

Abstract
A short ischemic period induced by the transient occlusion of major brain arteries induces neuronal damage in selectively vulnerable regions of the hippocampus. Adenosine is considered to be one of the major neuroprotective substances produced in the ischemic brain. It can be released from damaged cells, but it also could be generated extracellularly from released ATP via a surface-located enzyme chain. Using the rat model of global forebrain ischemia, we applied a short (10 min) transient interruption of blood flow and studied the distribution of ectonucleotidase activities in the hippocampus. Northern hybridization of mRNA isolated from hippocampi of sham-operated and ischemic animals revealed an upregulation of ectoapyrase (capable of hydrolyzing nucleoside 5′-tri- and diphosphates) and ecto-5′-nucleotidase (capable of hydrolyzing nucleoside 5′-monophosphates). A histochemical analysis that used ATP, UTP, ADP, or AMP as substrates revealed a strong and selective increase in enzyme activity in the injured areas of the hippocampus. Enhanced staining could be observed first at 2 d. Staining increased within the next days and persisted at 28 d after ischemia. The spatiotemporal development of catalytic activities was identical for all substrates. It was most pronounced in the CA1 subfield and also could be detected in the dentate hilus and to a marginal extent in CA3. The histochemical staining corresponded closely to the development of markers for reactive glia, in particular of microglia. The upregulation of ectonucleotidase activities implies increased nucleotide release from the damaged tissue and could play a role in the postischemic control of nucleotide-mediated cellular responses.
Keywords: astrocyte, ectoapyrase, ecto-ATPase, ecto-5′-nucleotidase, ischemia, microglia
Following global ischemia, neurons in specific subfields of the hippocampus reveal selective vulnerability. Neuronal death takes a characteristic delayed time course and becomes evident at the light microscopic level 2–4 d after ischemia. It is accompanied by a typical glial reaction involving both astrocytes and microglia (Kraig et al., 1995). Reactive astrocytes display hypertrophy, increased staining for the intermediate filament glial fibrillary acidic protein (GFAP), and hyperplasia. In regions of permanent ischemic injury astrocytes also express vimentin. From postischemic day 1 onward, microglia proliferate and show increased expression of major histocompatibility complex (MHC) class I and II immunomolecules at their surface and finally transform into intrinsic brain phagocytes, removing neuronal debris. Reactive astrocytes and microglia undergo a considerable number of additional specific functional changes, including the upregulation of many protein species and the secretion of cytokines and growth factors (Eddleston and Mucke, 1993; Gehrmann et al., 1995). Some of these may mediate tissue damage. Others play an important role in limiting the extent of damage or also a role in tissue repair.
A loss of energy charge resulting from the interrupted blood supply results in a massive release of excitatory neurotransmitters. Glutamate is considered to play a major role in cytotoxicity by inducing an intracellular and lethal increase in neuronal Ca2+. Extracellular adenosine is increased as an early response to ischemia and may, in turn, exert neuroprotective actions by inhibiting glutamate release via presynaptic receptors (Schubert and Kreutzberg, 1993). It can be released via carrier-mediated mechanisms as a result of the hypoxia-induced massive breakdown of intracellular nucleotides, but hypoxia may result also in the direct release of nucleotides such as ATP (Dubyak and El-Moatassim, 1993). Both neurons and glia possess receptors for ATP and other nucleotides (ADP, UTP, UDP) either that function as ion channels (P2X receptors) or that are G-protein-coupled (P2Y receptors) (Boarder et al., 1995; Buell et al., 1996). P2X receptors are permeable not only for Na+ and K+ but also for Ca2+. Their hyperactivation is, therefore, potentially cytotoxic. Extracellular ATP is inactivated by a surface-located enzyme chain that results in the extracellular formation of adenosine (Zimmermann, 1996). The chain potentially includes the recently cloned ecto-ATP diphosphohydrolase (ectoapyrase) and ecto-ATPase as well as ecto-5′-nucleotidase (Kegel et al., 1997). Whereas ecto-ATPase has a high preference for ATP, ectoapyrase hydrolyzes ATP and ADP, resulting in the formation of AMP. Ecto-5′-nucleotidase catalyzes the formation of adenosine from AMP. An upregulation of these enzymes in the damaged tissue could serve the rapid elimination of any cytotoxic effects of ATP and in addition provide extracellular adenosine for purine salvage or as a neuroprotective agent. We previously have observed increased activity of ecto-5′-nucleotidase in the volume bordering the infarcted tissue after permanent middle cerebral artery occlusion (MCAO) (Braun et al., 1997). In the present study we induced transient forebrain ischemia by bilateral common carotid artery occlusion and hypertension in rats, and we probed for the expression of the entire ectonucleotidase chain.
MATERIALS AND METHODS
Transient forebrain ischemia. Forebrain ischemia was induced in 28 male Wistar rats (300–350 gm, Charles River, Sulzfeld, Germany). Rats were fasted overnight with free access to water. They were anesthetized with halothane (1.5%) in a mixture of N2O/O2 (70%/30%). Body and skull temperatures were maintained at 37.0–37.5°C by a temperature control feedback system (Technical and Scientific Equipment, Bad Homburg, Germany). The skull temperature was monitored with a thermocouple placed subcutaneously over the midline of the skull. Ischemia was induced for 10 min by clamping both common carotid arteries and lowering the mean blood pressure to 40 mm Hg by trimethaphan camphor sulfonate (5 mg/kg, i.v.) and central venous exsanguination (Smith et al., 1984). After 10 min the clips were removed, and the shed blood was reinjected rapidly. Arterial pH, pCO2, pO2, blood pressure, and plasma glucose concentration were measured before ischemia. Sham-operated animals were prepared as described above without clamping carotid arteries and lowering blood pressure. They were compared with untreated animals. After ischemia, animals were kept at an environmental temperature of 30°C for 2 hr and then at 20°C in their home cages. At 6 hr and at 2, 3, 7, 14, and 28 d after ischemia the rats received a lethal dose of chloral hydrate and were decapitated. Brains were removed, snap-frozen in prechilled (−20°C) 2-methylbutane, and stored at −70°C until sectioning.
Nucleotidase histochemistry. For localization of nucleotidase activity a lead phosphate method (Braun et al., 1997) was applied. Frozen sections (12 μm) were deposited on 3-aminopropyltriethoxy Silane-coated (Sigma, Deisenhofen, Germany) slides and allowed to dry for 1 hr. The dry tissue sections were stored at −80°C until further processing. Sections warmed to room temperature were fixed with 0.05 m cacodylate-buffered 4% paraformaldehyde, pH 7.4, for 12 min at room temperature and subsequently were washed with 0.05 m cacodylate buffer, pH 7.4, containing 0.25 m sucrose. Preincubation was performed for 30 min at room temperature in a Tris-maleate sucrose buffer (TMS; 0.25 m sucrose and 50 mm Tris-maleate, pH 7.4) containing 2 mm MgCl2 when 5′-nucleotidase activity was to be analyzed or 2 mm CaCl2 for the subsequent detection of hydrolysis of ATP, UTP, or ADP. Nucleotides (ATP, UTP, ADP, and AMP) (all from Sigma) were added to the buffer solution at a concentration of 1 mm. The enzyme reaction for the detection of ATPase, ADPase, or UTPase activity was performed for 2.4 hr at room temperature in a Tris-maleate-buffered substrate solution [composition (in mm): 2 Pb(NO3)2, 5 MnCl2, 2 CaCl2, and 50 Tris-maleate, plus 0.25 m sucrose, pH 7.4]. AMP was applied in the same buffer solution except that CaCl2 was omitted. After the sections were washed with demineralized water, the lead orthophosphate that was precipitated as a result of nucleotidase activity was visualized as a brown deposit by incubating sections in an aqueous solution of ammonium sulfide (2% v/v). To control for nonspecific phosphatase activity, we substituted nucleotides byp-nitrophenyl phosphate (Sigma). To control for nonspecific lead precipitation, we omitted nucleotides from the incubation medium. Subsequently, the sections were processed as for immunocytochemistry.
Immunocytochemical procedures. For immunocytochemical labeling of microglia, the monoclonal antibody MRC OX42 (Camon, Wiesbaden, Germany) was applied (Milligan et al., 1991). This antibody recognizes the rat complement receptor type 3 (Robinson et al., 1986). A monoclonal antibody G-A-5 (Sigma) against the GFAP from pig spinal cord was used as an astroglial marker. Vimentin was detected by using a monoclonal antibody (VIM 3B4, Progen Biotechnik, Heidelberg, Germany) against vimentin from bovine lens. Frozen sections were prepared as described for enzyme histochemistry. For immunoperoxidase labeling, frozen sections were warmed to room temperature, fixed with absolute methanol (7 min at −20°C), and rinsed with PBS [composition (in mm): 137 NaCl, 3 KCl, and 15 Na+/K+-phosphate buffer, pH 7.4]. Nonspecific binding was suppressed with 5% bovine serum albumin (BSA; Roth, Karlsruhe, Germany). Antibodies were diluted with PBS containing 1% BSA. Sections were incubated for 80 min at room temperature with antibodies against GFAP (1:100), OX42 (1:50), or vimentin (1:10). All further steps were performed at room temperature. After being washed with PBS for 25 min, the sections were incubated for 1 hr with a peroxidase-linked affinity-purified secondary antibody against mouse-IgG (Amersham, Braunschweig, Germany). After excess washing with PBS, the peroxidase reaction was performed with a diaminobenzidine (DAB) substrate solution (0.25 mg of DAB/ml, 0.03% NiCl2, 0.01% H2O2, and 100 mm Tris-HCl, pH 7.5). In control experiments only the secondary antibody was applied. After dehydration in graded ethanol, the sections were mounted with Permount (Fisher Scientific, Springfield, NY). They were examined with a Zeiss Axiophot microscope (Oberkochen, Germany) and photographed on Ilford Pan F film developed in Ultrafin (Tetenal).
Northern analysis. For Northern blotting, hippocampi were excised from rat brains 7 d after vessel occlusion or sham operation. Polyadenylated RNA was isolated by using the Oligotex direct kit (Qiagen, Hilden, Germany). Polyadenylated RNA (0.5 μg), as determined photometrically, was separated on a 1% agarose formaldehyde gel. Subsequently, the RNA was transferred by pressure (PosiBlot, Stratagene, Germany) onto a Hybond N membrane (Amersham). Using various constructs of DIG-labeled RNA as a probe, we hybridized blots overnight at 68°C in the presence of 5× SSC and 50% formamide. Filters were washed twice at room temperature with 2× SSC containing 0.1% SDS and twice at 68°C for 15 min with 0.2× SSC containing 0.1% SDS. The hybridization signal was visualized by using an alkaline phosphatase-conjugated anti-DIG antibody and a chemiluminescent substrate (CSPD; both from Boehringer Mannheim, Mannheim, Germany) according to the manufacturer’s instructions. For obtaining riboprobes, the mouse ectoapyrase construct CD39-5b (Maliszewski et al., 1994) and the rat ecto-ATPase construct rat9.4 (Kegel et al., 1997) were digested with SalI and with BamHI, respectively. The insert of the rat liver ecto-5′-nucleotidase construct λcNT34 (Misumi et al., 1990) was subcloned into pBluescript II SK and digested with BamHI. DIG-labeled antisense RNAs (ectoapyrase, 2.3 kb; ecto-ATPase, 1.6 kb; ecto-5′-nucleotidase, 1.8 kb) were transcribed by using T3 or T7 RNA polymerase according to the manufacturer’s instructions (Boehringer Mannheim). For detection of β-actin mRNA, a riboprobe from a cDNA clone coding for mouse β-actin (Stratagene, Heidelberg, Germany) was used.
RESULTS
Northern analysis
To probe for the expression of ectonucleotidases identified in molecular terms, we isolated polyadenylated RNA from hippocampi 7 d after transient forebrain ischemia or 7 d after sham operation. Equal amounts (0.5 μg) of mRNA were analyzed for ecto-5′-nucleotidase, ectoapyrase, and ecto-ATPase (Fig.1). The probe for rat ecto-5′-nucleotidase hybridized with a 3.9 kb mRNA species (Misumi et al., 1990). Corresponding to previous results (Kegel et al., 1997), the probe for ectoapyrase detected two major bands of 4.9 and 5.4 kb, which may represent the use of different polyadenylation sites at the 3′-uncoding region, whereas the riboprobe for ecto-ATPase yielded a prominent mRNA band at 2.2 kb. The hybridization signal for β-actin (2.2 kb) was determined as a control. As compared with sham-operated controls, the hybridization signals for both ecto-5′-nucleotidase and ectoapyrase were increased distinctly 7 d after ischemia. In contrast, the hybridization signal for ecto-ATPase was not affected by forebrain ischemia. Actin mRNA levels were evaluated by rehybridization. They did not differ between controls and postischemic hippocampi.
Fig. 1.

Northern blot analysis of ectonucleotidases. Equal amounts (0.5 μg) of polyadenylated RNA isolated from hippocampi 7 d after sham operation (a) or 7 d after 10 min of global ischemia (b) were subjected to electrophoresis, blotted, and hybridized with rat ecto-5′-nucleotidase (e-5′-nucl.), ectoapyrase (e-apyrase), and ecto-ATPase (e-ATPase) antisense riboprobes. A significant postischemic enhancement of the hybridization signals is detectable for ecto-5′-nucleotidase and ectoapyrase. The riboprobe for ectoapyrase hybridizes with two major bands at 4.9 and 5.4 kb. A riboprobe for mouse β-actinwas used as an internal control.
Increase in the activity of ATPase, UTPase, and ADPase in lesioned regions
The postischemic reaction was investigated at the septodorsal level of the hippocampus. Brain sections were analyzed at 6 hr and at 2, 3, 7, 14, and 28 d after ischemia. The extent of neuronal degeneration was visualized by Nissl staining (Figs.2A, 3A,4A). In the pyramidal cell layer of CA1, damaged neurons first became apparent at 2 d after ischemia. At 3 d the cell death of pyramidal cells was increased (Fig.3A), and from day 7 onward most neurons of CA1 were disintegrated (Fig.4A). No neuronal damage was detected in hippocampal sections of sham-operated animals (see Fig.2A).
Fig. 2.
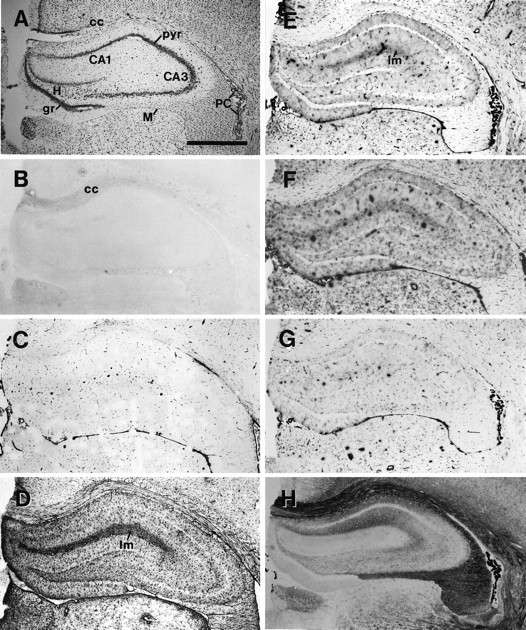
Corresponding hippocampal sections 7 d after sham operation. Comparison of Nissl staining (A), immunocytochemistry (B–D), and nucleotidase histochemistry (E–H). A, Nissl staining revealed no neuronal damage in the hippocampus. B, OX42 immunostaining is significant only in fiber tracts, as for example in the corpus callosum. C, Vimentin is detectable in capillaries. D, GFAP immunoreaction is strongest in the stratum lacunosum moleculare. E–G, ATPase (E), UTPase (F), and ADPase (G) reaction product in the hippocampus is restricted mainly to capillaries. Staining is enhanced slightly in the stratum lacunosum. H, 5′-Nucleotidase activity is highest in CA1 and in the stratum lacunosum moleculare of CA1 and CA3. The pyramidal cell layers and the dentate granule cell layer are free of reaction product. CA1, CA3, Subfields of the hippocampus; cc, corpus callosum;gr, granular cell layer of the dentate gyrus;H, hilus of the dentate gyrus; lm, stratum lacunosum moleculare; pyr, stratum pyramidale;PC, plexus choroideus; M, meninx. Scale bar, 1 μm.
Fig. 3.

Corresponding hippocampal sections 3 d after global ischemia. Comparison of Nissl staining (A), immunocytochemistry (B–D), and nucleotidase histochemistry (E–H). A, Reduced Nissl staining in CA1 (arrowheads) indicates damaged pyramidal neurons. Immunostaining for OX42 (B), vimentin (C), and GFAP (D) is upregulated most significantly in CA1 and the dentate hilus.E–H, ATPase (E), UTPase (F), ADPase (G), and 5′-nucleotidase activity (H) is enhanced in the pyramidal cell layer of CA1(arrows). Abbreviations as in Figure 2. Scale bar, 1 μm.
Fig. 4.

Corresponding hippocampal sections 7 d after global ischemia. Comparison of Nissl staining (A), immunocytochemistry (B–D), and nucleotidase histochemistry (E–H). A, Reduced Nissl staining in the CA1 subfield (arrowheads) indicates damaged pyramidal cells. Immunolabeling for OX42 (B), vimentin (C), and GFAP (D) is strongly enhanced in CA1 and the dentate hilus. In contrast to GFAP immunoreactivity, OX42 and vimentin immunostaining is very distinct in the layer of damaged pyramidal cells.E–H, Strong upregulation of ATPase (E), UTPase (F), ADPase (G), and 5′-nucleotidase (H) activity in all layers of CA1 and the dentate hilus. The reaction product is most intense in the layer of damaged pyramidal cells and the stratum lacunosum moleculare. Abbreviations as in Figure 2. Scale bar, 1 μm.
Because suitable antibodies for the immunodetection of ectonucleotidases were not available, we analyzed the hippocampi by enzyme cytochemistry. The histochemical staining pattern in the hippocampal formation was practically identical for the hydrolysis of ATP, UTP, and ADP and demonstrated a parallel and strong postischemic increase in staining intensity. UTP was included in the analysis to probe for the presence of ectoapyrase that hydrolyzes both ATP and UTP. The enzyme reaction in hippocampal sections of sham-operated animals (see Fig. 2E–G) was very low and identical to the staining pattern obtained for untreated rats. There was a faint staining of the entire tissue section, except for fiber tracts, the pyramidal cell layer, and the granule cell layer of the dentate gyrus. These neuronal layers became apparent as a light and unstained band. Reaction product was enhanced slightly in the stratum lacunosum moleculare. Intense lead staining was restricted to the endothelium of brain capillaries, the meninges covering the hippocampal formation, and the choroid plexus. These observations suggest that the histochemical staining performed at alkaline pH does not represent the activity of intracellular nucleoside 5′-triphosphate or 5′-diphosphate-hydrolyzing enzymes that are present at high activity in every cell.
At 2 d after global ischemia the unstained band corresponding to the pyramidal cell layer of CA1 had disappeared. It now revealed the same faint staining as the stratum oriens and stratum pyramidale. At 3 d the histochemical reaction for all three substrates clearly was enhanced in the stratum pyramidale of CA1 (see Fig.3E–G). There was also a small increase in staining intensity in the dentate hilus. The postischemic enzyme reaction was developed strongly at 7 d (see Fig. 4E–G). All layers of CA1 exhibited a distinct upregulation of ATP, UTP, and ADP hydrolysis. The pyramidal cell layer revealed the strongest enhancement. Staining was increased in the stratum lacunosum moleculare and in the dentate hilus. There were small variations in overall staining intensity between individual experiments and also between individual days of experimentation. This did not affect, however, the qualitative staining pattern. The intense staining in CA1 persisted 14 and 28 d after global ischemia. The reaction in the dentate hilus was still present at 14 d but had disappeared at 28 d. No reaction product was obtained in the control experiments in which nucleotides were replaced by p-nitrophenyl phosphate or when nucleotides were omitted from the incubation medium.
Lesion-induced increase in 5′-nucleotidase activity
When AMP was used as a substrate, the staining pattern differed clearly from that obtained with the other nucleotides, but the time course of the postischemic increase was comparable. 5′-Nucleotidase activity was high in all layers of CA1 of sham-operated (see Fig.2H) or untreated animals. Also, the stratum lacunosum moleculare of CA3 displayed a distinct reaction. The other layers of CA3 and the hilus and the molecular layer of the dentate gyrus were stained only faintly. The pyramidal cell layer and the dentate granule cell layer were unlabeled. Fiber tracts were labeled intensely (for example, fimbria, alveus, and corpus callosum). This also applied to the choroid plexus. As for the other nucleotides, the previously unstained pyramidal cell layer in CA1 had disappeared by 2 d. At 3 d a slight enhancement of 5′-nucleotidase activity became apparent in the CA1 pyramidal cell layer and in the dentate hilus (see Fig. 3H). At 7 d the upregulation of enzyme activity was apparent in all layers of CA1, whereby the pyramidal cell layer and the stratum lacunosum moleculare displayed the strongest staining. A clear enhancement of the enzyme reaction also was observed in the dentate hilus. The enhanced staining of the dentate hilus had disappeared at 28 d, but the increase in CA1 persisted. No reaction product was obtained when 5′-AMP was replaced byp-nitrophenyl phosphate as a substrate or when 5′-AMP was omitted from the incubation medium.
Comparison with the distribution of astrocytes and microglia
To identify cell types that could be responsible for the increase in nucleotidase activities, we compared—within corresponding sections—the pattern obtained by enzyme histochemistry with that for immunostaining for GFAP (astrocytes), vimentin (endothelial cells, reactive astrocytes, and activated microglia) (Graeber et al., 1988;Schmidt-Kastner et al., 1990), and complement receptor 3 (OX42, microglia). In the hippocampal formation of sham-operated (see Fig.2B,C) and untreated animals the immunostaining for vimentin and OX42 was very low. The immunoreaction for vimentin was confined to capillaries (Fig.5D). GFAP-labeled astrocytes were frequent throughout the entire hippocampus of sham-operated (see Fig. 2D) or control animals. GFAP staining was strongest in the stratum lacunosum moleculare. The pyramidal cell layer (see Figs. 2D, 5G) and the granule cell layer of the dentate gyrus (see Fig. 2D) were mainly free of GFAP positive astrocytes. GFAP-positive astroglial cells displayed a normal morphology, with small cell bodies and fine branched processes (see Figs. 2D, 5G).
Fig. 5.

Immunocytochemical detection of the postischemic glial reaction in CA1. A–C, OX42 immunoreactivity 7 d after sham operation (A) and 3 d (B) and 7 d (C) after ischemia. Arrows depict activated microglial cells that are apparent at 3 and 7 d. At 7 d the accumulation of the OX42 immunoreaction is strongest in the pyramidal cell layer.D–F, Vimentin immunoreactivity 7 d after sham operation (D) and 3 d (E) and 7 d (F) after ischemia. Capillaries are strongly labeled for vimentin (arrowheads). Vimentin-expressing cells with radiate ramifications (arrows) presumably represent reactive astrocytes, whereas rod-shaped cells presumably represent microglia.G–I, GFAP immunoreactivity 7 d after sham operation (G) and 3 d (H) and 7 d (I) after ischemia. Reactive and swollen astrocytes are clearly detectable at 3 and 7 d. Note that astrocytes do not accumulate in the pyramidal cell layer after the disappearance of neurons. ori, Stratum oriens;pyr, pyramidal cell layer; rad, stratum radiatum. Scale bar, 100 μm.
The postischemic microglial activation was detectable first at 2 d as a faint enhancement in OX42 staining of the pyramidal layer. At 3 d a distinct microglial activation appeared in the layer of damaged pyramidal cells within CA1 and in the dentate hilus (see Fig.3B). At higher magnification, activated microglia also could be detected in the stratum oriens and the stratum radiatum of CA1 (see Fig. 5B). At 7 d immunostaining for OX42 was strongest in the pyramidal cell layer and the stratum lacunosum moleculare (see Fig. 4B). Rod-shaped microglial cells were frequent in the stratum radiatum (see Fig. 5C). This general staining pattern persisted up to 28 d, with the exception of the dentate hilus, where the microglial reaction was reduced.
Ischemia-induced immunostaining for vimentin appeared first at 2 d in sparsely distributed astroglia-like processes of CA1. At 3 d vimentin expression was clearly detectable in cells with radiating processes that could represent activated astrocytes and also in smaller elongated cells that presumably represent microglia (see Figs.3C, 5E). Staining for vimentin was enhanced significantly at 7 d in CA1 and the dentate hilus (see Fig.4C). The pyramidal cell layer containing damaged neurons expressed strong vimentin immunoreactivity (see Fig.5F). At 28 d after ischemia the reaction clearly was reduced in the dentate hilus.
GFAP staining of the hippocampus was enhanced at 2 d, and at 3 d positive astrocytes displayed the swollen morphology typical for reactive astrocytes (see Figs. 3D,5H). The staining for GFAP was enhanced further at 7 d, particularly in CA1, dentate hilus, and CA3 (see Fig.4D). There was a significant difference in the postischemic expression within the pyramidal cell layer of CA1 between GFAP on the one hand and OX42 and vimentin on the other hand. The band corresponding to the dead pyramidal cells displayed a very strong immunoreactivity for OX42 and vimentin (see Figs.5C,F). GFAP-reactive astrocytes invaded this layer with increasing time but did not accumulate there (see Fig.5I). This pattern persisted at 28 d. In control experiments no immunoreaction was obtained when only the secondary antibody was applied.
Spatial distribution and the time course of the increased immunoreactivity for OX42 and vimentin within the layer of damaged pyramidal cells of CA1 correspond closely to the increase in postischemic nucleotidase activity. This suggests that the expression of nucleotidases in this region is mainly by activated microglia. Considering the lack of cellular resolution for the nucleotidase histochemistry, we also cannot, however, exclude an expression of these enzymes by reactive astrocytes. Nucleotidase activity also is upregulated distinctly in the stratum oriens, stratum radiatum, and the dentate hilus. This increase in enzyme activity is not paralleled by a comparable increase in immunoreactivity of OX42 and, rather, corresponds to the distribution of GFAP immunoreactivity (see Figs.4D, 5I).
DISCUSSION
Neural death and upregulation of glial markers after transient two-vessel occlusion
The two-vessel or the four-vessel occlusion models are used when transient interruption of blood flow and reperfusion are required. The two models yield similar neuronal injury in the hippocampus, including the CA1 and CA4 pyramidal cells and the subiculum (Ginsberg and Busto, 1989). Our results obtained for the markers GFAP, vimentin, and OX42 correspond closely to those previously described by others, using the four-vessel occlusion model and similar occlusion times. The immunoreactivity for the microglial marker OX42 develops progressively during the postischemic time period. It is restricted mainly to the band of injured pyramidal cell bodies and to a lesser extent to the stratum lacunosum moleculare and the dentate gyrus (Morioka et al., 1992; Jørgensen et al., 1993; Kato et al., 1995). The hippocampal distribution of vimentin is very similar to that of OX42 immunoreactivity. Several studies on transient cerebral ischemia have demonstrated an increase in immunoreactivity for vimentin in the lesioned neuronal layers and have interpreted it as an astrocytic reaction. In contrast to vimentin, the immunoreactivity for the astrocytic marker GFAP also is prominent throughout the hippocampus in unlesioned animals. Ischemia causes a time-dependent increase in GFAP immunoreactivity within the entire hippocampus, particularly in CA1 and dentate hilus (Petito et al., 1990; Schmidt-Kastner et al., 1990;Jørgensen et al., 1993). Prolonged postischemic survival time results in the immigration of GFAP-positive astrocytes into the damaged pyramidal cell layer of CA1, but the immunoreactivity never becomes as intense as that for OX42 and vimentin.
Glial upregulation of ectonucleotidases
Northern analysis demonstrates an ischemia-induced upregulation of ectoapyrase and ecto-5′-nucleotidase. Together, the two enzymes can account for the entire hydrolysis chain from ATP to adenosine. Ecto-5′-nucleotidase is bound to the membrane via a glycosyl phosphatidylinositol (GPI) anchor. Ectoapyrase represents a transmembrane protein with two hydrophobic domains at the N- and C-terminal ends and the catalytic site facing the extracellular space. Both enzymes have a very broad substrate specificity toward nucleoside 5′-di- and tri- and monophosphates, respectively (Zimmermann, 1996). ATP, UTP, and ADP thus could be hydrolyzed by a single enzyme (ectoapyrase). These results suggest that the histochemical staining pattern observed in the injured regions is attributable to the activity of two defined enzymes, ectoapyrase and ecto-5′-nucleotidase.
Hydrolysis of nucleoside 5′-tri- and diphosphates was very low in the hippocampal subfield of unlesioned animals. Staining for 5′-nucleotidase activity was strong in CA1 and within fiber tracts. This corresponded to earlier histochemical analyses of 5′-nucleotidase in the rat hippocampus (Lee et al., 1986; Fastbom et al., 1987). With all substrate nucleotides, ischemia induced an increase in staining intensity in select hippocampal regions. Its temporal development corresponded to that of markers for activated glia. There was a strong and continued increase in the entire CA1 subfield, with particular emphasis on the layer of damaged pyramidal cell bodies and the stratum lacunosum moleculare. Enzyme activity also was upregulated within the dentate hilus and to a small extent in CA3. CA1 neurons, along with some hilar neurons, are considered to represent the most vulnerable neurons of the hippocampus. The strong focus of histochemical reaction product in the pyramidal layer and the stratum lacunosum moleculare suggests that enzyme activity there is attributable mainly to activated microglia. The more diffuse upregulation in the other areas could result from both activated microglia and astrocytes. In our previous study applying permanent MCAO, staining for 5′-nucleotidase at the immediate rim of the infarcted tissue corresponded to the distribution of binding sites for OX42, whereas the more distant and less intensive staining corresponded to the distribution of GFAP (Braun et al., 1997).
Hydrolysis of inosine 5′-diphosphate (IDP) previously has been allocated to the surface of microglia and subsequently has been used as a microglial marker. The ischemia-induced increase in staining for IDP as a substrate (Finsen et al., 1993) is similar to that observed for ATP, UTP, and ADP as substrates. This would be expected if all substrates were hydrolyzed by the same enzyme (ectoapyrase). The presence of the extracellular hydrolysis chain from ATP to adenosine previously has been demonstrated in astrocytic cultures (Lai and Wong, 1991) and hippocampal slices (Wieraszko et al., 1989; Cunha et al., 1996; Dunwiddie et al., 1997). The activity of ectonucleotidases also is associated with isolated synaptosomes (Nagy et al., 1986) and cultured neural cells (Stefanovic et al., 1976). We have no indication for an upregulation of ectoapyrase and ecto-5′-nucleotidase on neural cells or their processes.
Upregulation of ectonucleotidases and nucleotide release
The upregulation of ectonucleotidases implies increased nucleotide release. ATP can be released from a large variety of cells and mechanisms of membrane passage other than exocytosis may be involved (Cantiello, 1997). Tissue injury or hypoxia alone can release ATP and other adenine nucleotides from a variety of tissues, including the brain (Dubyak and El-Moatassim, 1993; Lutz and Kabler, 1997). Transient global ischemia resulting in the alteration of both composition and flow rate of the blood may induce osmotic stress with changes in cell volume (van der Toorn et al., 1996). Increases in cell volume can lead, in turn, to the cellular efflux of ATP. ATP is released from HTC rat hepatoma cells by hypotonic exposure and subsequently is required for recovery from swelling (Wang et al., 1996). Because this effect is attenuated by enzymes hydrolyzing ATP, an increase in ectonucleotidase activity would be damaging rather than protective. The increase in ectonucleotidase activity may be induced directly by neural cell damage or indirectly by general homeostatic shifts. Because the increase is not global but is restricted to sites of intense neural damage, it appears to be correlated directly with neural cell death. Its onset coincides with that of significant neural cell death, which becomes apparent at the microscopic level after 2 d (Sweeney et al., 1995).
Significance of increased ectonucleotidase activity
A major factor for neuron survival is the efficient replenishment of the depleted ATP stores. An effective extracellular hydrolysis of released ATP to adenosine would provide the nucleoside for purine salvage via carrier-mediated cellular uptake. There is a wealth of literature in support of a neuroprotective function of extracellular adenosine in attenuating the release of cytotoxic amino acids or as a vasodilating agent (Rudolphi et al., 1992; Sweeney, 1997). ATP, on the other hand, may be cytotoxic. A role of ATP as a fast neurotransmitter as well as a neuromodulator has been demonstrated for several central and peripheral synapses (Gibb and Halliday, 1996). When released in large amounts and for an extended period of time, it may cause a P2X receptor-mediated buildup of intracellular calcium that would be equally damaging as that induced by excess glutamate (Edwards, 1996). Studies on hippocampal neurons suggest that glutamate itself may stimulate the release of ATP, which in turn can act as a transmitter and induce an increase in [Ca]i via P2 receptors. This increase also can be elicited in the presence of high Mg2+ concentrations and thus can evoke membrane depolarization that removes Mg2+ blocking of the NMDA receptor. ATP therefore even could potentiate the cytotoxic effects of glutamate (Inoue et al., 1995). Upregulated ectonucleotidases would counteract this effect.
Ectonucleotidase activity also could be relevant for glial cells. The reactive glial cells may have a demand for increased energy supply and thus may reinforce the extracellular purine salvage pathway. In addition, nucleotides can affect glial cells directly. Cultured astrocytes and microglia express metabotropic as well as ionotropic receptors for nucleotides (Walz et al., 1994; Nörenberg et al., 1997). ATP or also UTP mediates an increase in intracellular Ca2+ concentrations in cultured astrocytes in vitro (Czubayko and Reiser, 1996; Centemeri et al., 1997).In vitro ischemia has been found to promote Ca2+ influx and intracellular Ca2+ release in hippocampal astrocytes (Duffy and MacVicar, 1996). ATP or adenosine can stimulate the proliferation of astrocytes (Abbracchio, 1996). ATP induces astrogliosis in vitro, including an increase in expression of GFAP, promotion of astrocytic hypertrophy, and elongation of cellular processes (Bolego et al., 1997). Extracellular GTP increases the synthesis and release of NGF from cultured astrocytes (Middlemiss et al., 1995).
Microglial cells are endangered by ATP. They express the cytolytic P2z (P2X7) subtype of ATP receptors as do peripheral macrophages (Collo et al., 1997; Ferrari et al., 1997a). Activation of this receptor mediates a reversible permeabilization of the plasma membrane that causes the opening of a pore associated with ion fluxes and the leakage of small metabolites such as nucleotides. Sustained exposure to ATP would be a potent cytolytic stimulus. Microglial cell lines release mediators of inflammation, including interleukin-1β, when stimulated with bacterial endotoxins, and this release can be inhibited by the blockade of P2X7 receptors (Ferrari et al., 1997b). The upregulation of ectoapyrase on activated microglia may be a protective reaction.
In summary, our data suggest that transient forebrain ischemia results in an upregulation of the enzyme chain for the complete hydrolysis of extracellular ATP and other nucleoside 5′-triphosphates in the damaged hippocampal areas. Enzyme activity appears to be associated mainly with reactive glia. The increase in ectonucleotidase activity implies increased nucleotide release and could play a role in the postischemic control of nucleotide-mediated cellular responses.
Footnotes
This study was supported by Grants from the Deutsche Forschungsgemeinschaft (SFB 269, A4), the European Community (BMH4-CT96-0676), and the Fonds der Chemischen Industrie. We are grateful to Klaus Hammer for expert technical support. Rat ecto-5′-nucleotidase cDNA was kindly provided by Dr. Y. Ikehara, Fukuoka University, Japan.
Correspondence should be addressed to Dr. Norbert Braun, Biozentrum der J.W. Goethe-Universität, AK Neurochemie, Zoologisches Institut, Marie-Curie-Strasse 9, D-60439 Frankfurt am Main, Germany.
REFERENCES
- 1.Abbracchio MP. P1 and P2 receptors in cell growth and differentiation. Drug Dev Res. 1996;39:393–406. [Google Scholar]
- 2.Boarder MR, Weisman GA, Turner JT, Wilkinson GF. G-protein-coupled P2 purinoceptors: from molecular biology to functional responses. Trends Pharmacol Sci. 1995;16:133–139. doi: 10.1016/s0165-6147(00)89001-x. [DOI] [PubMed] [Google Scholar]
- 3.Bolego C, Ceruti S, Brambilla R, Puglisi L, Cattabeni F, Burnstock G, Abbracchio MP. Characterization of the signalling pathways involved in ATP and basic fibroblast growth factor-induced astrogliosis. Br J Pharmacol. 1997;121:1692–1699. doi: 10.1038/sj.bjp.0701294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braun N, Lenz C, Gillardon F, Zimmermann M, Zimmermann H. Focal cerebral ischemia enhances glial expression of 5′-nucleotidase. Brain Res. 1997;766:213–226. doi: 10.1016/s0006-8993(97)00559-3. [DOI] [PubMed] [Google Scholar]
- 5.Buell G, Collo G, Rassendren F. P2X receptors: an emerging channel family. Eur J Neurosci. 1996;8:2221–2228. doi: 10.1111/j.1460-9568.1996.tb00745.x. [DOI] [PubMed] [Google Scholar]
- 6.Cantiello HF. Nucleotide transport through the cystic fibrosis transmembrane conductance regulator. Biosci Rep. 1997;17:147–171. doi: 10.1023/a:1027381412574. [DOI] [PubMed] [Google Scholar]
- 7.Centemeri C, Bolego C, Abbracchio MP, Cattabeni F, Puglisi L, Burnstock G, Nicosia S. Characterization of the Ca2+ responses evoked by ATP and other nucleotides in mammalian brain astrocytes. Br J Pharmacol. 1997;121:1700–1706. doi: 10.1038/sj.bjp.0701293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collo G, Neidhart S, Kawashima E, Kosco-Vilbois M, North RA, Buell G. Tissue distribution of the P2X7 receptor. Neuropharmacology. 1997;36:1277–1283. doi: 10.1016/s0028-3908(97)00140-8. [DOI] [PubMed] [Google Scholar]
- 9.Cunha RA, Vizi ES, Ribeiro JA, Sebastiao AM. Preferential release of ATP and its extracellular catabolism as a source of adenosine upon high- but not low-frequency stimulation of rat hippocampal slices. J Neurochem. 1996;67:2180–2187. doi: 10.1046/j.1471-4159.1996.67052180.x. [DOI] [PubMed] [Google Scholar]
- 10.Czubayko U, Reiser G. P2u nucleotide receptor activation in rat glial cell line induces [Ca2+]i oscillations which depend on cytosolic pH. Glia. 1996;16:108–116. doi: 10.1002/(SICI)1098-1136(199602)16:2<108::AID-GLIA3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Dubyak GR, El-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 12.Duffy S, MacVicar BA. In vitro ischemia promotes calcium influx and intracellular calcium release in hippocampal astrocytes. J Neurosci. 1996;16:71–81. doi: 10.1523/JNEUROSCI.16-01-00071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunwiddie TV, Diao L, Proctor WL. Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J Neurosci. 1997;17:7673–7682. doi: 10.1523/JNEUROSCI.17-20-07673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards FA. Features of P2X receptor-mediated synapses in the rat brain: why doesn’t ATP kill the postsynaptic cells? In: Chadwick DJ, editor. P2 purinoceptors: localization, function, and transduction mechanisms. Wiley; Chichester, UK: 1996. pp. 278–289. [PubMed] [Google Scholar]
- 16.Fastbom J, Pazos A, Palacios JM. The distribution of adenosine A1 receptors and 5′-nucleotidase in the brain of some commonly used experimental animals. Neuroscience. 1987;22:813–826. doi: 10.1016/0306-4522(87)92961-7. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Collo G, Buell G, Di Virgilio F. ATP-mediated cytotoxicity in microglial cells. Neuropharmacology. 1997a;36:1295–1301. doi: 10.1016/s0028-3908(97)00137-8. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1β release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997b;159:1451–1458. [PubMed] [Google Scholar]
- 19.Finsen BR, Jørgensen MB, Diemer NH, Zimmer J. Microglial MHC antigen expression after ischemic and kainic acid lesions of the adult rat hippocampus. Glia. 1993;7:41–49. doi: 10.1002/glia.440070109. [DOI] [PubMed] [Google Scholar]
- 20.Gehrmann J, Banati RB, Wiessner C, Hossmann KA, Kreutzberg GW. Reactive microglia in cerebral ischaemia: an early mediator of tissue damage? Neuropathol Appl Neurobiol. 1995;21:277–289. doi: 10.1111/j.1365-2990.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 21.Gibb AJ, Halliday FC. Fast purinergic transmission in the central nervous system. Semin Neurosci. 1996;8:225–232. [Google Scholar]
- 22.Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- 23.Graeber MB, Streit WJ, Kreutzberg GW. The microglial cytoskeleton: vimentin is localized within activated cells in situ. J Neurocytol. 1988;17:573–580. doi: 10.1007/BF01189811. [DOI] [PubMed] [Google Scholar]
- 24.Inoue K, Koizumi S, Nakazawa K. Glutamate-evoked release of adenosine 5′-triphosphate causing an increase in intracellular calcium in hippocampal neurones. NeuroReport. 1995;6:437–440. doi: 10.1097/00001756-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Jørgensen MB, Finsen BR, Jensen MB, Castellano B, Diemer NH, Zimmer J. Microglial and astroglial reactions to ischemic and kainic acid-induced lesions of the adult rat hippocampus. Exp Neurol. 1993;120:70–88. doi: 10.1006/exnr.1993.1041. [DOI] [PubMed] [Google Scholar]
- 26.Kato H, Kogure K, Araki T, Itoyama Y. Graded expression of immunomolecules on activated microglia in the hippocampus following ischemia in a rat model of ischemic tolerance. Brain Res. 1995;694:85–93. doi: 10.1016/0006-8993(95)00769-m. [DOI] [PubMed] [Google Scholar]
- 27.Kegel B, Braun N, Heine P, Maliszewski CR, Zimmermann H. An ecto-ATPase and an ecto-ATP diphosphohydrolase are expressed in rat brain. Neuropharmacology. 1997;36:1189–1200. doi: 10.1016/s0028-3908(97)00115-9. [DOI] [PubMed] [Google Scholar]
- 28.Kraig RP, Lascola CD, Caggiano A. Glial response to brain ischemia. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; Oxford: 1995. pp. 964–976. [Google Scholar]
- 29.Lai KM, Wong PCL. Metabolism of extracellular adenine nucleotides by cultured rat brain astrocytes. J Neurochem. 1991;57:1510–1515. doi: 10.1111/j.1471-4159.1991.tb06345.x. [DOI] [PubMed] [Google Scholar]
- 30.Lee KS, Schubert P, Reddington M, Kreutzberg GW. The distribution of adenosine A1 receptors and 5′-nucleotidase in the hippocampal formation of several mammalian species. J Comp Neurol. 1986;246:427–434. doi: 10.1002/cne.902460402. [DOI] [PubMed] [Google Scholar]
- 31.Lutz PL, Kabler S. Release of adenosine and ATP in the brain of the freshwater turtle (Trachemys scripta) during long-term anoxia. Brain Res. 1997;769:281–286. doi: 10.1016/s0006-8993(97)00719-1. [DOI] [PubMed] [Google Scholar]
- 32.Maliszewski CR, DeLepesse GJT, Schoenborn MA, Armitage RJ, Fanslow WC, Nakajima T, Baker E, Sutherland GR, Poindexter K, Birks C, Alpert A, Friend D, Gimpel SD, Gayle RB. The CD39 lymphoid cell activation antigen: molecular cloning and structural characterization. J Immunol. 1994;153:3574–3583. [PubMed] [Google Scholar]
- 33.Middlemiss PJ, Gysbers JW, Rathbone MP. Extracellular guanosine and guanosine-5′-triphosphate increase: NGF synthesis and release from cultured mouse neopallial astrocytes. Brain Res. 1995;677:152–156. doi: 10.1016/0006-8993(95)00156-k. [DOI] [PubMed] [Google Scholar]
- 34.Milligan CE, Cunningham TJ, Levitt P. Differential immunochemical markers reveal the normal distribution of brain macrophages and microglia in the developing rat brain. J Comp Neurol. 1991;314:125–135. doi: 10.1002/cne.903140112. [DOI] [PubMed] [Google Scholar]
- 35.Misumi Y, Ogata S, Hirose S, Ikehara Y. Primary structure of rat liver 5′-nucleotidase deduced from the cDNA. Presence of the COOH-terminal hydrophobic domain for possible post-translational modification by glycophospholipid. J Biol Chem. 1990;265:2178–2183. [PubMed] [Google Scholar]
- 36.Morioka T, Kalehua AN, Streit WJ. Progressive expression of immunomolecules on microglial cells in rat dorsal hippocampus following transient forebrain ischemia. Acta Neuropathol (Berl) 1992;83:149–157. doi: 10.1007/BF00308474. [DOI] [PubMed] [Google Scholar]
- 37.Nagy AK, Shuster TA, Delgado-Escueta AV. Ecto-ATPase of mammalian synaptosomes: identification and enzymic characterization. J Neurochem. 1986;47:976–986. doi: 10.1111/j.1471-4159.1986.tb00707.x. [DOI] [PubMed] [Google Scholar]
- 38.Nörenberg W, Cordes A, Blohbaum G, Fröhlich R, Illes P. Coexistence of purino- and pyrimidinoceptors on activated rat microglial cells. Br J Pharmacol. 1997;121:1087–1098. doi: 10.1038/sj.bjp.0701241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petito CK, Morgello S, Felix JC, Lesser ML. The two patterns of reactive astrocytosis in postischemic rat brain. J Cereb Blood Flow Metab. 1990;10:850–859. doi: 10.1038/jcbfm.1990.141. [DOI] [PubMed] [Google Scholar]
- 40.Robinson AP, White TM, Mason DW. Macrophage heterogeneity in the rat as delineated by two monoclonal antibodies MRC OX-41 and MRC OX-42, the latter recognizing complement receptor type 3. Immunology. 1986;57:239–247. [PMC free article] [PubMed] [Google Scholar]
- 41.Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol Sci. 1992;13:439–445. doi: 10.1016/0165-6147(92)90141-r. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt-Kastner R, Szymas J, Hossmann K-A. Immunohistochemical study of glial reaction and serum–protein extravasation in relation to neuronal damage in rat hippocampus after ischemia. Neuroscience. 1990;38:527–540. doi: 10.1016/0306-4522(90)90048-9. [DOI] [PubMed] [Google Scholar]
- 43.Schubert P, Kreutzberg GW. Cerebral protection by adenosine. Acta Neurochir Suppl (Wien) 1993;57:80–88. doi: 10.1007/978-3-7091-9266-5_12. [DOI] [PubMed] [Google Scholar]
- 44.Smith M-L, Bendek G, Dahlgren IR, Wieloch T, Siesjö BK. Models for studying long-term recovery following forebrain ischemia in the rat. II. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- 45.Stefanovic V, Ledig M, Mandel P. Divalent cation-activated ecto-nucleoside triphosphatase activity of nervous system cells in tissue culture. J Neurochem. 1976;27:799–805. doi: 10.1111/j.1471-4159.1976.tb10411.x. [DOI] [PubMed] [Google Scholar]
- 46.Sweeney MI. Neuroprotective effects of adenosine in cerebral ischemia, window of opportunity. Neurosci Biobehav Rev. 1997;21:207–217. doi: 10.1016/s0149-7634(96)00011-5. [DOI] [PubMed] [Google Scholar]
- 47.Sweeney MI, Yager JY, Walz W, Juurlink BHJ. Cellular mechanisms involved in brain ischemia. Can J Physiol Pharmacol. 1995;73:1525–1535. doi: 10.1139/y95-211. [DOI] [PubMed] [Google Scholar]
- 48.van der Toorn A, Sykova E, Dijkhuizen RM, Vorisek I, Vargova L, Skobisova E, van Lookeren Campagne M, Reese T, Nicolay K. Dynamic changes in water ADC, energy metabolism, extracellular space volume, and tortuosity in neonatal brain during global ischemia. Magn Reson Med. 1996;36:52–60. doi: 10.1002/mrm.1910360110. [DOI] [PubMed] [Google Scholar]
- 49.Walz W, Gimpl G, Ohlemeyer C, Kettenmann H. Extracellular ATP-induced currents in astrocytes—involvement of a cation channel. J Neurosci Res. 1994;38:12–18. doi: 10.1002/jnr.490380104. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y, Roman R, Lidovsky S, Fitz JG. Autocrine signaling through ATP release represents a novel mechanism for volume regulation. Proc Natl Acad Sci USA. 1996;93:12020–12025. doi: 10.1073/pnas.93.21.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wieraszko A, Goldsmith G, Seyfried TN. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989;485:244–250. doi: 10.1016/0006-8993(89)90567-2. [DOI] [PubMed] [Google Scholar]
- 52.Zimmermann H. Extracellular purine metabolism. Drug Dev Res. 1996;39:337–352. [Google Scholar]