibition of the LG neuron, and therefore it did not fire.
The AB neuron can initiate an LG neuron burst only after sufficient accumulation of MCN1 excitation of LG
In this section, we formulate the ideas qualitatively described in the previous section in a more rigorous manner. The pyloric latency of the onset of the LG neuron burst was approximately constant (Fig.5C). This constant latency suggested that the LG neuron burst was initiated by the AB neuron-evoked disinhibition of the LG neuron from Int1. However, not every AB neuron burst triggered an LG neuron burst. Rather, the ability of the AB neuron to evoke an LG neuron burst was gated by a sufficient accumulation of MCN1 excitation to the LG neuron. To demonstrate this, we compared the effect of a single AB neuron burst delivered at different times after the onset of the MCN1 to LG neuron synapse (Fig.8A). We started the simulation with both MCN1 and the AB neuron off. In this condition, Int1 fired tonically (data not shown), and the LG neuron was quiescent. We then stimulated MCN1 (shown by the bar) and elicited a single AB neuron burst at varying delays (d) after the start of the MCN1 stimulation. Figure 8A shows the LG neuron membrane potential when a single AB neuron burst was evoked at delays of 1 sec (bottom trace) to 6 sec (top trace) after the start of the MCN1 stimulation. With short delays, the AB neuron burst triggered a transient subthreshold depolarization; with longer delays, it generated a burst in the LG neuron. Notice that the amplitude of the AB neuron-evoked depolarization in the LG neuron increased as the LG neuron membrane potential depolarized. This increase occurred because the LG neuron depolarization is caused by a conductance decrease (the removal of Int1’s inhibition), and therefore depolarization increases the driving force on the synaptic potential.
Fig. 8.
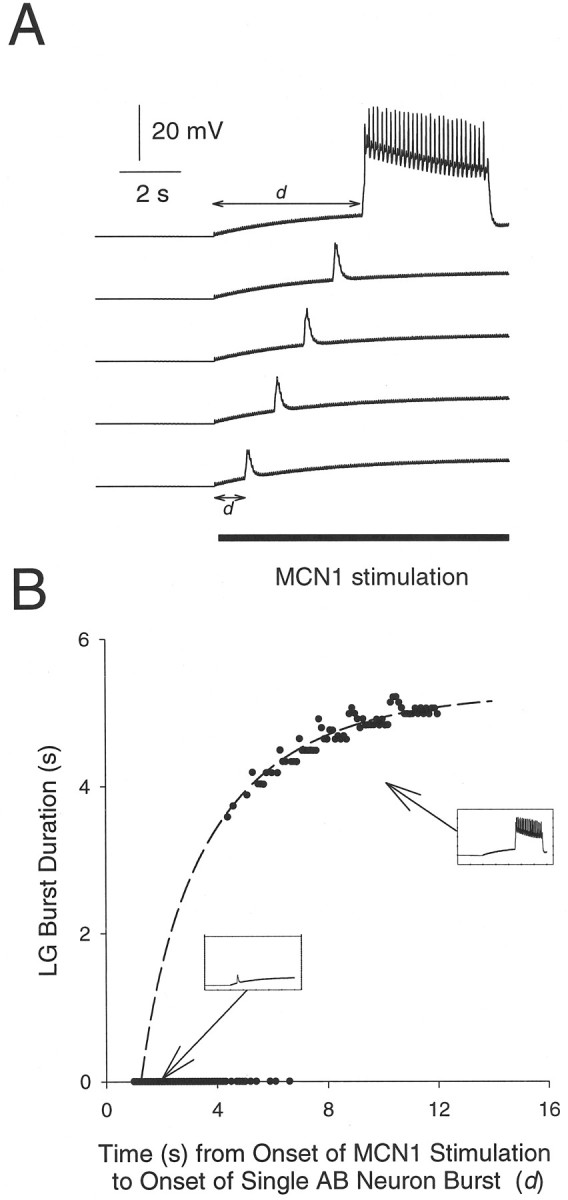
The onset of the LG neuron burst depends on the accumulation of MCN1 to LG neuron excitation (gMCN1→LG). A, Thesolid bar indicates MCN1 stimulation. Traces show the response of the model LG neuron when a single AB neuron burst was generated at 1, 2, 3, 4, or 5 sec after the start of MCN1 stimulation.B, The LG neuron burst duration plotted against the delay (d in A) between the start of MCN1 excitation and the single AB neuron burst. The dashed line is the plot of Equation 1 as derived in the.
In Figure 8B, the LG neuron burst duration is plotted against d. No burst was initiated when an AB neuron burst was evoked with d shorter than 4.4 sec. For dbetween 4.4 and 6.1 sec, the AB neuron burst resulted in either a subthreshold depolarization in the LG neuron or an LG neuron burst. With d longer than 6.1 sec, the AB neuron burst consistently resulted in an LG neuron burst. The duration of these LG neuron bursts increased gradually with d. The gradual increase in the LG neuron burst durations can be explained as follows. From the start of the MCN1 stimulation to the start of the LG neuron burst,gMCN1→LG grew from 0 and exponentially approached gmax, its maximum possible value for this level of MCN1 stimulation. WhengMCN1→LG reached some valuegburst, the LG neuron started to fire and inhibited the MCN1 terminals. When the LG neuron started to fire,gMCN1→LG exponentially decayed fromgburst to gmin. Unlikegmin, gburst was dependent on the strength and timing of the AB neuron to Int1 synapse. On the basis of these facts, we derived the following equation to describe the LG neuron burst duration B as function ofd:
| Equation 1 |
The derivation is given in the . This function (plotted as a dashed line in Fig. 8B) saturates to a maximum of 5.3–5.9 sec (depending ongmin) as t → ∞.
The results shown in Figure 8B imply that as the pyloric period is changed from its canonical value, the model LG neuron burst duration is approximately restricted between a minimum of 3.5 sec and a maximum of 5.9 sec.
Full parameter sweeps
To explore fully the effect of larger parameter variations on the gastric mill period, we did a complete parameter sweep by varying each maximal synaptic conductance from 0 to at least twice its canonical value. At each value of the parameter, the simulation was run for a stretch of 200 sec, and the gastric mill periods were measured. The parameter value was then increased incrementally by a small amount, and the procedure was repeated.
The interplay between MCN1→LG andAB→Int1
Figure 9 explores further the interaction of parameter variations ofMCN1→LG andAB→Int1 on the gastric mill period. When MCN1→LG was decreased from its canonical value (dotted lines), the gastric mill period increased sharply (Fig. 9A) because it took longer for the LG neuron to escape from the Int1 inhibition. The gastric mill rhythm was disrupted when MCN1→LG was <0.2 nS/cm2. IncreasingMCN1→LG from its canonical value did not alter the rhythm appreciably. WhenAB→Int1 was varied fourfold, the gastric mill period varied 14-fold (Fig. 9B). AsAB→Int1 was decreased, the gastric mill period increased. Gastric mill oscillations were disrupted whenAB→Int1 was <1.25 nS/cm2.
Fig. 9.

The interaction between the MCN1 to LG neuron excitation and the AB neuron input. Dotted lines inA and B represent the values of the canonical model. A, An increase inMCN1→LG results in a moderate decrease in gastric mill period. B, An increase inAB→Int1 results in a sharp decrease in gastric mill period. C, The duration of the LG neuron interburst interval (and therefore the gastric mill period) depends on the strength of both the conductance of the MCN1 chemical excitation of the LG neuron (MCN1→LG) and the Int1 pyloric-timed inhibition (AB→Int1). AsAB→Int1 increases (left to right, from 1.35 to 1.8 nS/cm2), the time it takes the LG neuron to reach its burst threshold decreases, fewer pyloric disinhibitions occur, and the period decreases. AsMCN1→LG increases (top to bottom, from 0.3 to 0.45 nS/cm2), for a given amplitude of AB→Int1, again it takes less time to reach the burst threshold of the LG neuron, and therefore the period decreases.
Figure 9C summarizes the effects of changes in the two conductances that are responsible for the generation of the MCN1-elicited gastric mill rhythm. Four cases are shown: the top two cases are with MCN1→LG held constant and AB→Int1 varied; underneath are the results of increasing MCN1→LG. This figure shows that when both AB→Int1 andMCN1→LG were small, the gastric mill period was long; increasing either of them shortened the period. The transition of the LG neuron from off to on occurred at the peak of the pyloric-timed disinhibition of the LG neuron. Consequently, when the LG neuron was depolarized more rapidly by the stronger MCN1-evoked depolarization, the period decreased for a givenAB→Int1. Also when the size ofAB→Int1 increased, the LG neuron reached its burst threshold earlier for a givenMCN1→LG sooner, and the period decreased.
It is important to note that sensitivity of the gastric mill period to many of the model parameters is the result of the interaction between the slow excitatory input of MCN1 to the LG neuron and the intermittent disinhibition by the AB neuron. Therefore, when the strength of the MCN1 input to the LG neuron is significantly varied, this will in turn alter the specifics of the sensitivity to other model parameters.
The effects of the other MCN1 inputs
To observe the complete range of periods obtained by varyingMCN1→Int1, we changed this parameter by three orders of magnitude (note logarithmic scale on thex-axis of Fig.10A). The period did not vary with MCN1→Int1 for values less than the canonical value, but steeply rose at large values ofMCN1→Int1 (Fig. 10A). At very large values of MCN1→Int1, the strong excitation of Int1 produced a strong inhibition from Int1 to the LG neuron, which competed with the MCN1 excitation of the LG neuron and slowed down the gastric mill rhythm (data not shown). AtMCN1→Int1 values >29 nS/cm2, the MCN1 excitation of the LG neuron was insufficient to overcome the inhibition from Int1, and the rhythm was disrupted.
Fig. 10.
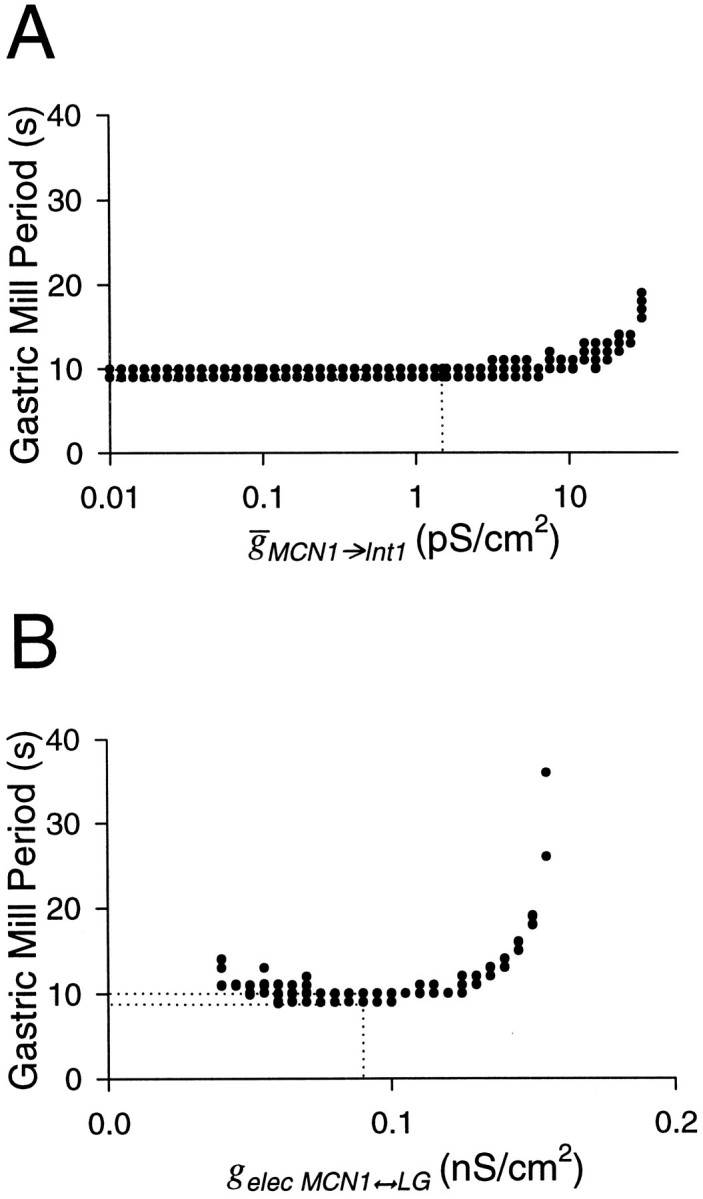
The effect of changing maximal synaptic conductances of the MCN1 to Int1 synapse (A) and the MCN1 to LG electrical coupling (B) on the model gastric mill period. Dotted lines represent the values of the canonical model. The x-axis inB is plotted on a logarithmic scale because the MCN1 to Int1 conductance is varied 2000-fold.
All parameters discussed so far affected the gastric mill period by altering the duration of the interburst phase of the LG neuron (which coincides with the Int1 burst). In contrast, the strength of the electrical coupling gelec determines the LG neuron burst duration. As mentioned in Materials and Methods, the canonical value of gelec was tuned so that the LG neuron could not maintain a burst of action potentials solely attributable to MCN1 electrical excitation. An increase ingelec increased the duration of the LG neuron burst and therefore the rhythm period (Fig. 10B). Forgelec > 0.155 nS/cm2, the gastric mill oscillations were disrupted, with the LG neuron firing tonically and continually inhibiting Int1. The disruption of the gastric mill rhythm in this case was qualitatively different from the other cases described above, in which Int1 fired continuously and inhibited the LG neuron.
The effect of the reciprocally inhibitory LG neuron to Int1 synapses
Figure 11A shows the effect of the maximal synaptic conductance of the LG neuron to Int1 synapse. When LG→Int1 was small, the rhythm had a period equal to the pyloric period (left inset). In this case, whenever the AB neuron inhibited Int1, the LG neuron fired an action potential. However, because the LG neuron to Int1 inhibition was weak, at the end of the AB neuron inhibition Int1 produced a burst. In this case, the transition from the LG neuron burst to Int1 burst was determined by the properties of the nonactive Int1. AsLG→Int1 increased, the gastric mill period sigmoidally increased and reached a saturation level of 10 sec. This saturation was caused by the fact that no matter how strong the LG neuron to Int1 inhibition, the LG neuron burst duration was determined by the waning of the chemical excitation from MCN1 to LG neuron. At these large values of LG→Int1, the LG neuron burst terminated independently of whether Int1 fired (data not shown). Therefore, the transition from the LG neuron burst to Int1 burst was determined only by the active LG neuron (right inset). The canonical model (dotted lines) is closer to the extremity at which the active LG neuron determines the transitions from LG neuron burst to Int1 burst.
Fig. 11.
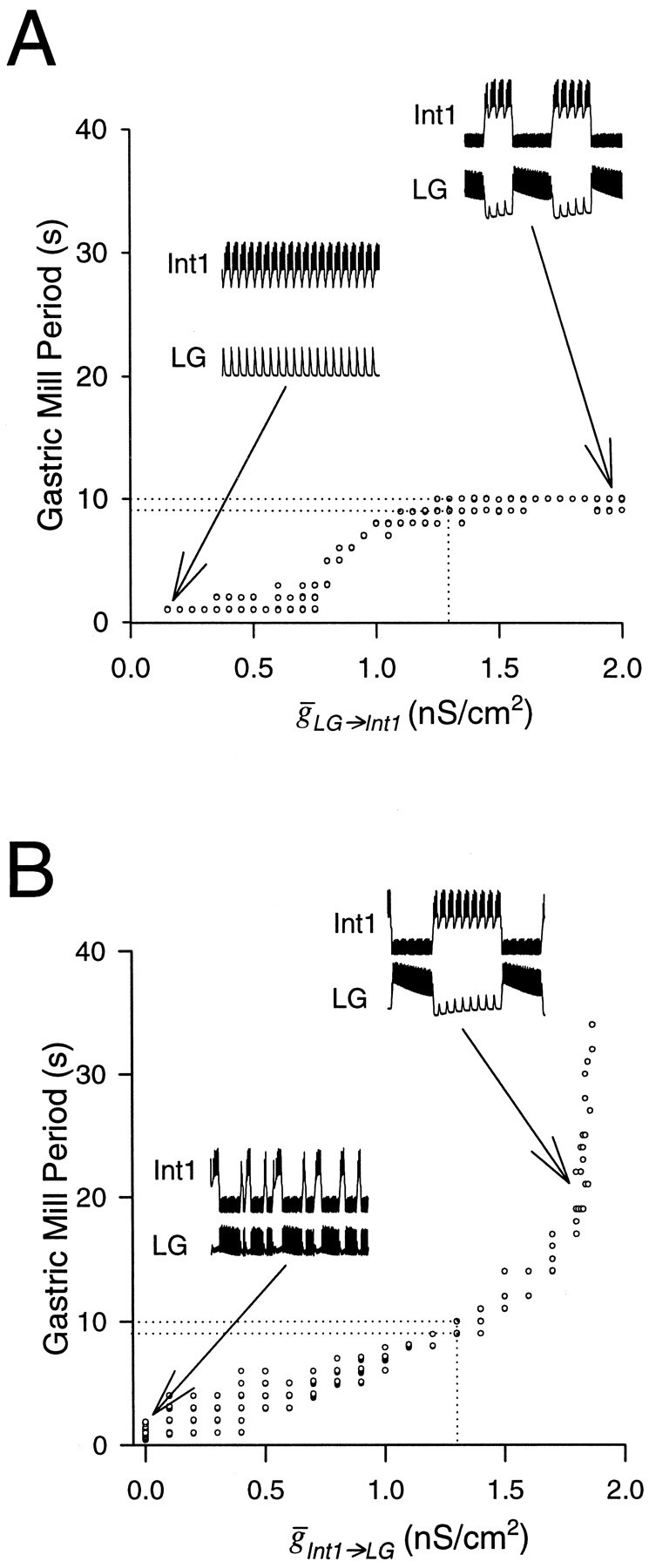
The effect of changing the maximal synaptic conductances of the reciprocally inhibitory pair. Dotted lines represent the values of the canonical model.A, The model gastric mill period as a function ofLG→Int1. Insets show Int1 and LG neuron activity for LG→Int1= 0.15 nS/cm2 andLG→Int1 = 2.0 nS/cm2. B, The model gastric mill period as a function ofInt1→LG. Insets show Int1 and LG neuron activity for Int1→LG= 0 nS/cm2 andInt1→LG = 1.8 nS/cm2.
Figure 11B shows the effect of varyingInt1→LG on the rhythm period. WhenInt1→LG = 0, the AB neuron did not play any role in the model oscillation. Consequently, the gastric mill rhythm was not locked to the pyloric rhythm but was produced merely by the interaction between MCN1 and the LG neuron (left inset). AsInt1→LG increased, the MCN1 to LG neuron excitation had to overcome more of Int1’s inhibition, and thus it took longer for the LG neuron to start a burst (right inset). Beyond the value of 1.87 nS/cm2, the MCN1 excitation of the LG neuron did not accumulate enough to overcome Int1’s inhibition of the LG neuron. Consequently, the gastric mill rhythm was disrupted and replaced by pyloric-timed firing of Int1 and subthreshold oscillations in the LG neuron (data not shown).
Figure 11 illustrates that the transitions from Int1 burst to LG neuron burst and from the LG neuron burst to Int1 burst are generated by different mechanisms. When Int1→LG = 0 (left inset) or was very small (data not shown), the transition from Int1 burst to LG neuron burst was completely independent of the presynaptic neuron. This changed with larger values ofInt1→LG. However, in contrast to the case of LG→Int1 (Fig.11A), the transition from Int1 burst to LG neuron burst was never purely dependent on the presynaptic neuron (there was no saturation of period as Int1→LGincreased).
DISCUSSION
As animals move through the world, their nervous systems generate various oscillatory discharges that generate movement or may be important for sensory processing (Gray, 1995; Marder and Calabrese, 1996). Often, meaningful movement demands coordination among several central pattern-generating circuits that produce rhythmic motor patterns of different frequencies. However, relatively little is known about the biological mechanisms by which rhythmic neural circuits of different frequencies interact (Bartos and Nusbaum, 1997). The stomatogastric nervous system generates several behaviorally relevant rhythmic motor patterns of significantly different periods. Uncovering the mechanisms by which the coupling among these subcircuits of the stomatogastric nervous system is achieved should provide insights into various mechanisms used in biological systems for coordinating the activity of different-period oscillators.
There is a significant amount of both experimental and theoretical work on oscillators formed by two reciprocally inhibitory neurons (Perkel and Mulloney, 1974; Satterlie, 1985; Wang and Rinzel, 1992, 1993;Friesen, 1994; Skinner et al., 1994; Van Vreeswijk et al., 1994;Calabrese, 1995; Nadim et al., 1995; Olsen et al., 1995; Gerstner et al., 1996; Sharp et al., 1996). In these cases, the two neurons are either identical or close in physiological properties, and the transitions are generally symmetrical. At the center of the circuit we are studying is a reciprocally inhibitory pair of neurons, the LG neuron and Int1, that have distinct properties. Indeed, under one set of conditions, Int1 exhibits pyloric-timed activity, and the LG neuron is silent. Here, the transformation of the Int1/LG neuron pair into an anti-phase oscillator requires that the level of excitation of the two neurons be made more equivalent, and this is accomplished by the activation of the modulatory neuron. This is similar to the results ofMiller and Selverston (1982), who obtained stable alternating bursts from the LP and PD neurons in the stomatogastric ganglion only when the appropriate level of current injection was chosen carefully. However, unlike the case of anti-phase oscillators formed from similar neurons, the asymmetry of this circuit raises the possibility that the transitions from off to on and from on to off may occur by different cellular mechanisms. To the extent to which the MCN1-activated gastric mill rhythm can be thought of as an asymmetric anti-phase oscillator, one would expect—and our simulations show (Fig. 11)—that the period of the gastric mill rhythm depends strongly on the strengths and time courses of the reciprocally inhibitory synapses between Int1 and the LG neuron.
In the original work on the MCN1-activated gastric mill rhythm, the emphasis was placed on the interaction between the LG neuron and the MCN1 axon terminals. In this formulation, Int1 is a “follower” and fires in anti-phase to the LG neuron, but the essential burst-generating mechanism is the LG neuron inhibition of MCN1 and the slow MCN1 excitation of the LG neuron (Coleman et al., 1995; Marder, 1996). The naive prediction of this “word model” was that the period of the MCN1-activated gastric mill rhythm would depend primarily on the strength and time course of the MCN1 excitation of LG. One of our motivations in constructing the biophysical model of the MCN1-elicited gastric mill rhythm was to determine the relative role of these two mechanisms in producing a stable rhythm. By exploring the sensitivity of each of the components of the model to parameter changes, we hoped to obtain insight into how the MCN1-activated gastric mill rhythm was produced and controlled.
To our surprise, in the process of building the model, we discovered the hitherto unexpected crucial role that the pyloric-timed inhibition of Int1 from the AB neuron plays in the construction of the MCN1-activated gastric mill rhythm. In this new mechanism, the AB neuron inhibition of Int1 provides a phasic disinhibition of the LG neuron, which acts synergistically with the slow depolarization from MCN1 (Fig. 9C). In effect, the LG neuron receives an enabling depolarization that is then phasically gated by the pyloric rhythm.
Functional significance of the pyloric–gastric interaction
The mechanism that we describe here produces obligatory coupling between the pyloric and gastric mill rhythms. Given that food must move from the gastric mill, where chewing occurs, to the pylorus, which filters the chewed food, and that these regions of the crustacean foregut are mechanically coupled, it is easy to imagine that it is advantageous to provide a fixed temporal relationship between the pyloric and gastric rhythms. Indeed, this model predicts, and our data show, that there is a fixed pattern of coupling between pyloric and gastric mill rhythms, and that there is a fixed latency between the burst discharge of the AB neuron and the onset of the LG neuron burst in the MCN1-activated gastric mill rhythm. Moreover, we predict that analyses of gastric mill activity stimulated by MCN1 should reveal the same kind of integer coupling as seen in the model.
There are myriad cases in which it may be important to produce some kind of obligatory coupling between fast and slow rhythms that ensures that the slower rhythm occurs at a fixed time or latency with respect to a faster rhythm. For example, breathing and locomotory patterns must be appropriately coupled to achieve optimal biomechanics (Bramble and Carrier, 1983; Graves et al., 1983; Ramirez and Pearson, 1989;Baudinette, 1991; Syed and Winlow, 1991; Young et al., 1992; Bramble and Jenkins, 1993; Funk et al., 1993; Lafortuna et al., 1996). The mechanism revealed here is one of a family of similar mechanisms (LoFaro et al., 1994) that allows a fast rhythm to influence the period of a slower rhythm over a long dynamic range. In this light, it is important to reiterate that the variations in gastric mill period produced by alterations in the strength of the coupling from the pyloric to gastric mill can be significantly larger than the pyloric period itself. This “gearing” effect may be critical for maintaining appropriate coupling between two rhythms of a very different period that nonetheless must be kept in register.
Consequences of the locus of neuromodulatory control
The gastric mill and pyloric rhythms are influenced by many known modulatory neurons that release various amines and neuropeptides (Marder and Calabrese, 1996). An immediate and interesting consequence of the organization described in this paper is that modulation of the gastric mill rhythm could be produced directly by modulation of the gastric mill circuit, or indirectly by modulation of the strength of the AB neuron to Int1 synapse, the amplitude and duration of the AB neuron burst, or the period of the pyloric rhythm. Figure12 is a schematic drawing that illustrates the potential consequences of modulation at various loci in this coupled network. Figure 12A shows that the pyloric and gastric mill rhythms are coupled. In principle, modulatory substances could act directly on the neurons of the gastric mill network. For example, neuromodulatory substances could alter the properties of Int1 and the LG neuron (or their connections) to produce a gastric mill rhythm that was more independent of pyloric timed inhibition (Fig. 12B), in which case the obligatory coupling between the pyloric and gastric rhythms might be lost. Alternatively, modulatory substances could act directly to alter the period of the pyloric rhythm (Fig. 12C) to produce changes in the gastric mill period, or modulatory substances might directly influence the strength of the AB neuron to Int1 synapse (Fig.12D). The work presented in this paper argues that a very effective way to modulate the gastric mill rhythm would be to target the strength of this synapse, which would produce modifications of the gastric mill period without altering the pyloric rhythm.
Fig. 12.
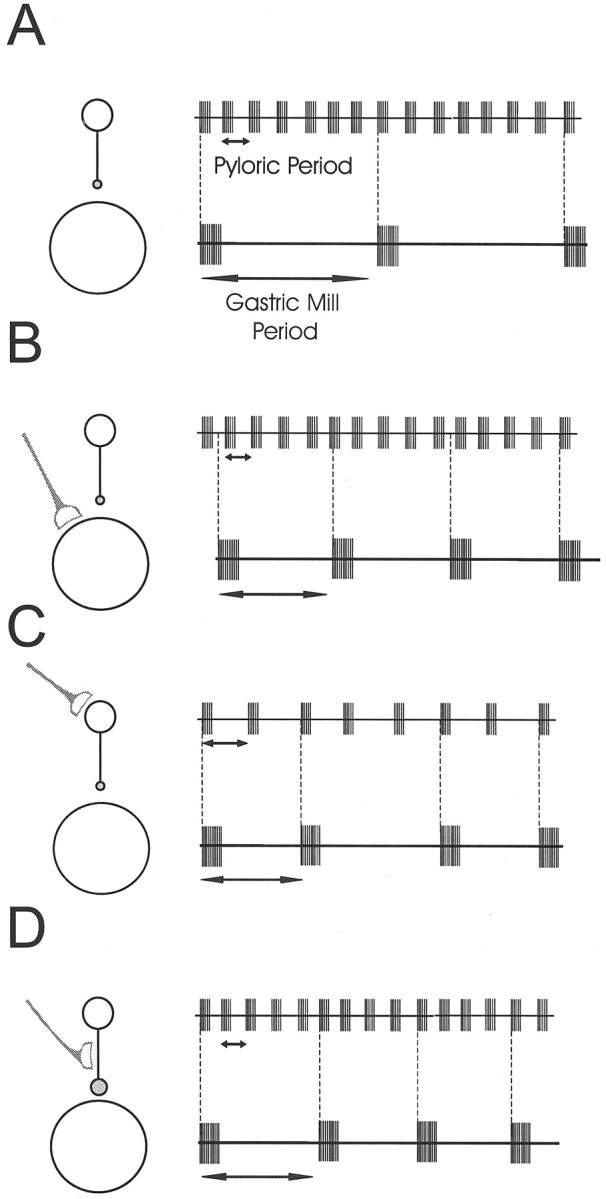
Schematic drawing showing the possible loci of neuromodulation. In all panels the inhibitory connection is shown as acircle; the size of the circle indicates its strength. The modulatory input is indicated as the half-moon symbol. A, The slow gastric mill rhythm (bottom trace) and fast pyloric rhythm (top trace) in control conditions. Vertical dashed lines show that the two rhythms are time-locked.B, When modulatory inputs act directly on the gastric mill circuit, the two rhythms may become uncoupled. C, Modulation could affect the gastric mill rhythm indirectly by acting on the pyloric circuit. D, Modulation of the connection from the pyloric circuit to the gastric mill circuit can effectively change the gastric mill period without any effect on the pyloric rhythm.
It is important to reiterate that the work described here points out unambiguously that modulation of the period of an oscillator may be more selectively controlled not by direct actions of neuromodulatory substances on the neurons of that network but by altering the extent of coupling between that network and another.
Smooth segues between mechanisms
An attractive speculation is that in some modulatory environments the Int1/LG neuron pair may be very close to being able to produce a self-sustaining half-center type oscillation and that the inputs from MCN1 and the AB neuron need only to be relatively weak to obtain a gastric mill rhythm. Under other modulatory conditions it is possible that the MCN1 to the LG neuron synapse is the dominant mechanism producing the gastric rhythm and that Int1 is almost a “follower.” Under yet other modulatory conditions, it is possible that the gastric mill rhythm depends primarily on the AB neuron to Int1 inhibition and is only weakly dependent on MCN1. We conjecture that the rich modulatory environment of the stomatogastric ganglion (Marder and Calabrese, 1996), by altering independently the intrinsic membrane properties of all of these participant neurons and their synaptic interactions, could provide the substrate for each of these different mechanisms to prevail under disparate physiological conditions.
: DERIVATION OF EQUATION 1
We have shown that after sufficient accumulation of the MCN1 to LG neuron excitation, a single AB neuron burst can initiate an LG neuron burst (Fig. 8). The time of this single AB neuron burst calculated from the beginning of the MCN1 stimulation is denoted by d. Equation 1 describes the relationship between the LG neuron burst duration (B) and d based on the dynamics of the synaptic conductance from MCN1 to the LG neuron.
This conductance gMCN1→LG is given byMCN1→LG S, whereS obeys the differential equation:
| Equation A1 |
and VMCN1 is the potential of the MCN1 axonal terminals (see Materials and Methods).
Because τS(VMCN1) is several orders of magnitude longer than the duration of MCN1 action potentials (Table 2), in calculating S we can ignore these action potentials and use the average MCN1 membrane potentialMCN1. During the time intervald, S grows from its initial value of 0 and exponentially approaches Smax(≈S∞(MCN1)) with time constant τrise = τS(MCN1). During the LG neuron burst, S decays exponentially toward 0 with time constant τfall = τS(MCN1). We denote the value of S when it grows bySrise(t) and when it is decays bySfall(t). From Equation EA1,Srise and Sfall obey the following differential equations:
| Equation A2 |
| Equation A3 |
Solving A2 for an arbitrary initial valueSrise(0) we obtain:
For conditions described in the discussion of Equation 1, the MCN1 to LG neuron synaptic conductance is initially zero, henceSrise(0) = 0. The value ofSrise at the onset of the LG neuron burst (t = d) is:
| Equation A4 |
During the LG neuron burst, Sfall decays from this initial value Srise(d) toward 0. However, the LG neuron burst terminates beforeSfall reaches 0, at some valueSmin. The LG neuron burst duration (B) is the time it takes Sfallto decay from Srise(d) toSmin. Solving A3 for an arbitrary initial condition Sfall(0) we obtain:
Substituting Sfall(0) =Srise(d) and using A4 we get:
| Equation A5 |
Solving A5 for B and substitutingSfall(B) =Smin we obtain:
Footnotes
This research was supported by National Institutes of Health Grants NS17813 (E.M.), MH46742 (E.M.), and NS29436 (M.P.N.), and the Human Frontier Science Program Organization. F.N. was a fellow of the Sloan Center for Theoretical Neurobiology at Brandeis University. We acknowledge the support of the W. M. Keck Foundation to Brandeis University. We thank Nancy Kopell, Marlene Bartos, and Steve Epstein for many helpful discussions. Marlene Bartos participated in collecting the data shown in Figure 5.
F.N. and Y.M. contributed equally to this work.
Correspondence should be addressed to Dr. Eve Marder, Volen Center, Brandeis University, 415 South Street, Waltham, MA, 02254.
REFERENCES
- 1.Alonso A, Llinás RR. Subthreshold Na+ dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:175–177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- 2.Ayers JL, Selverston AI. Monosynaptic entrainment of an endogenous pacemaker network: a cellular mechanism for von Holt’s magnet effect. J Comp Physiol. 1979;129:5–17. [Google Scholar]
- 3.Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. J Physiol (Lond) 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartos M, Nusbaum MP. Intercircuit control of motor pattern modulation by presynaptic inhibition. J Neurosci. 1997;17:2247–2256. doi: 10.1523/JNEUROSCI.17-07-02247.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baudinette R. The energetics and cardiorespiratory correlates of mammalian terrestrial locomotion. J Exp Biol. 1991;160:209–231. doi: 10.1242/jeb.160.1.209. [DOI] [PubMed] [Google Scholar]
- 6.Bramble D, Carrier D. Running and breathing in mammals. Science. 1983;219:251–256. doi: 10.1126/science.6849136. [DOI] [PubMed] [Google Scholar]
- 7.Bramble D, Jenkins FJ. Mammalian locomotor-respiratory integration: implications for diaphragmatic and pulmonary design. Science. 1993;262:235–240. doi: 10.1126/science.8211141. [DOI] [PubMed] [Google Scholar]
- 8.Calabrese RL. Oscillations in motor pattern-generating networks. Curr Opin Neurobiol. 1995;5:816–823. doi: 10.1016/0959-4388(95)80111-1. [DOI] [PubMed] [Google Scholar]
- 9.Coleman MJ, Meyrand P, Nusbaum MP. A switch between two modes of synaptic transmission mediated by presynaptic inhibition. Nature. 1995;378:502–505. doi: 10.1038/378502a0. [DOI] [PubMed] [Google Scholar]
- 10.Corio M, Palisses R, Viala D. Origin of the central entrainment of respiration by locomotion facilitated by MK 801 in the decerebrate rabbit. Exp Brain Res. 1993;95:94–90. doi: 10.1007/BF00229657. [DOI] [PubMed] [Google Scholar]
- 11.Friesen WO. Reciprocal inhibition: a mechanism underlying oscillatory animal movements. Neurosci Biobehav. 1994;18:547–553. doi: 10.1016/0149-7634(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 12.Funk G, Shlomenko G, Valenzuela I, Steeves J, Milsom W. Coordination of wing beat and respiration in Canada geese during free flight. J Exp Biol. 1993;175:317–323. [Google Scholar]
- 13.Gerstner W, van Hemmen JL, Cowan JD. What matters in neuronal locking? Neural Comp. 1996;8:1653–1676. doi: 10.1162/neco.1996.8.8.1653. [DOI] [PubMed] [Google Scholar]
- 14.Graubard K. Synaptic transmission without action potentials: input-output properties of a non-spiking presynaptic neuron. J Neurophysiol. 1978;41:1014–1025. doi: 10.1152/jn.1978.41.4.1014. [DOI] [PubMed] [Google Scholar]
- 15.Graubard K, Raper JA, Hartline DK. Graded synaptic transmission between spiking neurons. Proc Natl Acad Sci USA. 1980;77:3733–3735. doi: 10.1073/pnas.77.6.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graubard K, Raper JA, Hartline DK. Graded synaptic transmission between identified spiking neurons. J Neurophysiol. 1983;50:508–521. doi: 10.1152/jn.1983.50.2.508. [DOI] [PubMed] [Google Scholar]
- 17.Graves C, Glass L, Laporta D, Meloch R, Grassino A. Respiratory phase locking during mechanical ventilation in anesthetized human subjects. Am J Physiol. 1983;250:R902–909. doi: 10.1152/ajpregu.1986.250.5.R902. [DOI] [PubMed] [Google Scholar]
- 18.Gray CM. Synchronous oscillations in neuronal systems: mechanisms and functions. J Comput Neurosci. 1995;1:11–38. doi: 10.1007/BF00962716. [DOI] [PubMed] [Google Scholar]
- 19.Hines M. NEURON: A program for simulation of nerve equations. In: Eeckman F, editor. Neural systems: Analysis and modeling. Kluwer; Boston: 1993. pp. 127–136. [Google Scholar]
- 20.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawahara K, Kumagai S, Nakazono Y, Myamoto Y. Coupling between respiratory and stepping rhythms during locomotion in decerebrate cats. J Appl Physiol. 1989;67:110–115. doi: 10.1152/jappl.1989.67.1.110. [DOI] [PubMed] [Google Scholar]
- 22.Kopell N. Toward a theory of modelling central pattern generators. In: Cohen AH, Rossignol S, Grillner S, editors. Neural control of rhythmic movements in vertebrates. Wiley; New York: 1988. pp. 369–413. [Google Scholar]
- 23.Kopell N, Ermentrout GB. Coupled oscillators and the design of central pattern generators. Math Biosci. 1988;90:87–109. [Google Scholar]
- 24.Lafortuna C, Reinach E, Saibene F. The effects of locomotor-respiratory coupling on the pattern of breathing in horses. J Physiol (Lond) 1996;492:587–596. doi: 10.1113/jphysiol.1996.sp021331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llinás RR, Yarom Y. Oscillatory properties of guinea-pig inferior olivary neurones and their pharmacological modulation: an in vitro study. J Physiol (Lond) 1986;376:163–182. doi: 10.1113/jphysiol.1986.sp016147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LoFaro T, Kopell N, Marder E, Hooper SL. Subharmonic coordination in networks of neurons with slow conductances. Neural Comp. 1994;6:69–84. [Google Scholar]
- 27.Manor Y, Nadim F, Marder E, Nusbaum MP. A model for the MCN1-activated gastric mill rhythm in the crab. Soc Neurosci Abstr. 1996;22:1438. [Google Scholar]
- 28.Marder E. Neural modulation: following your own rhythm. Curr Biol. 1996;6:119–121. doi: 10.1016/s0960-9822(02)00438-4. [DOI] [PubMed] [Google Scholar]
- 29.Marder E, Calabrese RL. Principles of rhythmic motor pattern generation. Physiol Rev. 1996;76:687–717. doi: 10.1152/physrev.1996.76.3.687. [DOI] [PubMed] [Google Scholar]
- 30.McFarland DH, Lund JP. An investigation of the coupling between respiration, mastication and swallowing in the awake rabbit. J Neurophysiol. 1993;69:95–108. doi: 10.1152/jn.1993.69.1.95. [DOI] [PubMed] [Google Scholar]
- 31.Meda P, Atwater I, Goncalves A, Bangham A, Orci L, Rojas E. The topography of electrical synchrony among β-cells in the mouse islet of Langerhans. Q J Exp Physiol. 1984;69:719–735. [PubMed] [Google Scholar]
- 32.Miller JP, Selverston AI. Mechanisms underlying pattern generation in lobster stomatogastric ganglion as determined by selective inactivation of identified neurons. IV. Network properties of pyloric system. J Neurophysiol. 1982;48:1416–1432. doi: 10.1152/jn.1982.48.6.1416. [DOI] [PubMed] [Google Scholar]
- 33.Nadim F, Olsen ØH, Calabrese RL. Modeling the leech heartbeat elemental oscillator. I. Interactions of intrinsic and synaptic currents. J Comput Neurosci. 1995;2:215–235. doi: 10.1007/BF00961435. [DOI] [PubMed] [Google Scholar]
- 34.Olsen ØH, Nadim F, Calabrese RL. Modeling the leech heartbeat elemental oscillator. II. Exploring the parameter space. J Comput Neurosci. 1995;2:237–257. doi: 10.1007/BF00961436. [DOI] [PubMed] [Google Scholar]
- 35.Perkel DH, Mulloney BM. Motor pattern production in reciprocally inhibitory neurons exhibiting postinhibitory rebound. Science. 1974;185:181–183. doi: 10.1126/science.185.4146.181. [DOI] [PubMed] [Google Scholar]
- 36.Pinsker HM. Aplysia bursting neurons as endogenous oscillators. I. Phase-response curves for pulsed inhibitory synaptic input. J Neurophysiol. 1977a;40:527–543. doi: 10.1152/jn.1977.40.3.527. [DOI] [PubMed] [Google Scholar]
- 37.Pinsker HM. Aplysia bursting neurons as endogenous oscillators. II. Synchronization and entrainment by pulsed inhibitory synaptic input. J Neurophysiol. 1977b;40:544–556. doi: 10.1152/jn.1977.40.3.544. [DOI] [PubMed] [Google Scholar]
- 38.Ramirez J, Pearson K. Alteration of the respiratory system at the onset of locust flight. J Exp Biol. 1989;142:401–424. [Google Scholar]
- 39.Rand RH, Cohen AH, Holmes PJ. Systems of coupled oscillators as models of central pattern generators. In: Cohen AH, Rossignol S, Grillner S, editors. Neural control of rhythmic movements in vertebrates. Wiley; New York: 1988. pp. 333–367. [Google Scholar]
- 40.Satterlie RA. Reciprocal inhibition and postinhibitory rebound produce reverberation in a locomotor pattern generator. Science. 1985;229:402–404. doi: 10.1126/science.229.4711.402. [DOI] [PubMed] [Google Scholar]
- 41.Sharp AA, Skinner FK, Marder E. Mechanisms of oscillation in dynamic clamp constructed two-cell half-center circuits. J Neurophysiol. 1996;76:867–883. doi: 10.1152/jn.1996.76.2.867. [DOI] [PubMed] [Google Scholar]
- 42.Skinner FK, Kopell N, Marder E. Mechanisms for oscillation and frequency control in reciprocal inhibitory model neural networks. J Comput Neurosci. 1994;1:69–87. doi: 10.1007/BF00962719. [DOI] [PubMed] [Google Scholar]
- 43.Somers D, Kopell N. Rapid synchronization through fast threshold modulation. Biol Cybern. 1993;68:393–407. doi: 10.1007/BF00198772. [DOI] [PubMed] [Google Scholar]
- 44.Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 45.Syed N, Winlow W. Coordination of locomotor and cardiorespiratory networks of Lymnaea stagnalis by a pair of identified interneurones. J Exp Biol. 1991;158:37–62. doi: 10.1242/jeb.158.1.37. [DOI] [PubMed] [Google Scholar]
- 46.Van Vreeswijk C, Abbott LF, Ermentrout GB. When inhibition not excitation synchronizes neural firing. J Comput Neurosci. 1994;1:313–321. doi: 10.1007/BF00961879. [DOI] [PubMed] [Google Scholar]
- 47.Wang XJ, Rinzel J. Alternating and synchronous rhythms in reciprocally inhibitory model neurons. Neural Comp. 1992;4:84–97. [Google Scholar]
- 48.Wang X-J, Rinzel J. Spindle rhythmicity in the reticularis thalami nucleus: synchronization among mutually inhibitory neurons. Neuroscience. 1993;53:899–904. doi: 10.1016/0306-4522(93)90474-t. [DOI] [PubMed] [Google Scholar]
- 49.Welsh JP, Lang EJ, Sugihara I, Llinás RR. Dynamic organization of motor control within the olivocerebellar system. Nature. 1995;374:453–457. doi: 10.1038/374453a0. [DOI] [PubMed] [Google Scholar]
- 50.Young I, Alexander RM, Woakes A, Butler P, Anderson L. The synchronization of ventilation and locomotion in horses (Equus caballus). J Exp Biol. 1992;166:19–31. doi: 10.1242/jeb.166.1.19. [DOI] [PubMed] [Google Scholar]