

Abstract
In this report, we have tested the hypothesis that brain-derived neurotrophic factor (BDNF) is an anterograde neurotrophic factor in the CNS and have focused on central noradrenergic neurons that synthesize BDNF. Double-label immunocytochemistry for BDNF and dopamine-β-hydroxylase (DBH), a marker for noradrenergic neurons, demonstrated that BDNF is partially localized to noradrenergic nerve fibers and terminals in the adult rat brain. To test the functional importance of this anterograde BDNF, we analyzed transgenic mice carrying a DBH–BDNF minigene. Increased synthesis of BDNF in noradrenergic neurons of DBH–BDNF mice caused elevated TrkB tyrosine kinase activation throughout postnatal life in the neocortex, a noradrenergic target region. This afferently regulated increase in TrkB receptor activity led to long-lasting alterations in cortical morphology. To determine whether noradrenergic neuron-expressed BDNF also anterogradely regulated neuronal survival, we examined a second noradrenergic target, neonatal facial motoneurons. One week after axotomy, 72% of facial motoneurons were lost in control animals, whereas only 30–35% were lost in DBH–BDNF transgenic mice. Altogether, these results indicate that BDNF is anterogradely transported to fibers and terminals of noradrenergic neurons, that anterogradely secreted BDNF causes activation of TrkB in target regions, and that this secretion has functional consequences for target neuron survival and differentiation. This presynaptic secretion of BDNF may provide a cellular mechanism for modulating neural circuitry, in either the developing or mature nervous system.
Keywords: BDNF, noradrenergic neurons, anterograde neurotrophic factors, cerebral cortex, motor neurons, neuronal regeneration, neuronal development
The neurotrophic factor hypothesis postulates that trophic factors produced and released by target neurons regulate the survival and differentiation of their innervating neurons (for review, see Oppenheim, 1991). Developing neurons, however, may be equally dependent for survival and differentiation on afferent neuronal input (for review, see Linden, 1994). For example, deafferentation can increase the number of neurons lost during the period of naturally occurring cell death (Sohol and Narayahan, 1975; Clarke, 1985; Furber et al., 1987; Linden and Pinon, 1987), and hyperinnervation can increase the survival of target neurons (Cunningham et al., 1979). These findings suggest the existence of anterogradely released trophic factors that are functionally similar to retrogradely acting growth factors such as nerve growth factor (for review, see Levi-Montalcini, 1987).
One neuronal population that may play an afferent trophic role during development is brainstem noradrenergic neurons, which innervate a large number of CNS structures, including the cerebral cortex (Jones and Moore, 1977; for review, see Foote et al., 1983). These central noradrenergic neurons are born from embryonic day (E) 10 to E13 (Lauder and Bloom, 1974), and they project axons into targets such as the neocortex while cortical neurons are still undergoing birth, migration, and differentiation (Altman and Bayer, 1990; Bayer and Altman, 1990). A trophic role for these early afferents is supported by lesion studies that demonstrate that the loss of noradrenergic afferents during development alters both the number and morphology of cortical neurons (Maeda et al., 1974; Felten et al., 1982).
The nature of the signal responsible for the trophic effects of noradrenergic afferents is still unknown. However, locus coeruleus neurons make brain-derived neurotrophic factor (BDNF) (Castren et al., 1995; Conner et al., 1997), a member of the neurotrophin family (Barde et al., 1982; Leibrock et al., 1989) known to have effects on many CNS populations, including the developing cortex (Ghosh et al., 1994; Nawa et al., 1994; Cabelli et al., 1995; McAllister et al., 1995, 1996). Moreover, recent evidence indicates that neurotrophins, including BDNF, can be anterogradely transported in neurons. Specifically, (1) exogenous neurotrophins can be anterogradely transported in the developing chick CNS (von Bartheld et al., 1996); (2) BDNF is present in axons and potentially in terminals of peripheral (Zhou and Rush, 1996; Michael et al., 1997), and central neurons (Fawcett et al., 1997;Conner et al., 1997); (3) BDNF is localized to vesicles in presynaptic terminals (Fawcett et al.,1997) in the CNS; and (4) neurotrophins can be synthesized and released in an activity-dependent manner (for review, see Thoenen, 1995). Finally, TrkB tyrosine kinase receptors have recently been localized to neuronal dendrites in both the hippocampus and cortex (Fryer et al., 1996; Yan et al., 1997a).
On the basis of these recent findings, we hypothesized that BDNF may be a trophic signal derived from noradrenergic afferents. In this paper, we have tested this hypothesis and demonstrate that BDNF is localized to axons and terminals of noradrenergic neurons, that increased BDNF secretion within the cortex from noradrenergic afferents can lead to long-lasting changes in cortical organization, and that increased secretion of BDNF onto developing target neurons can modify neuronal survival itself.
MATERIALS AND METHODS
Animals and surgical procedures. The DBH–BDNF mice used in these studies, lines D481 and D498, have been described previously (Causing et al., 1997), and were bred and genotyped as described. As controls for these transgenic mice, we used either littermates or animals of the same genetic background that were null for the DBH–BDNF transgene. Mice heterozygous for a targeted mutation in the BDNF gene (Ernfors et al., 1994) were obtained from Jackson Laboratory (Bar Harbor, ME) and maintained, bred, and genotyped as described previously (Causing et al., 1997).
For the TrkB and BDNF biochemical analysis, adult or 1 week old DBH–BDNF, BDNF+/−, or appropriate wild-type controls were decapitated. For immunocytochemistry, animals were killed with sodium pentobarbital (65 mg/kg) and then transcardially perfused with 4% paraformaldehyde or 4% paraformaldehyde/15% picric acid in phosphate buffer (PB), pH 7.4. Brains were then removed and post-fixed in 4% paraformaldehyde or 4% paraformaldehyde/15% picric acid in PB for 30 min at 4°C and subsequently cleared of picric acid with PBS, pH 7.4. Alternatively, rats were transcardially perfused with 37°C heparinized PBS followed by 2% paraformaldehyde in 0.1 mPB, pH 7.3, and were then post-fixed for 2 hr at 4°C. All sections were cryoprotected in graded sucrose solutions and sectioned on the cryostat before immunocytochemical analysis.
For the facial motoneuron studies, postnatal day 5 wild-type or transgenic animals were anesthetized by inhalation with metofane to affect. Alternatively, adult female BDNF heterozygote mice ranging in age from 2.5 to 3 months were anesthetized using sodium pentobarbital (35 mg/kg). After complete anesthetization (in accordance with an animal care protocol meeting the standards of the Canadian Council on Animal Care), the facial nerve was transected unilaterally at the stylomastoid foramen. One week after nerve transection, young or adult animals were perfused with PBS, pH 7.4, and 4% paraformaldehyde in 0.1m NaH2PO4, after which the brains were removed and post-fixed overnight in the same fixative at 4°C. Brains were then cryoprotected overnight in 30% sucrose and sectioned on a cryostat.
Histological and morphometric analysis. For morphometric analysis of the neocortex, 16 μm sections were stained using cresyl violet, and neuronal counts were performed in 528-μm-wide strips extending from corpus callosum to pia, in two separate regions of the forebrain.
For the analysis of facial motoneuron size and number, 16 μm serial coronal sections were collected throughout the extent of the facial motor nuclei and stained with cresyl violet. For size determination, neuronal profiles containing a nucleus with a distinct nucleolus were displayed on a video screen, and the cellular cross-sectional areas were measured using a computer-based image analysis system (Biocom). For neuronal numbers, all neurons containing a nucleus in the facial nuclei were counted on every fifth serial 16 μm section using the same image analysis system, which prevents double measurements of profiles. The numbers obtained using this approach are not corrected for split nucleoli. Results were expressed as mean values ± SEM and were tested for significance using the one-tailed Student’st test.
Antibodies, immunoprecipitations, and Western blot analysis.The following, previously described antibodies were used for the biochemical studies described here: anti-TrkBin, which specifically recognizes the intracellular domain of TrkB (Fryer et al., 1996); anti-TrkBout, which specifically recognizes the extracellular domain of full-length and truncated TrkB (Knusel et al., 1994); anti-panTrk 203, which recognizes the intracellular domain of all Trk family receptors (Hempstead et al., 1992); anti-BDNF, which recognizes mature BDNF (Santa Cruz Biotechnology, Santa Cruz, CA) (Causing et al., 1997;Fawcett et al., 1997); and antiphosphotyrosine 4G10 (UBI).
For biochemical analysis of TrkB, different regions of 1-week-old and adult mouse brains were dissected out, homogenized, and lysed in Tris-buffered saline (TBS) lysis buffer (Knusel et al.,1994) containing 137 mm NaCl, 20 mm Tris, pH 8.0, 1% (v/v) NP-40, 10% (v/v) glycerol, 1 mm phenylmethyl sulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, and 1.5 mm sodium vanadate. Total Trk protein was immunoprecipitated using 3 μl of anti-panTrk 203. The immunoprecipitates were collected with Protein A-Sepharose (Pharmacia, Dorval, Québec, Canada) for 1.5 hr at 4°C followed by centrifugation. Immunoprecipitates were then washed three times with cold lysis buffer, boiled in sample buffer (2% SDS, 100 mmdithiothreitol, 10% glycerol, and 0.05% bromophenol blue) for 5 min, and electrophoresed on 7.5% SDS-polyacrylamide minigels. After electrophoresis, proteins were transferred to 0.2 μm nitrocellulose for 1 hr at 0.5 amps, and the membranes were washed 2 × 10 min in TBS. For all antibodies except antiphosphotyrosine, for which membranes were blocked in 2% BSA (Sigma, St. Louis), membranes were blocked in 5% nonfat dry milk in TBS for 1.5 hr. Membranes were then washed 2 × 10 min in TBS, and the primary antibodies were used overnight at 4°C at dilutions of 1:10,000 for antiphosphotyrosine 4G10 (UBI), 1:2000 for anti-panTrk 203, 1:5000 for anti-TrkBout, and 1:2000 for anti-TrkBin. Secondary antibodies were incubated for 1.5 hr at room temperature and were used at dilutions of 1:10,000 for a goat anti-mouse horse radish peroxidase (HRP) antibody and 1:10,000 for a goat anti-rabbit HRP antibody (Boehringer Mannheim, Mannheim, Germany). Detection was performed using enhanced chemiluminescence (Amersham, Oakville, Ontario, Canada) and XAR x-ray film (Kodak). Results were quantitated by image analysis, and statistical significance was determined using Student’s t test.
For biochemical analysis of BDNF, brain lysates were boiled in sample buffer, and 20–100 μg of protein was separated on a 15% SDS-polyacrylamide minigel, as described previously (Causing et al., 1997; Fawcett et al., 1997). After transfer, the nitrocellulose was blotted with an antibody specific to BDNF (Santa Cruz) at a 1:5000 dilution, using the protocols described above. As controls, we used human recombinant BDNF (obtained from Amgen, Thousand Oaks, CA), and an extract of PC12 cells infected with vaccinia virus encoding BDNF (Causing et al., 1997).
Immunocytochemistry. Two different BDNF antibodies were used for the immunocytochemical studies (the Santa Cruz antibody that was used for Western blots does not work for immunocytochemistry). One, an anti-peptide antibody, was used for detection of BDNF synthesis in the DBH–BDNF transgenic mice and has previously been shown to work for immunocytochemistry (Patterson et al., 1996) (W. Friedman and D. Kaplan, unpublished data). A second, more sensitive BDNF antibody (RAB) was used for the BDNF–DBH double-labeling studies. This antibody has previously been characterized (Yan et al., 1997b) and demonstrated to detect BDNF in nerve fibers and terminals (Conner et al., 1997; Fawcett et al., 1997; Yan et al., 1997b) and was the kind gift of Dr. A. Welcher (Amgen). A commercially available antibody (EugeneTech) was used to detect DBH immunocytochemically. For fluorescence immunocytochemical detection of BDNF and DBH in locus coeruleus neurons, 14-μm-thick cryosections on slides were post-fixed with 4% paraformaldehyde in 0.1 m phosphate buffer for 2 min at room temperature and washed 2 × 10 min in 1 mHEPES-buffered saline (HBS), pH 7.4. Sections were permeabilized with HBS plus 0.2% Triton X-100, blocked with 3% serum for 45 min at room temperature, and incubated with the primary antibody in HBS plus 0.2% Triton X-100 plus 3% serum overnight at 4°C at a dilution of 1:500 for anti-BDNF and 1:600 for anti-DBH. Slides were then washed 3 × 15 min in HBS. Binding of the primary antibody was visualized with a rhodamine-conjugated secondary antibody (goat anti-rabbit IgG at a 1:200 dilution; Jackson). Slides were then washed, mounted with a 10% glycerol, 90% PBS solution, and coverslipped. In all cases, control and transgenic tissue were prepared at the same time and analyzed on the same slides.
For immunoperoxidase detection of DBH, the primary antibody was used as above (1:600 dilution; Eugene Tech), with the exception that DBH was visualized using a biotin-conjugated secondary antibody (goat anti-rabbit dilution 1:200; Vector Laboratories, Burlingame, CA), and PBS, pH 7.4, was used as a buffer. Slides were then washed as before and incubated with the ABC complex (Vector) for 1 hr at room temperature and then with DAB for ∼5 min, or until desired staining was reached. In all cases, control and transgenic tissue were prepared at the same time and analyzed on the same slides.
For the double-label analysis of BDNF and DBH, 10-μm-thick sections were blocked in PB, pH 7.4, containing 10% normal goal serum and 0.2% Triton X-100 for 1 hr. BDNF (Amgen) and DBH antibodies were then added to the blocking solution at dilutions of 1:5000 and 1:1000, respectively, for 24 hr at 4°C. Sections were washed three times in the blocking solution and then incubated for 1 hr in blocking solution containing an anti-rabbit CY3-conjugated antibody to detect anti-BDNF and an anti-mouse CY2-conjugated antibody to detect anti-DBH, both used at a dilution of 1:1000. Sections were then washed in PBS and mounted in 50% glycerol in PBS containing 2% Dabco. Sections were analyzed and photographed on a Zeiss Axioscope, using filters that were designed for double-label analysis using these two fluorochromes.
Quantitation of the relative level of immunoreactivity for BDNF in locus coeruleus neurons from DBH–BDNF versus control animals was performed using image analysis. Five different pairs of line D481 versus control animals were analyzed. Specifically, transgenic and control neurons were analyzed by immunofluorescence at the same time (and in many cases, on the same slide) and were photographed using the same exposure and developing times. These photographic images were scanned, and the relative fluorescence intensity was measured and expressed as mean optical density per neuron. The background level of fluorescence, measured lateral to the locus coeruleus neuron cluster, was subtracted from the values obtained for BDNF-positive cells. Statistical significance was determined using Student’s ttest.
Measurement of tissue monoamines. Animals were decapitated swiftly with a guillotine, and their brains were removed quickly and frozen at −80°C. The brains were later placed on a cold plate to thaw, and a series of 1.0- to 1.5-mm-thick sections (usually four to five) were cut. The brains from six wild-type and six transgenic mice were dissected over the cold plate under microscopic observation, as described previously (Reader and Grondin, 1987; Reader et al., 1989), and the following eight regions were taken for the monoamine assays: frontal and entorhinal-piriform cortices, neostriatum (caudate-putamen), thalamus, hypothalamus, hippocampus, cerebellum, and brainstem. The discrete tissue samples (10–20 mg wet weight) were placed in tubes already containing 1–2 ml of 0.1N cold monochloroacetic acid with 2.15 mm Na2EDTA and disrupted in a glass homogenizer with a Teflon pestle, and the homogenate was centrifuged at 39,000 × g for 45 min at 4°C. The pellets were dissolved overnight in 0.5 ml of 1N NaOH for protein determinations (Lowry et al., 1951), and the supernatants were assayed by HPLC with electrochemical detection, following well established procedures (Lakhdar-Ghazal et al., 1986; Sauve and Reader, 1988). Briefly, the supernatants were filtered through 0.45 μm pores (GS; Millipore, Bedford, MA) and injected into the 3-μm-particle-size chromatographic column (100.0 × 4.1 mm) (Adsorbosphere Catecholamine; Alltech Associates, Deerfield, IL). The isocratic mobile phase was 0.1N monochloroacetic acid adjusted to pH 3.3 with 1N NaOH and containing 800 mg/l of Na2EDTA, 300 mg of sodium octyl sulfate, and 10% of HPLC-grade methanol. The flow was set at 0.6 ml/min, the temperature of the column was kept at 37°C, and the electrochemical detector (Model M-400; Princeton Applied Research, Princeton, NJ) was set at a gain of 50 nA full scale for neostriatum and brainstem and at 20 nA full scale for the remaining regions. The eluted compounds were oxidized with a glassy carbon electrode at a potential of 0.68 V relative to the Ag/AgCl reference electrode. The peaks generated by the compounds were recorded, and their surfaces were integrated with a Hewlett Packard 3392A integrator. For every chromatographic run, external standards containing 1.25 ng of each of the authentic monoamines were injected to quantify peak area as well as retention times. Both parameters showed very good reproducibility, with a coefficient of variation never exceeding 5%. Although the method allows for the separation of catecholamines, indoleamines, and their major metabolites (Reader and Grondin, 1987), only the tissue levels of noradrenaline (NA), dopamine (DA), 3,4-dihyroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 3-methyoxytyramine (3-MT), serotonin (5-HT), and 5-hydroxyindole-3-acetic acid (5-HIAA) are reported here, in nanograms per milligram of protein.
RESULTS
BDNF and dopamine-β-hydroxylase are partially colocalized in fibers and nerve terminals in the adult brain
To determine whether BDNF, which is synthesized by central noradrenergic neurons (Castren et al., 1995; Conner et al., 1997), is anterogradely transported into noradrenergic axons and nerve terminals, we performed double-label immunocytochemical analysis for BDNF and dopamine-β-hydroxylase (DBH), a marker for noradrenergic and adrenergic neurons and fibers (Figs. 1,2), using a specific BDNF antibody (RAB) from Amgen that has been characterized previously (Conner et al., 1997;Fawcett et al., 1997; Yan et al., 1997b) and a commercially available monoclonal (DBH) antibody. We chose to focus on the brainstem for these studies, because both BDNF- and DBH-positive fibers are abundant in this brain region (Moore and Card, 1984; Conner et al., 1997).
Fig. 1.
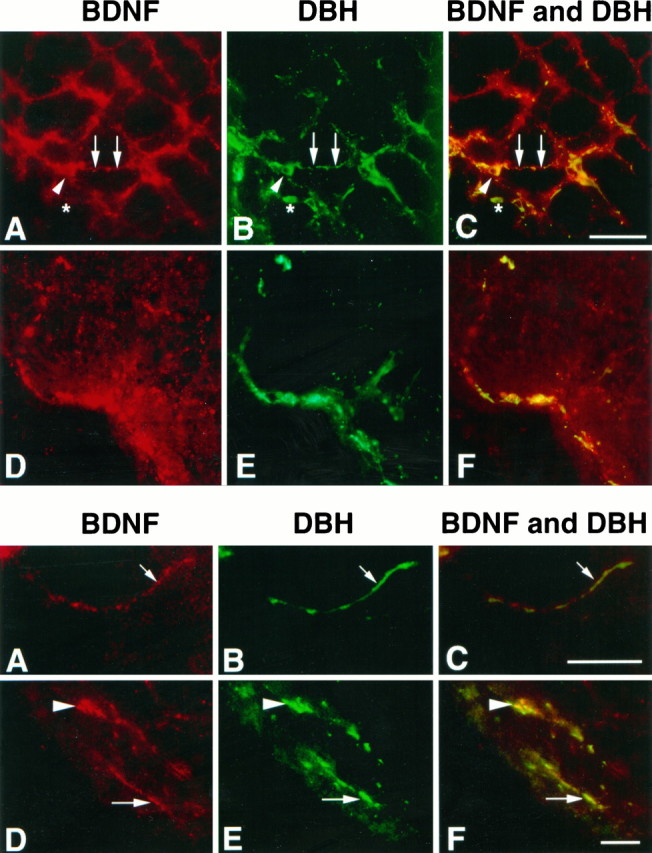
Colocalization of BDNF and dopamine-β-hydroxylase (DBH) in fibers and nerve terminals in the adult rat brain. Fluorescence photomicrographs of the rat brainstem double-labeled with antibodies to BDNF (A, D, visualized using a CY3-labeled secondary antibody) and DBH (B, E, visualized using a CY2-labeled secondary antibody). C and F are photographic double exposures; in these panels, yellow indicates regions of double labeling. A–C, The region of noradrenergic innervation rostral to the ventrolateral reticular region. Noradrenergic neuron cell bodies are DBH-positive (asterisk) but are largely negative for BDNF, with some cells exhibiting faint BDNF immunoreactivity (large arrowhead). However, the processes of these noradrenergic neurons are largely positive for both BDNF and DBH (arrows). D–F, The ventrolateral periolivary region is immunoreactive for both the BDNF (D) and DBH antibody (E), with punctate staining that may represent terminals or cut fibers. Space bar, 50 μm.
Fig. 2.
BDNF and DBH are colocalized in axons in the brainstem. Fluorescence photomicrographs of brainstem sections double-labeled for BDNF and DBH as described in Figure 1.A–C, BDNF and DBH are colocalized in fine processes in the rostral ventrolateral reticular region (arrows).D–F, BDNF and DBH are colocalized in fibers (arrows) in the ventrolateral periolivary region. The occasional single double-labeled punctate structure can be visualized in this region (arrowhead). Space bars, 25 μm.
This analysis revealed that although many DBH-positive fibers were not immunoreactive for BDNF and many BDNF-positive fibers were not immunoreactive for DBH, these two proteins were highly colocalized in certain brainstem structures (Fig. 1). As reported previously (Conner et al., 1997), noradrenergic neuron cell bodies were largely negative for BDNF immunoreactivity, although faint immunostaining could be seen occasionally in the soma of DBH-positive noradrenergic neurons (Fig.1A–C). However, BDNF immunoreactivity was detected in the network of DBH-positive processes that surround noradrenergic cell groups, such as those of the ventrolateral reticular group (Fig.1A–C). In these locations, DBH-positive fibers were also BDNF-positive, and in some cases BDNF immunoreactivity was detected in DBH-positive processes as they exited the noradrenergic cell soma (Fig. 1A–C). At higher magnification, this colocalization could be seen to occur in single noradrenergic fibers (Fig. 2A–C). These data, together with previousin situ hybridization data (Castren et al., 1995), indicate that BDNF is synthesized by noradrenergic neurons and is transported from the cell soma into noradrenergic processes.
BDNF was also colocalized to DBH-positive fibers rostral and lateral to noradrenergic cell groups. In particular, BDNF was colocalized with DBH-positive fibers, presumably axons, in the rostroventrolateral periolivary region (Figs. 1D–F). Higher-resolution analysis (Fig. 2D–F) revealed that in some cases, this colocalization occurred in fibers coursing through the tissue section (Fig.2D–F), whereas in others the colocalized immunoreactivity was punctate in nature, reflecting either noradrenergic terminals or axons cut in cross-section (Fig.2D–F, arrowheads). Both DBH and BDNF terminal staining was also seen in the raphi pallidus, a terminal field of noradrenergic innervation (data not shown). Thus, BDNF can be immunocytochemically detected in noradrenergic axons and nerve terminals.
DBH–BDNF mice overexpress BDNF in central noradrenergic neurons
These data indicate that BDNF is anterogradely transported by noradrenergic afferents, and they suggest that it may be secreted from noradrenergic terminals, thereby regulating the development and maintenance of their target neurons. To test this hypothesis, we took advantage of transgenic mice that overexpress BDNF in noradrenergic neurons from the dopamine-β-hydroxylase promoter (DBH–BDNF mice) (Causing et al.,1997). We have demonstrated previously that in two lines of DBH–BDNF mice, D498 and D481, BDNF synthesis is increased approximately two- to fourfold in sympathetic noradrenergic neurons, with no ectopic expression or secretion of BDNF into the circulation. To determine whether BDNF is also overexpressed in central noradrenergic neurons, as predicted (Hoyle et al., 1994), we examined the locus coeruleus. Immunocytochemical analysis using a BDNF-specific antibody (Patterson et al., 1996) (W. Friedman and D. Kaplan, unpublished data) demonstrated BDNF-immunoreactivity detectable in a few neuronal cell bodies within the control locus coeruleus (Fig.3A). In contrast, BDNF immunoreactivity was evident in most locus coeruleus neuron bodies in DBH–BDNF animals from lines D481 (Fig. 3B) and D498 (data not shown). Image analysis of the fluorescence intensity per locus coeruleus neuron revealed that BDNF immunoreactivity was reproducibly increased in four different pairs of control versus line D481 DBH–BDNF animals (5.52 ± 0.29 vs 11.19 ± 0.38, n = 52 and 78, p < 0.0001; 11.27 ± 0.57 vs 16.09 ± 0.72, n = 41 and 90, p < 0.0001; 9.57 ± 0.35 vs 17.04 ± 0.87, n = 141 and 71, p < 0.0001; 9.88 ± 1.78 vs 21.57 ± 1.61, n = 15 and 43, p < 0.0002; in all four pairs, the data represents the mean fluorescence intensity ± the SE, and n represents the number of neurons analyzed). Thus, the DBH–BDNF transgene was expressed in both central and peripheral (Causing et al., 1997) noradrenergic neurons.
Fig. 3.
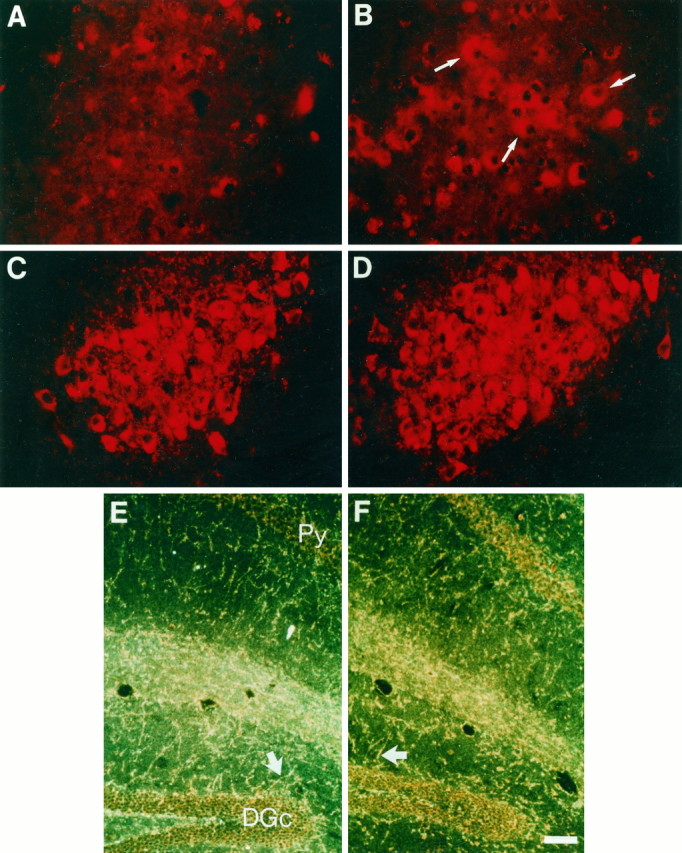
A, B, BDNF expression is increased in the locus coeruleus of DBH–BDNF mice. Fluorescence photomicrographs at the level of the locus coeruleus in control (A) and line D481 DBH–BDNF (B) animals that were immunostained with an antibody specific to BDNF. Note that in control locus coeruleus, the level of immunostaining is not above background, whereas in the DBH–BDNF locus coeruleus, there are many BDNF-immunoreactive cell bodies (arrows). C–F, The level and pattern of dopamine-β-hydroxylase immunostaining is not altered in DBH–BDNF mice. C, D, Fluorescence micrographs at the level of the locus coeruleus in control (C) and line D481 DBH–BDNF (D) animals that were immunostained with an antibody specific to dopamine-β-hydroxylase, which recognizes noradrenergic and adrenergic neurons and fibers. The pattern and level of DBH immunostaining are similar in both cases.E, F, Dark-field micrographs of coronal sections of the hippocampus from control (E) and line D481 DBH–BDNF (F) brains that have been immunostained with anti-DBH and visualized with peroxidase. Both photographs derive from a similar level of the hippocampus, and the dentate granule cell (DGc) and pyramidal cell (Py) layers are marked. Note that in dark field the DBH-positive nerve fibers appear bright silver–yellow (arrows), and there are no apparent differences in the pattern or density of DBH-immunoreactive fibers in transgenic versus control animals. Scale bar, 100 μm.
To confirm this conclusion, we also quantitated BDNF in the brains of control versus DBH–BDNF mice by Western blot analysis. We initially examined the brains of 1-week-old mice, after the onset of expression from the dopamine-β-hydroxylase promoter (Kapur et al., 1991), at a time when endogenous BDNF levels are low (Maisonpierre et al., 1990). Analysis of Western blots revealed that brainstem BDNF levels increased ∼1.8-fold (n = 5 each; p < 0.003) in 1-week-old transgenic animals of line D498 relative to their control littermates (Fig. 4C,top). BDNF levels similarly increased ∼2.4-fold (n = 5 each; p < 0.0007) in the brainstem of adult animals of line D498 (Fig. 4C,bottom) and D481 (data not shown). In contrast, BDNF levels were unchanged in the cortex of adult line D481 animals (data not shown) or in the neonatal or adult cortex of line D498 DBH–BDNF mice (n = 5 each; p > 0.05) (Fig.4A,B, bottom), as reported previously in the spinal cord (Causing et al., 1997). Both the cortex and spinal cord are locations in which there are no resident noradrenergic or adrenergic neurons. Together with our previous findings (Causing et al., 1997), this analysis indicates that BDNF synthesis is increased in developing and mature noradrenergic neurons of DBH–BDNF mice and that this overexpression is confined to regions of the nervous system predicted by previous analysis of the 1.6 kb DBH promoter (Hoyle et al., 1994).
Fig. 4.

A, B, Endogenous levels of TrkB autophosphorylation are increased in the cortex of DBH–BDNF mice, whereas BDNF levels are unchanged. Top, Cortical lysates from individual adult (A) and 1-week-old (B) control (−) and line D498 DBH–BDNF (+) animals were immunoprecipitated with anti-panTrk, and then analyzed by Western blots with antiphosphotyrosine (4G10). To ensure that the observed increases reflected an increase in the activation of TrkB, the blots were reprobed with anti-TrkBout (TrkB). Image analysis quantitation was used to normalize the level of autophosphorylation of the 145 kDa TrkB band relative to levels of TrkB protein. The normalized data (shown in the graphs, with ann of at least 3 individual animals in each case) were analyzed statistically for significance using a Student’st test. *p < 0.05; **p < 0.005. The size of the two TrkB isoforms is indicated by 145 and 190. Bottom, Western blot analysis of BDNF protein in the cortex of individual adult (A) and 1-week-old (B) control (−) and line D498 DBH–BDNF (+) mice. Thegraphs represent image analysis quantification of the data obtained on the same Western blot of three individual control and transgenic animals, with the optical density (O. D.) being arbitrary numbers. C, BDNF levels are increased in the brainstem of DBH–BDNF mice. Western blot analysis of BDNF protein in the brainstem of individual 1-week-old (top) and adult (bottom) control (−) versus line D498 DBH–BDNF (+) mice. The graphsrepresent image analysis quantification of the data obtained on the same Western blot of three individual control and transgenic animals, with the optical density (O. D.) being arbitrary numbers. Statistical analysis of these data demonstrates that BDNF is significantly increased in the brainstem of adult and neonatal DBH–BDNF animals. *p < 0.05; **p < 0.005.
Increased BDNF synthesis by noradrenergic neurons anterogradely regulates cortical development
If BDNF synthesized by noradrenergic neurons is anterogradely transported and secreted onto target neurons, then we would predict, in the DBH–BDNF mice, an increase in the baseline level of TrkB autophosphorylation in targets such as the neocortex. To test this prediction, we isolated cortical tissue from neonatal and adult mice, immunoprecipitated the lysates with anti-panTrk 203, and then analyzed the immunoprecipitates by Western blot analysis with antiphosphotyrosine. We reprobed the blots for total TrkB receptor levels using anti-TrkBout and normalized the level of increase in Trk autophosphorylation relative to total TrkB levels. This analysis indicated that levels of autophosphorylation of a 145 kDa Trk band were specifically increased 2.8-fold (n = 8 each;p < 0.003) in the cortex of 1-week-old DBH–BDNF versus control animals (Fig. 4B, top), although cortical BDNF levels were unchanged (Fig.4B, bottom). Similarly, in the adult DBH–BDNF cortex, the level of autophosphorylation of the 145 kDa Trk band was increased ∼2.5-fold relative to controls (n= 8 each; p < 0.05) (Fig. 4A,top). In the neonatal cortex, the increased Trk autophosphorylation was limited to this 145 kDa Trk band (Fig.4B), whereas in adult animals, we also observed increased autophosphorylation of a 190 kDa Trk-immunoreactive band (Fig. 4A). This phosphorylated Trk band likely corresponds to a novel, 190 kDa TrkB that has been reported previously in both the PNS (Bhattacharyya et al., 1997) and CNS (Roback et al., 1995) (R. Aloyz, D. Kaplan, and F. D. Miller, unpublished observations). Thus, increased BDNF expression within brainstem noradrenergic neurons led to increased Trk receptor autophosphorylation in the neocortex, a noradrenergic target region.
To determine whether the observed increase in TrkB receptor activation in the neocortex modified cortical development and maintenance, coronal sections of control versus DBH–BDNF brains were stained with cresyl violet, and the gross morphology of the neocortex was compared. Comparisons were made at two levels as shown in Figure5A. At the anterior level, the depth of the cortex from the corpus callosum to the pia was consistently smaller in animals of line D498 than in controls (Fig.5B, top panels). In contrast, cortical depth was similar in controls versus DBH–BDNF mice at the more posterior level (Fig. 5B, bottom panels). This observation was quantitated by determining the number of neurons in 528-μm-wide strips of cortex in control versus transgenic animals. At the anterior level, there were 2269.0 ± 41.45 cells per 528-μm-wide strip of transgenic cortex, compared with 2505 ± 21.55 in controls, a statistically significant decrease of 10% (p < 0.002; n = 5 each). At the posterior level, there were 1860.38 ± 54.44 cells in DBH–BDNF mice, compared with 1953.7 ± 68.1 cells in control mice, numbers that were statistically similar (p > 0.05);n = 5 each). Consistent with these observations, noradrenergic innervation is most dense in the rostral neocortex (Schlumpf et al., 1980). Thus, increased anterograde secretion of BDNF from noradrenergic neurons affects cortical development and maintenance, most probably through activation of endogenous TrkB receptors.
Fig. 5.
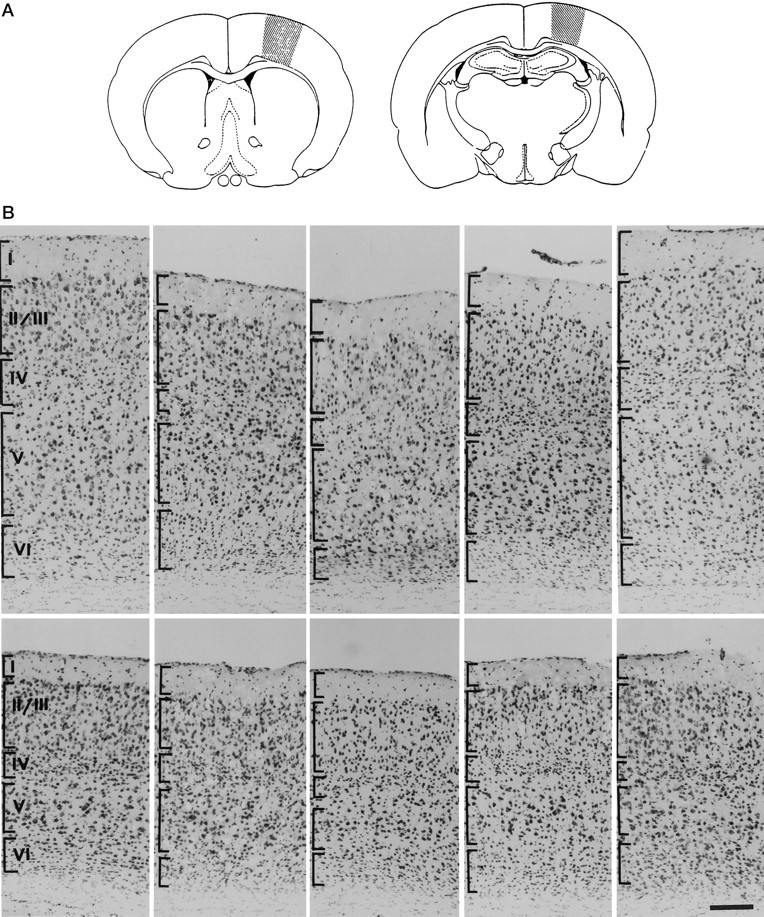
Overexpression of BDNF in noradrenergic neurons leads to decreased neuronal numbers in the anterior neocortex.A, Schematic drawing showing the rostral and caudal levels that were analyzed for morphology of the neocortex.B, Photomicrographs of coronal Nissl-stained sections of the neocortex of control and DBH–BDNF transgenic mice.Rightmost and leftmost panels are from control animals, whereas the three innermost panels are all from line D498 DBH–BDNF animals. The top setrepresents the rostral level of the neocortex indicated inA, and the bottom set represents the caudal level. Brackets define approximate boundaries of the different cortical layers indicated in the leftmost panel. Scale bar, 70 μm.
Increased synthesis of BDNF by noradrenergic neurons rescues neonatal facial motoneurons from axotomy-induced death
Our cortical results support the hypothesis that anterogradely secreted BDNF regulates the development and maintenance of noradrenergic target neurons. To further test this hypothesis, we turned to neonatal facial motoneurons because (1) motoneurons receive noradrenergic innervation (Pickel et al., 1974; McBride and Sutin, 1976) and (2) neonatal facial motoneurons, which normally die in response to facial nerve axotomy, can be rescued by application of exogenous BDNF (Koliatsos et al., 1993; Sendtner et al., 1993). We predicted that neonatal facial motoneurons would be rescued from axotomy-induced death in DBH–BDNF mice because of increased BDNF supplied by their noradrenergic input.
To perform these experiments, we initially characterized facial motoneuron size and number in DBH–BDNF mice. Image analysis of serial, cresyl violet-stained coronal sections from the brainstem of P12 control versus DBH–BDNF mice revealed that facial neuron numbers were unchanged by expression of BDNF in noradrenergic neurons (control, 2987 ± 514; D481, 3248 ± 200; D498, 2838 ± 471;n = 3 animals each; p > 0.3 for all comparisons). In contrast, the average cross-sectional area of facial motoneurons was increased ∼35 and 25%, respectively, in lines D481 and D498 relative to control animals (Fig.6A) (control, 256.8 ± 9.2 μm2; D481, 384.8 ± 8.5 μm2; D498, 333.8 ± 15.4 μm2; n = 4 animals each for D498 and control and 3 for D481; p < 0.002 for both comparisons).
Fig. 6.

A, Facial motorneurons are hypertrophied in neonatal DBH–BDNF transgenic mice. The average cross-sectional area of P12 facial motoneurons was determined by image analysis of cresyl violet-stained coronal sections of the appropriate brainstem level of DBH–BDNF mice of lines D498 and D481 versus their control littermates. In both lines of DBH–BDNF mice, facial motoneurons were significantly hypertrophied. ***p < 0.005. B, Neonatal facial motoneurons are rescued from axotomy-induced death in DBH–BDNF transgenic animals. The facial nerves of 5-d-old control and DBH–BDNF animals were unilaterally transected, and 7 d later, serial 16 μm sections were collected throughout the entirety of the facial nuclei. The number of facial motoneurons was then determined in the contralateral, control versus ipsilateral, transected facial nuclei by image analysis quantification of the number of facial motoneurons in every fifth section. The results of this analysis are presented as a ratio of the number of neurons in the transected versus untransected nuclei. The mean number of facial motoneurons within each group is presented within the text. Note that in control littermates, only 28% of facial motoneurons remain 1 week after a facial nerve transection, whereas in lines D498 and D481, 69 and 66% of facial motoneurons remain, respectively. ***p < 0.005;n = at least three animals in each group.C, Survival of transected facial motoneurons is reduced in adult BDNF+/− mice compared with their BDNF+/+ littermates. Methods similar to those described in B were used, with the exception that unilateral facial nerve transections were performed on 3-month-old BDNF+/− versus BDNF+/+ animals. The results of this analysis are presented as a ratio of the number of neurons in the transected versus untransected nuclei. Note that in BDNF+/+ littermates, 87% of motoneurons remain 1 week after a facial nerve transection, whereas in BDNF+/− mice, only 71% of facial motoneurons remain. ***p < 0.05; n = 3 animals in each group. D, BDNF levels are decreased in the BDNF+/− brain. Western blots of equal amounts of protein from the cortex of BDNF+/− versus BDNF+/+ mice were probed with an antibody to BDNF. A similar decrease was observed in other regions of the BDNF+/− brain (data not shown).
We next determined whether increased BDNF from noradrenergic neurons could rescue the death of axotomized, neonatal facial motoneurons, as can exogenous BDNF (Koliatsos et al., 1993; Sendtner et al., 1993). For these experiments, the facial nerve was unilaterally transected at postnatal day 5, and 1 week later the number of facial motoneurons in the contralateral control versus ipsilateral transected facial nuclei was determined. Specifically, serial 16 μm coronal sections were collected throughout the entirety of the facial motor nuclei, and the number of motoneurons was determined in every fifth cresyl violet-stained section (Fig. 6B). In control littermates, only 28% of axotomized neonatal facial motoneurons survived relative to their uninjured counterparts (control uninjured neuron number, 2579 ± 546; control axotomized neuron number, 631 ± 75; n = 4 each). In contrast, in animals of lines D481 and D498, 66 and 69% of facial motoneurons, respectively, remained in the axotomized versus uninjured facial nuclei (line D481 uninjured neuron number, 3248 ± 200; line D481 injured neuron number, 2133 ± 159; line D498 uninjured neuron number, 2521 ± 460; line D498 injured neuron number, 1721 ± 277;n = 3 for D481 and n = 4 for D498). This increase in facial motoneuron survival in the DBH–BDNF mice was highly significant relative to control animals (p < 0.006 for D498; p < 0.009 for D481) and is similar to the level of rescue reported previously for exogenous BDNF (Koliatsos et al., 1993; Sendtner et al., 1993).
Decreased synthesis of BDNF in BDNF+/− mice leads to increased death of axotomized adult facial motoneurons
Our data indicate that alterations in BDNF levels directly regulate the survival of injured motoneurons. To test this hypothesis further, we examined BDNF+/− mice, in which one BDNF allele has been mutated by targeted recombination. To confirm that BDNF levels are decreased in the brains of BDNF+/− mice relative to controls, we quantitated BDNF by Western blot analysis. This analysis revealed that BDNF levels were decreased approximately twofold in regions throughout the adult BDNF+/− brain (Fig. 6D), as reported previously using ELISAs (Altar et al., 1997). To determine whether this decrease in endogenous BDNF levels affected the survival of injured motoneurons, the facial nerve was unilaterally transected in adult BDNF+/− mice or their BDNF+/+ littermates, and 1 week later the number of surviving facial motoneurons was determined by morphological analysis (Fig. 6C). This analysis revealed that in BDNF+/+ littermates, 87 ± 1.6% of transected facial motoneurons remained relative to the control, untransected motoneurons, as observed previously in control mice (Majdan et al., 1997). In contrast, in BDNF+/− mice, only 71 ± 4% of transected facial motoneurons remained, relative to the untransected facial motoneurons in the same animals (Fig. 6C), a statistically significant decrease of 18% (p < 0.05; n = 3 animals each). These results, together with those obtained from the DBH–BDNF mice, support the hypothesis that the survival of injured facial motoneurons is at least partially dependent on BDNF and that relatively small alterations in BDNF levels can tightly regulate the survival of injured BDNF-responsive neurons.
Noradrenergic innervation is not altered in DBH–BDNF mice
Although our results indicate that anterogradely transported BDNF regulates neuronal development and maintenance by activation of the TrkB receptor, noradrenaline has been previously proposed to have trophic effects (Maeda et al., 1974; Felten et al., 1982; Foote et al., 1983), and it is formally possible that some of the observed biological effects in the DBH–BDNF mice are attributable to increased catecholaminergic innervation. To examine this possibility, we examined morphological and neurochemical measures of noradrenergic innervation density. Initially, we compared the level of immunostaining for dopamine-β-hydroxylase, which is specific to noradrenergic and adrenergic neurons, in the brains of control versus DBH–BDNF mice (Fig. 3C–F). As predicted, anti-DBH robustly stained the noradrenergic neurons of the locus coeruleus, with no significant qualitative different between control (Fig. 3C) and D481 (Fig. 3D) or D498 (data not shown) animals. To determine whether the level of noradrenergic target innervation was altered, we examined the hippocampus, which has a well defined pattern of noradrenergic innervation (Swanson and Hartman, 1975; Loy et al., 1980;Moudy et al., 1993). In control (Fig. 3E), D481 (Fig.3F), and D498 (data not shown) animals, the pattern and density of DBH-positive innervation was similar. Thus, the pattern of noradrenergic innervation was not apparently altered in DBH–BDNF mice.
To assess quantitatively catecholaminergic innervation, tissue levels were measured in discrete CNS regions, using HPLC with electrochemical detection. This method allowed for the measurement of noradrenaline (NA) and dopamine (DA), of the major metabolites of DA, namely DOPAC, HVA, and 3-MT, as well as of the indoleamines serotonin (5-HT) and 5-HIAA (Tables 1,2). In all regions examined, the levels of noradrenaline were similar in control versus transgenic animals (p > 0.05), confirming that noradrenergic innervation was not altered in these animals. In frontal cortex (Fro) the main catecholamine was NA, and although levels of this catecholamine were unaltered, this was the only CNS region in which endogenous DA levels were found to be somewhat higher in transgenic mice (Table 1). The entorhinal-piriform (EnPi) cortex had similar levels of NA and DA, as expected, and there were no differences detected. In hippocampus (Hipp), DA and its metabolites were not detected in the majority of samples; the main catecholamine was NA, which was unaltered. The main catecholamine in the neostriatum (NS; caudate-putamen) was DA, and there were also high levels of its metabolites DOPAC, HVA, and 3-MT; this was the only region in which significant levels of 3-MT were detected and measured in all samples. The indoleamines 5-HT and 5-HIAA were present in all of these regions, with values ranging from 3 to 5 ng/mg of protein, but there were no significant differences between control and transgenic mice, suggesting a normal 5-HT innervation of these forebrain areas. In the thalamus (Thal) of both control and transgenic mice there were moderate levels of NA, but also of DA and its metabolites DOPAC and HVA (Table2), and somewhat higher (5–7 ng/mg of protein) levels of 5-HT and 5-HIAA. Interestingly, in the hypothalamus (Hypo) the tissue levels of DA were lower in the transgenic mice relative to controls. Despite this reduction in endogenous DA, the levels of its metabolites DOPAC and HVA were unchanged, suggesting an increased turnover rate. The levels of the indoleamines 5-HT and 5-HIAA were similar in control and transgenic mice. In the brainstem the high contents of NA (4–5 ng/mg of protein), 5-HT (8 ng/mg of protein), and 5-HIAA (8–9 ng/mg of protein) reflect the fact that this region contains the nuclei of origin of noradrenergic and serotonergic projections. There were no differences between control and transgenic mice in levels of NA, DA, DOPAC, HVA, or the indoleamines in this region. Finally, in the cerebellum only moderate to low levels of NA, 5-HT, and 5-HIAA were measured, and the tissue contents were the same in control and transgenic mice.
Table 1.
Monoamines and metabolites in frontal (Fro) and entorhinal-piriform (EnPi) cortices, hippocampus (Hipp), and neostriatum (NS) of control and transgenic mice
| Compound | Fro | EnPi | Hipp | NS | |
|---|---|---|---|---|---|
| NA | Control | 1.12 ± 0.16 | 0.90 ± 0.23 | 2.85 ± 0.91 | 4.77 ± 0.92 |
| Transgenic | 1.72 ± 0.31 | 0.91 ± 0.18 | 3.63 ± 0.72 | 3.46 ± 0.94 | |
| DA | Control | 0.16 ± 0.05 | 1.16 ± 0.40 | n.d. | 58.26 ± 3.13 |
| Transgenic | 0.34 ± 0.06* | 0.77 ± 0.16 | n.d. | 64.50 ± 9.29 | |
| DOPAC | Control | 0.22 ± 0.05 | 0.75 ± 0.34 | n.d. | 19.09 ± 4.42 |
| Transgenic | 0.30 ± 0.04 | 0.72 ± 0.22 | n.d. | 16.72 ± 2.51 | |
| HVA | Control | n.d. | n.d. | n.d. | 5.13 ± 0.98 |
| Transgenic | n.d. | n.d. | n.d. | 5.74 ± 1.02 | |
| 3-MT | Control | n.d. | n.d. | n.d. | 4.42 ± 0.46 |
| Transgenic | n.d. | n.d. | n.d. | 5.46 ± 0.97 | |
| 5-HT | Control | 4.02 ± 1.31 | 5.25 ± 1.18 | 4.75 ± 1.18 | 3.25 ± 0.74 |
| Transgenic | 4.71 ± 0.69 | 4.53 ± 1.65 | 7.03 ± 1.57 | 3.73 ± 0.74 | |
| 5-HIAA | Control | 2.90 ± 0.80 | 3.38 ± 0.80 | 5.70 ± 1.29 | 4.32 ± 0.74 |
| Transgenic | 2.25 ± 0.33 | 3.04 ± 0.79 | 6.90 ± 1.18 | 4.77 ± 1.19 |
The values are the means ± SEM (n = 6) in nanograms per milligram of protein. The compounds are NA, noradrenaline; DA, dopamine; DOPAC, 3,4-dihydroxyphenylalanine; HVA, homovanillic acid; 3-MT, 3-methoxytyramine; 5-HT, serotonin; and 5-HIAA, 5-hydroxyindole-3-acetic acid. n.d., Not determined. Statistical comparisons were made by Student’s t test.
*p < 0.05.
Table 2.
Monoamines and metabolites in thalamus (Thal), hypothalamus (Hypo), brainstem (BS), and cerebellum (CB) of control and transgenic mice
| Compound | Thal | Hypo | BS | CB | |
|---|---|---|---|---|---|
| NA | Control | 3.74 ± 0.80 | 13.03 ± 1.35 | 5.51 ± 1.08 | 1.38 ± 0.18 |
| Transgenic | 3.56 ± 0.70 | 8.15 ± 1.76 | 4.69 ± 0.77 | 1.10 ± 0.45 | |
| DA | Control | 2.30 ± 0.74 | 2.13 ± 0.45 | 1.05 ± 0.18 | n.d. |
| Transgenic | 1.73 ± 0.61 | 0.99 ± 0.11* | 0.96 ± 0.21 | n.d. | |
| DOPAC | Control | 2.23 ± 0.54 | 6.72 ± 0.53 | 0.99 ± 0.16 | n.d. |
| Transgenic | 2.89 ± 0.75 | 6.39 ± 1.64 | 0.86 ± 0.17 | n.d. | |
| HVA | Control | 2.50 ± 0.75 | 2.82 ± 1.11 | 1.01 ± 0.18 | n.d. |
| Transgenic | 3.40 ± 2.51 | 3.23 ± 0.72 | 1.23 ± 0.27 | n.d. | |
| 5-HT | Control | 5.08 ± 0.63 | 6.61 ± 1.33 | 7.97 ± 1.09 | 1.83 ± 0.43 |
| Transgenic | 5.85 ± 1.03 | 7.76 ± 1.98 | 8.42 ± 0.97 | 1.89 ± 0.55 | |
| 5-HIAA | Control | 7.61 ± 0.42 | 8.12 ± 1.66 | 9.48 ± 1.20 | 1.59 ± 0.28 |
| Transgenic | 6.99 ± 1.25 | 7.37 ± 1.41 | 8.64 ± 1.31 | 1.95 ± 0.27 |
The values are the means ± SEM (n = 6) in nanograms per milligram of protein. The compounds are NA, noradrenaline; DA, dopamine; DOPAC, 3,4-dihydroxyphenylalanine; HVA, homovanillic acid; 5-HT, serotonin; and 5-HIAA, 5-hydroxyindole-3-acetic acid. n.d., Not determined. Statistical comparisons were made by Student’s t test.
*p < 0.05.
DISCUSSION
Evidence indicates that neuronal survival and differentiation is dependent on the appropriate establishment of afferent inputs (for review, see Linden, 1994). In this paper, we have tested the hypothesis that BDNF functions as an anterograde trophic factor for central noradrenergic neurons. The results reported here support our hypothesis and lead to three major conclusions. First, our data indicate that BDNF is synthesized by noradrenergic neurons and is anterogradely transported into noradrenergic axons and nerve terminal fields. Second, we demonstrate that overexpression of BDNF in noradrenergic neurons of DBH–BDNF mice has measurable anterograde effects on the cortex, leading to increased activation of cortical TrkB receptors, and differences in cortical morphology. Third, our studies demonstrating rescue of axotomized neonatal facial motoneurons in DBH–BDNF mice indicate that increased BDNF from noradrenergic afferents can directly regulate neuronal survival. Together, these data indicate that anterogradely secreted BDNF can modulate neuronal survival and differentiation and suggest that the effects of noradrenergic afferents on target neuron development may be at least partially mediated by BDNF.
Previous studies have demonstrated that BDNF is present in axons and nerve terminals in the mature CNS (Conner et al., 1997; Fawcett et al., 1997), and that TrkB, the preferred receptor for BDNF, is present on the dendrites of many central neurons (Fryer et al., 1996). Together, these data led us to hypothesize that BDNF might be an anterograde neurotrophic factor within the CNS and that it might mediate some of the previously described effects of afferent innervation on target neuron development. To test this hypothesis, we focused on central noradrenergic neurons, which synthesize BDNF mRNA (Castren et al., 1995) and have long been thought to play an organizational role in development of targets such as the neocortex (Maeda et al., 1974; Felten et al., 1982). The double-labeling data presented here indicate that BDNF may well be an anterograde trophic factor for these neurons; BDNF is localized to processes of noradrenergic neurons as they exit the cell soma, in noradrenergic axons in regions lateral to the cells themselves, and in noradrenergic terminal fields. These colocalization data, together with previous studies demonstrating synthesis of BDNF mRNA by noradrenergic neurons (Castren et al., 1995; Conner et al., 1997), strongly support the hypothesis that BDNF is a candidate anterograde neurotrophic factor for this class of neurons.
One noradrenergic target that might well be regulated by BDNF secreted from noradrenergic afferents is the neocortex. Previous studies indicate that a chemical lesion of developing noradrenergic neurons perturbs cortical development (Maeda et al., 1974; Felton et al., 1982), indicating a key regulatory role for early arriving afferents. Moreover, both BDNF and NT-4 regulate cortical development. Specifically, exogenous application of BDNF prevents the formation of ocular dominance columns (Cabelli et al., 1995) and regulates pyramidal neuron dendritic growth (McAllister et al., 1995), whereas exogenous NT-4 causes neuronal heterotopias, possibly by causing excess neurons to migrate into the developing marginal zone (Brunstrom et al., 1997). Moreover, endogenous BDNF is necessary for differentiation of cortical interneurons (Jones et al., 1994) and is thought to play an autocrine/paracrine role in cortical neuron survival (Ghosh et al., 1994). These effects presumably occur through activation of TrkB, which is present at high levels during corticogenesis (Escandon et al., 1994;Knusel et al., 1994), and which is expressed on cortical neurons throughout the developing and mature cortex (Cabelli et al., 1995;Fryer et al., 1996; Yan et al., 1997b).
Our studies with the DBH–BDNF mice support the hypothesis that noradrenergic afferents are likely to provide one endogenous source of BDNF for the developing cortex. Previous work demonstrates that the 1.6 kb DBH promoter used in these studies is highly specific for noradrenergic and adrenergic cells and is not ectopically expressed (Hoyle et al., 1994). Our data support this conclusion. The two lines of DBH–BDNF mice described here express increased BDNF in sympathetic neurons of the SCG and in noradrenergic neurons of the locus coeruleus, with no apparent increase in BDNF in the spinal cord or cortex (Causing et al., 1997; data shown here). Moreover, the effects described here cannot be ascribed to increased systemic BDNF, because (1) BDNF is undetectable in the circulation of these animals (Causing et al., 1997) and (2) sensory neurons of the dorsal root ganglia, which hypertrophy in response to BDNF, are unaffected in DBH–BDNF mice (C. Causing and F. Miller, unpublished data). In addition, we have demonstrated that the pattern and levels of noradrenergic innervation in the CNS (data shown here) and PNS (Causing et al., 1997) of these mice are apparently normal. Thus, the cortical effects we observe are best explained by increased secretion of BDNF from noradrenergic and potentially adrenergic afferents.
Interestingly, our data indicate that there is an increase in cortical Trk autophosphorylation without an apparent increase in cortical BDNF levels. The lack of a detectable increase in cortical BDNF is relatively easy to explain; it is likely that the amount of BDNF derived from noradrenergic afferents is small relative to the total amount of BDNF in the cortex, and therefore even a two- to fourfold increase in the amount of BDNF from this source is masked by endogenous cortical BDNF. Why, then, is there a two- to threefold increase in the basal level of Trk autophosphorylation? We propose that much of the BDNF present in cortical neurons and their afferents is packaged in secretory vesicles (Fawcett et al., 1997) and is therefore unavailable to neuronal TrkB receptors until it is released by neuronal activity, much like a neuropeptide. In contrast, although the amount of BDNF delivered by noradrenergic afferents is low relative to the total amount of BDNF in the cortex, it is likely that this BDNF is being secreted routinely, because noradrenergic neurons are constitutively active during waking hours (Harley, 1991). Thus, we propose that the amount of TrkB receptor activation in any given region of the cortex is not only a function of the amount of BDNF present in that region, but also of the amount of BDNF secretion that occurs in response to neuronal activity. Support for this model derives from our recent findings that pharmacological activation of cortical afferent systems leads to rapid and dramatic increases in cortical TrkB autophosphorylation, presumably as a consequence of activity-dependent BDNF release (R. Aloyz, J. Fawcett, and F. D. Miller, unpublished data).
What are the cortical perturbations observed in the DBH–BDNF mice in response to this increased TrkB receptor activation? In rats, noradrenergic neurons are born from E10 to E13 (Lauder and Bloom, 1974), and their afferents reach the developing cortex by E16 (Schlumpf et al., 1980), a time point when cortical neurons are actively being born, migrating, and differentiating (Altman and Bayer, 1990; Bayer and Altman, 1990). These afferents project through and branch into the marginal zone (Levitt and Moore, 1979), which is essential for the appropriate formation of cortical layers (Caviness, 1982) and which is one site of perturbed cortical development in response to NT-4 (Brunstrom et al., 1997). The DBH promoter is turned on in central noradrenergic neurons shortly after they are born (Hoyle et al., 1994), and our results confirm the neonatal expression of the DBH–BDNF transgene. The resultant increase in cortical Trk receptor activation throughout postnatal development presumably causes the decrease in neuronal number observed in the anterior DBH–BDNF cortex. This phenotype could result from any number of underlying mechanisms, including premature neurogenesis, inappropriate migration, and alterations in neuronal survival; our data do not discriminate among these possibilities.
Although the cellular mechanisms underlying the cortical phenotype reported here are unclear, the facial motoneuron studies indicate that afferent BDNF can directly regulate neuronal survival. Facial motoneurons in the DBH–BDNF mice are not rescued from developmental death, likely because they develop early relative to their noradrenergic innervation (Bayer and Altman, 1994). However, in neonates, when we know the DBH–BDNF transgene is on, increased BDNF from noradrenergic neurons causes facial motoneuron hypertrophy and rescues these neurons from axotomy-induced death. Whether endogenous afferent BDNF plays a similar role is unclear. However, our data with the BDNF+/− mice demonstrate that endogenous BDNF is essential for the survival of these injured neurons, although these studies do not directly determine the source of this endogenous BDNF. Moreover, these latter studies demonstrate that relatively small alterations in endogenous BDNF (approximately twofold) are sufficient to determine motoneuron survival after injury. Thus, these two studies together strongly support the idea that alterations in the amount of BDNF secreted from noradrenergic afferents could play a role in regulating the survival of developing or injured target neurons.
In summary, our immunocytochemical data indicate that BDNF is present in the axons and terminals of at least some noradrenergic neurons, and our studies with the DBH–BDNF mice indicate that such an afferent source of BDNF can affect the survival, differentiation, and maintenance of target neurons. We suggest that the previously documented trophic effects of noradrenergic afferents on cortical development are at least partially mediated by BDNF. Moreover, we suggest that activity-dependent presynaptic neurotrophin secretion and subsequent activation of postsynaptic Trk receptors may provide a cellular mechanism for modulating neural circuitry during development or in the mature nervous system or both.
Footnotes
This work was supported by a grant from the John D. and Catherine T. MacArthur Foundation Network on Psychopathology and Development to F.D.M. James Fawcett was supported by a studentship from the Rick Hansen Society, Shernaz Bamji by a Medical Research Council studentship, Carrie Causing by a Savoy Foundation studentship, and Raquel Aloyz by a fellowship from the Canadian NeuroSciences Network. Freda D. Miller is a Killam Scholar. We are grateful to Audrey Speelman and Rahul Varma for excellent technical assistance, to Tim Kennedy, Richard A. Murphy, Danny Baranes, Eddie Chang, and Marta Majdan for their advice and assistance with some of these experiments, to David Kaplan for reading this manuscript, and to Dr. Floyd Bloom and Dr. David Kupfer for their encouragement during the course of this work. We thank Dr. Qiao Yan and Dr. Andy Welcher for providing the BDNF antibody.
J.P.F. and S.X.B. contributed equally to this work.
Correspondence should be addressed to Freda D. Miller, Center for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 rue University, Montreal, Canada H3A 2B4.
REFERENCES
- 1.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay R, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Bayer SA. Vertical compartmentation and cellular transformations in the germinal matrices of the embryonic rat cerebral cortex. Exp Neurol. 1990;107:23–35. doi: 10.1016/0014-4886(90)90060-6. [DOI] [PubMed] [Google Scholar]
- 3.Barde Y-A, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bayer SA, Altman J. Development of layer I and the subplate in the rat neocortex. Exp Neurol. 1990;107:48–62. doi: 10.1016/0014-4886(90)90062-w. [DOI] [PubMed] [Google Scholar]
- 5.Bayer SA, Altman J. Neurogenesis and neuronal migration. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1994. pp. 1041–1078. [Google Scholar]
- 6.Bhattacharyya A, Watson FL, Bradlee TA, Pomeroy SL, Stiles CH, Segal RA. Trk receptors function as rapid retrograde signal carriers in the adult nervous system. J Neurosci. 1997;17:7007–7016. doi: 10.1523/JNEUROSCI.17-18-07007.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunstrom JE, Gray-Swain MR, Osborne PA, Pearlman AL. Neuronal heterotopias in the developing cerebral cortex produced by neurotrophin 4. Neuron. 1997;18:505–517. doi: 10.1016/s0896-6273(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 8.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 9.Castren E, Thoenen H, Lindholm D. Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience. 1995;64:71–80. doi: 10.1016/0306-4522(94)00386-j. [DOI] [PubMed] [Google Scholar]
- 10.Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- 11.Caviness VS., Jr Development of neocortical afferent systems: studies in the reeler mouse. Neurosci Res Prog Bull. 1982;20:560–569. [PubMed] [Google Scholar]
- 12.Clarke PGH. Neuronal death during development in the isthmo-optic nucleus of the chick: sustaining role of afferents from the tectum. J Comp Neurol. 1985;234:365–379. doi: 10.1002/cne.902340307. [DOI] [PubMed] [Google Scholar]
- 13.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunningham TJ, Huddleston C, Murray M. Modification of neuron numbers in the visual system of the rat. J Comp Neurol. 1979;184:423–434. doi: 10.1002/cne.901840213. [DOI] [PubMed] [Google Scholar]
- 15.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 16.Escandon E, Soppet D, Rosenthal A, Mendoza-Ramirez JL, Szonyi E, Burton LE, Henderson CE, Parada LF, Nikolics K. Regulation of neurotrophin receptor expression during embryonic and postnatal development. J Neurosci. 1994;14:2054–2068. doi: 10.1523/JNEUROSCI.14-04-02054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- 18.Felten DL, Hallman H, Jonsson G. Evidence for a neurotrophic role of noradrenaline neurons in the postnatal development of rat cerebral cortex. J Neurocytol. 1982;11:119–135. doi: 10.1007/BF01258008. [DOI] [PubMed] [Google Scholar]
- 19.Foote SL, Bloom FE, Aston-Jones G. Nucleus locus coeruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- 20.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Furber S, Oppenheim RW, Prevette D. Naturally occurring neuron death in the ciliary ganglion of the chick embryo following removal of preganglionic input: evidence for the role of afferents in ganglion survival. J Neurosci. 1987;7:1816–1832. doi: 10.1523/JNEUROSCI.07-06-01816.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 23.Harley C. Noradrenergic and locus coeruleus modulation of the perforant path-evoked potential in rat dentate gyrus supports a role for the locus coeruleus in attentional and memorial processes. Prog Brain Res. 1991;88:307–321. doi: 10.1016/s0079-6123(08)63818-2. [DOI] [PubMed] [Google Scholar]
- 24.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- 25.Hoyle GW, Mercer EH, Palmiter RD, Brinster RL. Cell-specific expression from the human dopamine-β-hydroxylase promoter in transgenic mice is controlled via a combination of positive and negative regulatory elements. J Neurosci. 1994;14:2455–2463. doi: 10.1523/JNEUROSCI.14-05-02455.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones BE, Moore RY. Ascending projections of the locus coeruleus in the rat. II. Autoradiographic study. Brain Res. 1977;127:23–53. [PubMed] [Google Scholar]
- 27.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kapur RP, Hoyle GW, Mercer EH, Brinster RL, Palmiter RD. Some neuronal cell populations express human dopamine β-hydroxylase-lacZ transgenes transiently during embryonic development. Neuron. 1991;7:717–727. doi: 10.1016/0896-6273(91)90275-5. [DOI] [PubMed] [Google Scholar]
- 29.Knusel B, Rabin S, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 31.Lakhdar-Ghazal N, Grondin L, Bengelloun WA, Reader TA. Alpha adrenoceptors and monoamine contents in the cerebral cortex of the rodent Jaculus orientalis: effects of acute cold exposure. Pharmacol Biochem Behav. 1986;25:903–911. doi: 10.1016/0091-3057(86)90405-3. [DOI] [PubMed] [Google Scholar]
- 32.Lauder JM, Bloom FE. Ontogeny of monoamine neurons in the locus coeruleus, raphe nuclei and substantia nigra of the rat. J Comp Neurol. 1974;155:469–4482. doi: 10.1002/cne.901550407. [DOI] [PubMed] [Google Scholar]
- 33.Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde Y-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 34.Levi-Montalcini R. The nerve growth factor: thirty-five years later. EMBO J. 1987;6:1145–1154. doi: 10.1002/j.1460-2075.1987.tb02347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levitt P, Moore RY. Development of the noradrenergic innervation of neocortex. Brain Res. 1979;162:242–259. doi: 10.1016/0006-8993(79)90287-7. [DOI] [PubMed] [Google Scholar]
- 36.Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 37.Linden R, Pinon LGP. Dual control by targets and afferents of developmental neuronal death in the mammalian central nervous system: a study in the parabigeminal nucleus of the rat. J Comp Neurol. 1987;266:141–149. doi: 10.1002/cne.902660202. [DOI] [PubMed] [Google Scholar]
- 38.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 39.Loy R, Koziell DA, Lindsey JD, Moore RY. Noradrenergic innervation of the adult hippocampal formation. J Comp Neurol. 1980;189:600–710. doi: 10.1002/cne.901890406. [DOI] [PubMed] [Google Scholar]
- 40.Maeda T, Tohyama M, Shimizu N. Modification of postnatal development of neocortex in rat brain with experimental deprivation of locus coeruleus. Brain Res. 1974;70:515–520. doi: 10.1016/0006-8993(74)90261-3. [DOI] [PubMed] [Google Scholar]
- 41.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 42.Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 44.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 45.McBride RL, Sutin J. Projections of the locus coeruleus and adjacent pontine tegmentum in the cat. J Comp Neurol. 1976;165:265–284. doi: 10.1002/cne.901650302. [DOI] [PubMed] [Google Scholar]
- 46.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennet DLH, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in trk-A expressing dorsal root ganglion cells and their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore RY, Card JP. Noradrenaline-containing neuron systems. In: Bjorkland A, Hokfelt T, editors. Handbook of chemical neuroanatomy, Vol 2: Classical transmitters in the CNS, Part 1. Elsevier; Amsterdam: 1984. pp. 123–156. [Google Scholar]
- 48.Moudy AM, Kunkel DD, Schwartzkroin PA. Development of dopamine-beta-hydroxylase-positive fiber innervation of the rat hippocampus. Synapse. 1993;15:307–318. doi: 10.1002/syn.890150407. [DOI] [PubMed] [Google Scholar]
- 49.Nawa H, Pelleymounter MA, Carnahan J. Intraventricular administration of BDNF increases neuropeptide expression in newborn rat brain. J Neurosci. 1994;14:3751–3765. doi: 10.1523/JNEUROSCI.14-06-03751.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 51.Patterson SL, Abel T, Deueul TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;15:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 52.Pickel VM, Segal M, Bloom FE. A radioautographic study of the efferent pathways of the nucleus locus coeruleus. J Comp Neurol. 1974;155:15–42. doi: 10.1002/cne.901550103. [DOI] [PubMed] [Google Scholar]
- 53.Reader TA, Grondin L. Distribution of catecholamines, serotonin and their major metabolites in rat cingulate, piriform-entorhinal, somatosensory and visual cortex: a biochemical survey using high-performance liquid chromatography. Neurochem Res. 1987;12:1099–1109. doi: 10.1007/BF00971709. [DOI] [PubMed] [Google Scholar]
- 54.Reader TA, Dewar KM, Grondin L. Distribution of monoamines and metabolites in rabbit neostriatum, hippocampus and cortex. Brain Res Bull. 1989;23:237–247. doi: 10.1016/0361-9230(89)90153-6. [DOI] [PubMed] [Google Scholar]
- 55.Roback JD, Marsh NH, Dowen M, Palfrey HC, Wainer BH. BDNF-activated signal transduction in rat cortical glial cells. Eur J Neurosci. 1995;7:849–862. doi: 10.1111/j.1460-9568.1995.tb01072.x. [DOI] [PubMed] [Google Scholar]
- 56.Sauve Y, Reader TA. Effects of a-methyl-p-tyrosine on monoamines and catecholamine receptors in rat cerebral cortex and neostriatum. Neurochem Res. 1988;13:807–815. doi: 10.1007/BF00970747. [DOI] [PubMed] [Google Scholar]
- 57.Schlumpf M, Shoemaker WJ, Bloom FE. Innervation of embryonic rat cerebral cortex by catecholamine-containing fibers. J Comp Neurol. 1980;192:361–376. doi: 10.1002/cne.901920210. [DOI] [PubMed] [Google Scholar]
- 58.Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde Y-A. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1993;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 59.Sohol GS, Narayahan CH. Effects of optic primordium removal on the development of the isthmo-optic nucleus in the duck. Exp Neurol. 1975;46:521–533. doi: 10.1016/0014-4886(75)90123-5. [DOI] [PubMed] [Google Scholar]
- 60.Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-β-hydroxylase as a marker. J Comp Neurol. 1975;163:467–506. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- 61.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 62.von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- 63.Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997a;378:135–157. [PubMed] [Google Scholar]
- 64.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennet L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997b;78:431–438. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 65.Zhou X-F, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]