

Abstract
When deprived of nerve growth factor (NGF), developing sympathetic neurons die by apoptosis. This death is associated with an increase in the level of c-Jun protein and is blocked by expression of a c-Jun dominant negative mutant. Here we have investigated whether NGF withdrawal activates Jun kinases, a family of stress-activated protein kinases that can stimulate the transcriptional activity of c-Jun by phosphorylating serines 63 and 73 in the transactivation domain and which can activate c-jun gene expression. We found that sympathetic neurons contained high basal levels of Jun kinase activity that increased further after NGF deprivation. In contrast, p38 kinase, another stress-activated protein kinase that can also stimulate c-jun gene expression, was not activated after NGF withdrawal. Consistent with Jun kinase activation, we found using a phospho-c-Jun-specific antibody that c-Jun was phosphorylated on serine 63 after NGF withdrawal. Furthermore, expression of a constitutively active form of MEK kinase 1 (MEKK1), which strongly activates the Jun kinase pathway, increased c-Jun protein levels and c-Jun phosphorylation and induced apoptosis in the presence of NGF. This death could be prevented by co-expression of SEKAL, a dominant negative mutant of SAPK/ERK kinase 1 (SEK1), an activator of Jun kinase that is a target of MEKK1. In contrast, expression of SEKAL alone did not prevent c-Jun expression, increases in c-Jun phosphorylation, or cell death after NGF withdrawal. Thus, activation of Jun kinase and increases in c-Jun phosphorylation and c-Jun protein levels occur at the same time after NGF withdrawal, but c-Jun levels and phosphorylation are regulated by an SEK1-independent pathway.
Keywords: apoptosis, c-Jun, Jun kinase, p38 kinase, signal transduction, sympathetic neurons, stress-activated protein kinases
Inhibitors of transcription or protein synthesis can protect developing sympathetic neurons against NGF withdrawal-induced death, suggesting that in this system new gene expression is required for death to occur (Martin et al., 1988). Consistent with this hypothesis, it was shown that NGF withdrawal led to a prolonged increase in the level of the transcription factor c-Jun, whereas the levels of other members of the Jun and Fos family did not increase, and microinjection of antibodies against c-Jun or expression of a c-Jun dominant negative mutant could protect sympathetic neurons against NGF withdrawal-induced death (Estus et al., 1994; Ham et al., 1995). Furthermore, overexpression of the wild-type c-Jun protein was sufficient to induce apoptosis in the presence of NGF (Ham et al., 1995).
The level of expression of c-Jun and its transcriptional activity are regulated by members of the MAP kinase superfamily (Karin, 1995). Jun kinases, also called Jun N-terminal kinase/stress-activated protein kinases (JNK/SAPKs), bind to the c-Jun transactivation domain and phosphorylate serines 63 and 73. Phosphorylation of c-Jun at these sites increases its ability to activate the transcription of target genes (Karin, 1995). Because c-Jun binds as a heterodimer with ATF-2 to specific sequences in the c-jun promoter, activation of Jun kinase can lead to an increase in the rate of transcription of the c-jun gene (Van Dam et al., 1995). Furthermore, Jun kinase also stimulates the transcriptional activity of ATF-2 by phosphorylating its transactivation domain (Gupta et al., 1995;Livingston et al., 1995; Van Dam et al., 1995). Jun kinase is itself activated by phosphorylation on threonine and tyrosine by SEK1 (Sanchez et al., 1994; Dérijard et al., 1995; Lin et al., 1995). SEK1 in turn is activated by the kinase MEKK1 (Yan et al., 1994). Another stress-activated protein kinase, p38 kinase, does not phosphorylate c-Jun but can increase c-jun gene expression by phosphorylating the ATF-2 transactivation domain (Raingeaud et al., 1995; Raingeaud et al., 1996).
Xia et al. (1995) showed that Jun kinase and p38 kinase were activated after NGF withdrawal in differentiated PC12 cells, and that the p38 pathway was necessary for cell death. Here we have investigated the role played by the Jun kinase and p38 kinase pathways in primary sympathetic neurons with the aim of determining the mechanisms by which NGF withdrawal leads to increased levels of c-Jun protein and apoptosis. We show that after NGF withdrawal, Jun kinase activity and the level of c-Jun phosphorylated on serine 63 increase, whereas p38 kinase is not activated. We also show that overexpression of MEKK1, a strong activator of the JNK pathway, increases c-Jun levels and induces apoptosis. Both effects are blocked by co-injection of SEKAL, a SEK1 dominant negative mutant. However, expression of SEKAL does not block induction of c-Jun or apoptosis after NGF withdrawal, suggesting that these processes are regulated by an SEK1-independent pathway. These results suggest that in sympathetic neurons the signaling pathways that activate c-jun gene expression and apoptosis after NGF withdrawal are different to those described in differentiated PC12 cells.
MATERIALS AND METHODS
Cell culture. Sympathetic neurons from the superior cervical ganglia (SCG) of 1-d-old Sprague Dawley rats (supplied by the Biological Services Unit, University College London) were isolated and cultured as described previously (Ham et al., 1995). After a final preplating step of 2 hr, which removed non-neuronal cells, the supernatant enriched in sympathetic neurons was plated in DMEM (Life Technologies, Gaithersburg, MD) containing 10% fetal calf serum (FCS), 2 mm glutamine, penicillin–streptomycin, and NGF at 50 ng/ml (SCG growth medium) on 3.5-cm-diameter dishes coated with poly-l-lysine (Sigma, Poole, UK) and laminin (Sigma) at a density of 5.5 × 104 cells per dish. For immunofluorescence and microinjection experiments, neurons were plated on 13-mm-diameter glass coverslips coated with poly-l-lysine and laminin (8000 neurons per coverslip). The antimitotic agents fluorodeoxyuridine and uridine (both from Sigma) were added to a final concentration of 20 μm to limit the proliferation of non-neuronal cells. NGF (2.5 S) was purified from adult male mouse submaxillary glands as described (Suda et al., 1978). The neurons were usually maintained for 6–8 d in the presence of NGF before being used for cell death experiments. NGF withdrawal was performed as follows: the medium was carefully removed from each dish, and the cells were very gently rinsed with prewarmed SCG growth medium lacking NGF and then refed with SCG medium lacking NGF supplemented with neutralizing anti-NGF antibody at 100 ng/ml (Boehringer Mannheim). The survival agents N-acetylcysteine and 8-(4-chlorophenylthio) cAMP (CPTcAMP) were purchased from Sigma.
PC12 cells were cultured in a defined medium supplemented with 2% FCS and 10 μg/ml insulin as described by Doherty et al. (1988) at a density of 2 × 106 cells per dish in 9 cm dishes coated with poly-l-lysine and collagen. Collagen was prepared from fresh rat tails according to standard procedures. PC12 cells were differentiated by plating them in defined medium supplemented with NGF (100 ng/ml) and were normally used after 7–8 d of NGF treatment. NGF withdrawal was performed as follows: the medium was removed, and the cells were quickly rinsed twice with prewarmed medium and then refed with defined medium supplemented with anti-NGF antibody at 100 ng/ml.
HeLa and Rat1 cells were cultured in DMEM with 10% FCS. For treatment with ultraviolet (UV) radiation, HeLa cells were grown to confluence and then were left in DMEM with 0.5% FCS overnight. The cells were exposed to short-wavelength UV radiation (254 nm) for 1 min using a hand-held UV lamp and were harvested 30 min later. Subconfluent Rat1 cells were treated with UV radiation in a similar manner, or anisomycin was added to the culture medium to a final concentration of 10 μg/ml, and the cells were harvested 30 min later.
Preparation of cell extracts for immune complex kinase assays. The growth medium from neuronal cultures was removed but kept to include detached, apoptotic neurons in the analysis. The remaining cells attached to the dish were then harvested by scraping, or rinsing in the case of sympathetic neurons, in a small volume of ice-cold PBS. The adherent and floating cells were pooled and spun down by centrifugation at 2000 rpm for 10 min at 4°C in a bench centrifuge. The cell pellet was resuspended in 1 ml of ice-cold PBS and transferred to a microfuge tube. The cells were then repelleted and resuspended in a small volume of ice-cold SAPK lysis buffer (20 mm HEPES, pH 7.4, 2 mm EGTA, 1% Triton X-100, 10% glycerol, 1 mm DTT, 50 mmβ-glycerophosphate, 1 mm sodium orthovanadate, 10 μg/ml leupeptin, 1 mm PMSF, 1 μg/ml pepstatin A, and 2 μg/ml aprotinin) and incubated on ice for 15 min. The lysate was then centrifuged at 13,000 × g for 30 min at 4°C. The supernatant was snap frozen in liquid nitrogen and stored at −80°C.
Jun kinase assay. Jun kinases were immunoprecipitated from cell extracts as follows. Protein A-agarose beads (Boehringer Mannheim) were equilibrated with SAPK lysis buffer. The extracts were then precleared by mixing up to 100 μg of extract in 750 μl of SAPK lysis buffer with 50 μl of a 1:1 suspension of protein A-agarose beads in SAPK buffer and rotating at 4°C for 1 hr. The beads were then spun down, and the supernatant was transferred to a fresh tube. One to 2 μl of anti-SAPK antibody (Kyriakis et al., 1994) was added together with 50 μl of washed protein A beads, and the tube was rotated at 4°C for 3 hr. The beads were then spun down at 4°C and washed three times with SAPK lysis buffer, three times with LiCl wash buffer (500 mm LiCl, 100 mm Tris-Cl, pH 7.6, 0.1% Triton X-100, and 1 mm DTT), and three times with 25 mm HEPES, pH 7.5, 0.2% Triton X-100, and 1 mmEDTA. The final supernatant was completely removed, and 30 μl of Jun kinase assay buffer (in mm: 25 HEPES, pH 7.5, 20 MgCl2, 20 β-glycerophosphate, 20p-nitrophenyl phosphate, 0.1 sodium orthovanadate, and 2 DTT) was added to 25 μl of beads together with 3 μg of glutathioneS-transferase (GST)c-Jun[1–169]. GSTc-Jun[1–169] was expressed in Escherichia coli transformed with the vector pGEX2Tc-Jun[1–169] and was purified from bacterial extracts on glutathione-Sepharose 4B beads (Pharmacia, Uppsala, Sweden) according to standard procedures (Smith and Johnson, 1988). The kinase reaction was started by adding 5 μCi of [γ-32P]ATP (>5000 Ci/mmol; Amersham, Little Chalfont, UK) and 0.5 μl of 2.3 mm ATP (to give a final concentration of 20 μm). The kinase mixtures were incubated at 30°C for 20 min and were stopped by adding 10 μl of 4× Laemmli sample buffer. The samples were heated at 90°C for 5 min, and the reaction products were separated on a 10% SDS-polyacrylamide gel. After electrophoresis, the gel was fixed and dried, and autoradiography was performed. Relative band intensity on the resulting autoradiographs was determined by scanning the films using an imaging densitometer (Bio-Rad). For peptide competition experiments, a 100:1 molar ratio of peptide to GSTc-Jun was used. The c-Jun δ peptide (mouse c-Jun amino acids 31–60) or a control peptide (mouse Bax amino acids 31–57) was added to the immunoprecipitated kinase and GSTc-Jun, and the mixture was incubated for 20 min at room temperature before starting the kinase reaction with ATP. Peptides were synthesized on a 431A synthesizer (Applied Biosystems) and were purified by HPLC. For peptide competition experiments, the peptides were dissolved in 20 mm HEPES, pH 7.9, at a concentration of 23 mg/ml.
p38 kinase assay. p38 kinase was immunoprecipitated from cell extracts using 2 μl of an antibody raised against theXenopus Mpk2/p38 protein (kindly provided by Dr. Angel Nebreda, European Molecular Biology Laboratory), which also recognizes mammalian p38 kinase (Rouse et al., 1994). Immune complex kinase assays were performed in the same way as the Jun kinase assays, except that the substrate was 2 μg of GSTATF-2[1–96] (Santa Cruz Biotechnology Inc., Santa Cruz, CA).
Immunoblotting. Immunoblotting was performed as described previously (Ham et al., 1995). c-Jun phosphorylated on serine 63 was detected using a phospho-c-Jun-specific monoclonal antibody (provided by D. Lallemand, Institut Pasteur, Paris, France), which had been raised against a 13 amino acid peptide corresponding to mouse c-Jun amino acids 57–68 and containing phosphoserine 63. The resulting antibody does not recognize unphosphorylated c-Jun or phosphorylated Jun B or Jun D (Watson et al., 1998) (D. Lallemand and M. Yaniv, unpublished observations). The phospho-c-Jun monoclonal antibody was diluted 1:500 for immunoblotting. In peptide competition experiments, the diluted antibody was incubated with a threefold excess (in micrograms) of either the phosphopeptide or corresponding unphosphorylated peptide for 1 hr at room temperature before being used for immunoblotting. p38 kinase and activated p38 kinase phosphorylated on tyrosine 182 were detected with p38 and phospho-p38-specific polyclonal antibodies, respectively, from New England Biolabs. These antibodies were used according to the manufacturer’s instructions.
Immunofluorescence analysis. c-Jun and Jun B were detected using specific affinity-purified rabbit polyclonal antibodies (Lallemand et al., 1997). c-Jun phosphorylated on serine 63 was detected using the phospho-c-Jun monoclonal antibody. For immunofluorescence experiments, neurons grown on glass coverslips were rinsed in PBS, fixed in 3% paraformaldehyde for 15–30 min at room temperature, rinsed again with PBS, and permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. After permeabilization, the coverslips were rinsed in PBS and incubated in a 1:1 mixture of goat serum and 1% BSA in PBS for 30 min at room temperature. After blocking, primary antibody was added for 1 hr at room temperature, followed by secondary antibody, also for 1 hr. Primary and secondary antibodies were diluted in 1% BSA in PBS. Fluorescein-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used at a dilution of 1:100. After the secondary antibody incubation, the cells were rinsed in PBS, and the nuclei were stained with Hoechst dye (H 33342; Calbiochem-Novabiochem) at 10 μg/ml in water and then given a final rinse with water. Coverslips were mounted in Citifluor. The slides were viewed on a Nikon Microphot FXA fluorescence microscope. Kodak (Rochester, NY) TMAX (TMY400) film was used for black-and-white photographs.
Plasmid constructions. To construct pGEX2Tc-Jun[1–169], an NcoI–AccI DNA fragment containing the mouse c-Jun coding sequences from amino acids 1–169 was cut from the plasmid T7-β-c-Jun (Hirai et al., 1989), made flush-ended by filling in with Klenow polymerase, and subcloned into pGEX2T cut withSmaI. The expression vector for the Myc epitope-tagged MEKK1 C terminus was constructed by Olson et al. (1995). The hemagglutinin (HA) SEKAL expression vector was described by Yan et al. (1994). pCDFLAGΔ169 and pCDBcl-2 were described by Ham et al. (1995). The reporter genes c-jun chloramphenicol acetyltransferase (CAT) and j1j2 CAT contain c-junpromoter sequences from −1600 to +170 cloned upstream of the bacterial CAT gene. In the case of j1j2 CAT, the jun1 and jun2 TPA response elements (TREs) have been mutated (Van Dam et al., 1995). SVeCAT is pCAT3 promoter (Promega, Southampton, UK) and contains the SV40 early promoter upstream of CAT. 6xjun2 SVeCAT was constructed by cloning six copies of the jun2 TRE element (5′-TTACCTCA-3′) upstream of the SV40 early promoter in pCAT3 promoter. This was accomplished by cloning three copies of an oligonucleotide that contained a tandem repeat of the jun2 TRE element into the unique BglII site in pCAT3 promoter. The sequence of the 2xjun2 oligonucleotide was upper strand, 5′-GATCAGCATTACCTCATCCCGATCAGCATTACCTCATCCC-3′; and lower strand, 5′-GA-TCGGGATGAGGTAATGCTGATCGGGATGAGGTA ATGCT-3′.
Microinjection. Neurons plated on glass coverslips were microinjected as described previously (Ham et al., 1995). All plasmids were purified on two cesium chloride gradients. DNA was injected in 0.5× PBS (−Ca and −Mg). CAT reporter gene assays were performed as follows. Sympathetic neurons were microinjected with c-jun CAT or j1j2 CAT (at 0.01 mg/ml) or 6xjun2 SVeCAT or SVeCAT (at 0.005 mg/ml) together with purified guinea pig IgG (Sigma) at 2.5 mg/ml. After injection, the cells were gently rinsed with SCG medium and then were refed with SCG medium lacking NGF supplemented with anti-NGF antibody or with fresh NGF-containing medium. Eighteen hours later, the injected cells were fixed and stained with a polyclonal anti-CAT antibody (5 Prime → 3 Prime, Inc., Boulder, CO) diluted 1:200. Secondary antibodies were a rhodamine-conjugated donkey anti-guinea pig IgG antibody and an FITC-conjugated anti-rabbit IgG antibody (both from Jackson ImmunoResearch) diluted 1:100. The percentage of injected cells (those that contained guinea pig IgG) that expressed CAT was then determined.
The MEKK1 and HA SEKAL expression vectors were injected at concentrations of 0.1 and 0.4 mg/ml, respectively. pCDFLAGΔ169 and pCDBcl-2 were injected at 0.05–0.2 mg/ml. Fifty to 80% of sympathetic neurons survived microinjection. Immunofluorescence analysis was performed to verify that the Myc epitope-tagged MEKK1 and HA-tagged SEKAL proteins were expressed and correctly localized within neurons after injection of the appropriate expression vectors. For these experiments, guinea pig IgG was added to the injection mix at a final concentration of 2.5 mg/ml. After injection, the neurons were fixed and stained with rhodamine-conjugated, anti-guinea pig IgG antibody to identify injected cells and the 9E10 and 12CA5 monoclonal antibodies (both from Boehringer Mannheim), which detect Myc-tagged and HA-tagged proteins, respectively, as described above, except that in the case of the 9E10 antibody the neurons were fixed and permeabilized with 50% methanol/50% acetone at −20°C for 20 min. In most cases, ∼90% of the neurons that had survived injection expressed the appropriate protein. The expression of FLAGΔ169 and Bcl-2 has been verified previously (Ham et al., 1995).
For analyzing the effect of MEKK1 overexpression, neurons were used 5–7 d after plating. Neutral, Mr70,000 Texas Red–dextran (Molecular Probes, Eugene, OR) was used to mark the injected cells in these experiments and was added to the injection mix at a final concentration of 5 mg/ml. After injection, the neurons were left for several hours and then were refed with fresh NGF-containing medium. The number of cells with Texas Red–dextran restricted to the nucleus was then determined, and their fate was followed over a period of days using a Zeiss Axiovert 100 inverted fluorescence microscope. Injected neurons with a normal morphology on phase optics were scored as viable. The Texas Red–dextran that had been injected into the nucleus of these cells remained within the nuclear envelope. In contrast, apoptotic neurons had a reduced nuclear and cytoplasmic volume, distorted nucleus, and fragmented neurites, and invariably the Texas Red–dextran that had originally been injected into the nucleus of these cells was distributed throughout the cell body. Thus, the percentage of viable neurons at a given time after injection was the percentage of the original number of injected cells that remained and that had a normal morphology. The effect of SEKAL and of pCDBcl-2 on neuronal survival in the absence of NGF was analyzed as described previously (Ham et al., 1995), except that Texas Red–dextran was added to the injection mix at 5 mg/ml rather than 20 mg/ml. All microinjection experiments were scored blind. Two hundred neurons were injected per construct, and each experiment was performed at least three times.
RESULTS
Transcriptional activation of the c-jun promoter in NGF-deprived sympathetic neurons requires the jun1 and jun2 TRE elements and activating protein 1 activity
The c-Jun protein can activate the transcription of the c-jun gene by binding as a heterodimer with ATF-2 to the jun1 and jun2 TRE elements in the c-jun promoter (Angel et al., 1988; Van Dam et al., 1995). To determine whether activating protein 1 (AP-1) activity was required for the increase in c-Jun protein levels that occurs after NGF withdrawal in sympathetic neurons, we investigated whether expression of the c-Jun dominant negative mutant FLAGΔ169 could prevent the appearance of c-Jun protein in NGF-deprived neurons. FLAGΔ169 lacks the c-Jun transactivation domain and functions as an inhibitor of AP-1 activity (Ham et al., 1995). Sympathetic neurons were microinjected with pCDFLAGΔ169 or with the empty cytomegalovirus (CMV) expression vector pcDNA1 as a negative control together with guinea pig IgG as a marker. After injection, the cells were rinsed and refed with medium lacking NGF supplemented with neutralizing anti-NGF antibody. Twenty-four hours later, the injected cells were fixed and stained with an antibody specific for guinea pig IgG, to identify the injected cells, and with an anti-c-Jun antibody. The percentage of injected cells that expressed c-Jun was then determined. Cells were only considered to be expressing c-Jun if nuclear staining with the c-Jun antibody was more intense than the background cytoplasmic staining. Uninjected cells on the same coverslips were also counted for comparison. In the presence of NGF, only a small percentage of sympathetic neurons (typically 5–10%) express detectable c-Jun protein (Ham et al., 1995). In contrast, 24 hr after NGF withdrawal, 65% of the uninjected neurons expressed c-Jun protein (Fig. 1A). Injection of the empty vector pcDNA1 reduced this value from 65 to 37%. This may be a nonspecific effect attributable to the presence of the strong CMV enhancer/promoter in pcDNA1, because we have not seen this reduction with vectors, such as pSG5, that contain the weaker SV40 promoter (J. Ham, unpublished observations) (see Fig. 7B). In contrast, microinjection of pCDFLAGΔ169 reduced the percentage of cells expressing c-Jun protein from 37% (the value obtained with pcDNA1) to 1.7%. To confirm that the effect of FLAGΔ169 on c-Jun expression was specific and not the result of a generalized repression of transcription, we investigated whether expression of FLAGΔ169 had any effect on the level of Jun B, another member of the AP-1 family. Jun B is expressed in sympathetic neurons, but its expression is regulated differently to that of c-Jun. In contrast to c-Jun, after NGF withdrawal, Jun B levels do not increase but remain more or less constant (Ham et al., 1995). Sympathetic neurons were microinjected with pcDNA1 or pCDFLAGΔ169 (at 0.2 mg/ml) together with guinea pig IgG as a marker. After injection, the cells were deprived of NGF for 24 hr and then were fixed and stained with an affinity-purified antibody specific for Jun B (Lallemand et al., 1997). Ninety-nine ± 0.6% (average of three independent experiments ± SE) of the uninjected cells expressed Jun B, which was located in the nucleus. Eighty-eight ± 5.5% of the cells injected with pcDNA1 and 86 ± 5.2% of the cells injected with pCDFLAGΔ169 expressed Jun B. Thus expression of FLAGΔ169 had no significant effect on Jun B protein levels. Because FLAGΔ169 inhibits AP-1-dependent gene expression in sympathetic neurons (Ham et al., 1995), these results indicate that AP-1 activity is specifically required for the increase in c-Jun protein levels that is caused by NGF withdrawal and suggests that c-Jun autoregulation may occur in sympathetic neurons.
Fig. 1.

Transcriptional activation of the c-jun promoter in NGF-deprived sympathetic neurons requires the jun1 and jun2 TRE elements and AP-1 activity.A, Sympathetic neurons were injected with pCDFLAGΔ169 (0.2 mg/ml) or the empty CMV expression vector pcDNA1 (0.2 mg/ml) together with guinea pig IgG (2.5 mg/ml). After injection, the cells were rinsed with medium lacking NGF and then were refed with −NGF medium supplemented with neutralizing anti-NGF antibody. Twenty-four hours later, the percentage of injected cells that expressed c-Jun protein was determined as described in Materials and Methods. Cells were only considered to be expressing c-Jun if nuclear staining with the c-Jun antibody was more intense than the background cytoplasmic staining. In each experiment 200 cells were injected per construct. Uninjected cells on the same coverslips were also scored for comparison. Coverslips were scored blind. The data shown represent the average of three independent experiments. Error bars indicate SE.B, Reporter gene structure. c-jun CAT contains wild-type c-jun promoter sequences from −1600 to +170 cloned upstream of the bacterial CAT gene. The position of the jun1 and jun2 TRE elements is indicated (j1j2). In j1j2 CAT, these have been mutated so that they are nonfunctional. 6xjun2 SVeCAT was constructed by cloning six copies of the jun2 TRE element upstream of the SV40 early promoter in SVeCAT (= pCAT3 promoter). C, c-jun CAT or j1j2 CAT was microinjected into sympathetic neurons at a concentration of 0.01 mg/ml together with guinea pig IgG (2.5 mg/ml). The injected cells were refed with medium containing (+NGF) or lacking (−NGF) NGF. Eighteen hours later, the percentage of cells expressing CAT was determined in immunofluorescence experiments with an anti-CAT antibody as described in Materials and Methods. The data shown represent the average of three independent experiments. Error bars indicate SE. D, 6xjun2 SVeCAT or SVeCAT was microinjected into sympathetic neurons at a concentration of 0.005 mg/ml together with guinea pig IgG. The injected cells were treated as described in C.
Fig. 7.

Expression of SEKAL does not prevent NGF withdrawal-induced death or the increase in c-Jun protein levels that occurs after NGF withdrawal. A, Expression of SEKAL does not protect sympathetic neurons against NGF withdrawal-induced death. Neurons were injected with pcDNA1 (0.05 mg/ml), pCDBcl-2 (0.05 mg/ml), pSG5 (0.4 mg/ml), or the SEKAL vector (0.4 mg/ml) together with Texas Red–dextran. pcDNA1 and pSG5 were control vectors for pCDBcl-2 and SEKAL, respectively. After injection, the cells were deprived of NGF. Seventy-two hours later, the percentage of viable injected neurons was determined as described in Materials and Methods. The results shown are the average of three independent experiments. Error bars indicate SE. B, SEKAL does not inhibit expression of c-Jun protein after NGF withdrawal. Sympathetic neurons were injected with pCDFLAGΔ169 (0.2 mg/ml), the SEKAL expression vector (0.4 mg/ml), or the empty vector pSG5 (0.4 mg/ml) together with guinea pig IgG at 5 mg/ml. After injection, the cells were withdrawn from NGF. Twenty-four hours later, the percentage of injected cells that expressed c-Jun was determined as described in Materials and Methods. c-Jun expression in uninjected cells on the same coverslips was scored for comparison. Only cells in which nuclear staining with the c-Jun antibody was more intense than the background cytoplasmic staining were considered to be expressing c-Jun. Two hundred cells were injected per construct. The data shown are the average of three independent experiments. Error bars indicate SE.
To test the autoregulation hypothesis further, we investigated whether the c-jun promoter was activated after NGF withdrawal and which c-jun promoter elements were required for c-jun gene expression. A construct in which c-junpromoter sequences from −1600 to +170 had been cloned upstream of the bacterial CAT gene (Fig. 1B, c-jun CAT) was microinjected into sympathetic neurons together with guinea pig IgG. The injected cells were then refed with medium containing or lacking NGF and after 18 hr were fixed and stained with an anti-CAT antibody and an antibody against guinea pig IgG. The percentage of cells expressing CAT was calculated, and the results are shown in Figure 1C. In the presence of NGF ∼10% of the cells injected with c-jun CAT expressed CAT protein. After 18 hr of NGF deprivation this had increased to 63%. Thus NGF deprivation causes a substantial (6.3-fold) increase in c-jun promoter activity, consistent with the previous observation that c-jun RNA levels increase after NGF withdrawal (Estus et al., 1994). In contrast, j1j2 CAT, a c-jun promoter construct in which the jun1 and jun2 TRE elements had been mutated (Fig. 1B), was not activated after NGF withdrawal (Fig. 1C). Furthermore, the basal level of j1j2 promoter activity in the presence of NGF was lower (Fig. 1C). This result indicates that the jun1 and jun2 TRE elements contribute to the basal level of c-jun promoter activity in sympathetic neurons and are also essential for activation of the promoter after NGF deprivation. To confirm the importance of these elements, we cloned six copies of the jun2 TRE sequence upstream of the heterologous SV40 early promoter linked to CAT (Fig.1B, 6xjun2 SVeCAT). When microinjected into sympathetic neurons, 6xjun2 SVeCAT was strongly activated by NGF withdrawal, whereas the control vector SVeCAT was not (Fig.1D). Thus multiple copies of the jun2 TRE can function as a transcriptional enhancer that is activated after NGF deprivation. These results suggest that in sympathetic neurons binding sites for c-Jun/ATF-2 heterodimers play a key role in the transcriptional response to NGF withdrawal.
Jun kinase activity and c-Jun phosphorylation increase after NGF withdrawal in sympathetic neurons
We previously observed that when sympathetic neurons were deprived of NGF there was a pronounced decrease in the mobility of the c-Jun protein in SDS-polyacrylamide gels (Ham et al., 1995). This gel shift has previously been shown to be the result of increased phosphorylation of the c-Jun transactivation domain at the serine and threonine residues that are phosphorylated by JNK/SAPKs (Pulverer et al., 1991;Papavassiliou et al., 1995). This result suggested that JNK/SAPKs may be activated by NGF withdrawal in sympathetic neurons. To measure JNK/SAPK activity directly, we used an immune complex Jun kinase assay (Fig. 2). Jun kinases were immunoprecipitated from whole-cell extracts using an anti-SAPK antibody that was raised against SAPKβ (JNK3) and that also recognizes other members of the rat SAPK family (Kyriakis et al., 1994). After immunoprecipitation, Jun kinase activity was assayed using purified GSTc-Jun[1–169] as substrate. As a control we used extracts prepared from HeLa cells that had been exposed to UV radiation, because it has been shown that Jun kinase is strongly activated by UV treatment in these cells (Hibi et al., 1993). Using the anti-SAPK antibody we were able to immunoprecipitate one or more kinases from UV-treated HeLa cells that phosphorylated GSTc-Jun (Fig. 2A). The kinase activity could be competed away by adding a 100-fold molar excess, compared with GSTc-Jun, of a peptide corresponding to the c-Jun δ domain (the JNK/SAPK binding site in c-Jun). In contrast, addition of an equal amount of a Bax peptide of identical molecular weight, but unrelated sequence, did not compete away the kinase activity, confirming the specificity of the immune complex kinase assay.
Fig. 2.
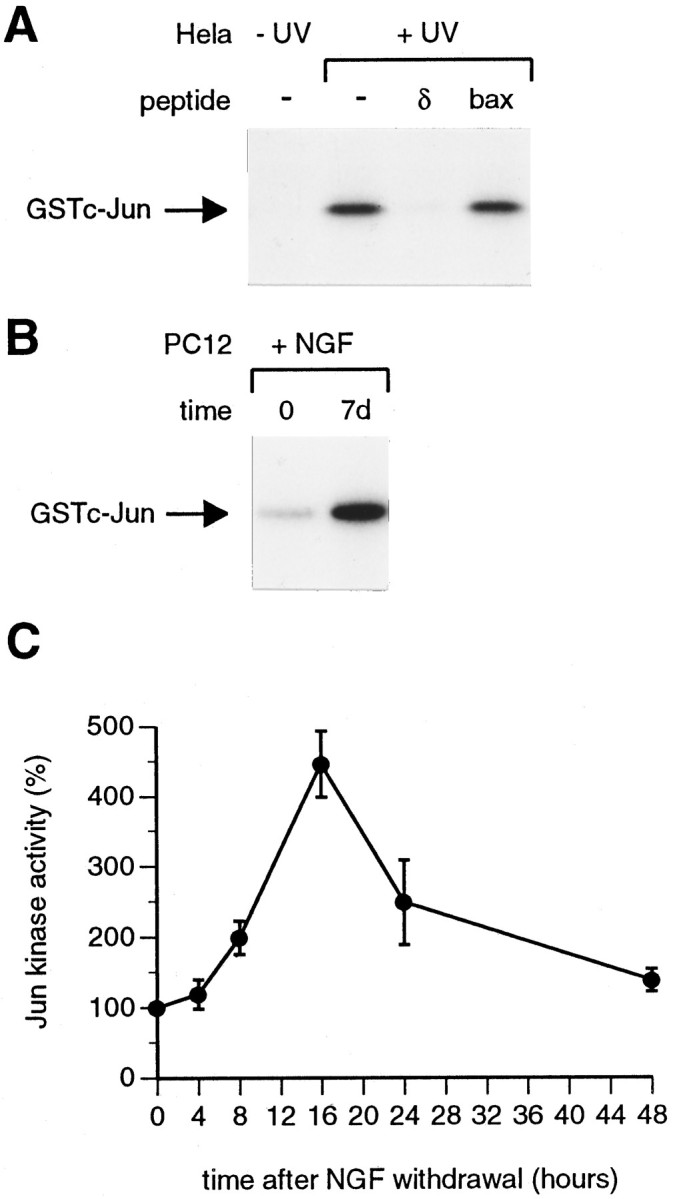
Jun kinase activity increases during PC12 differentiation and when differentiated PC12 cells are deprived of NGF.A, Control immune complex kinase assays were performed with extracts from quiescent (−UV) and UV-treated (+UV) HeLa cells as described in Materials and Methods. One hundred micrograms of extract were used for each immunoprecipitation. Competition with the c-Jun δ and Bax peptides was performed as described in Materials and Methods. The products of the Jun kinase assay were separated on a 10% SDS-polyacrylamide gel, which was fixed and dried, and autoradiography was performed. The position of phosphorylated GSTc-Jun[1–169] is shown. B, Jun kinase activity increases during PC12 differentiation. PC12 cells were treated with NGF, and extracts were prepared immediately after NGF addition (time 0) or 7 d later (7d). Jun kinase assays were performed using 100 μg of extract, as described in Materials and Methods. A representative result is shown. On average (7 experiments), Jun kinase activity increased fourfold during PC12 differentiation.C, Jun kinase is activated when differentiated PC12 cells are deprived of NGF. Differentiated PC12 cells were refed with defined medium lacking NGF, which had been supplemented with anti-NGF antibody. Extracts were made at the times indicated, and immune complex kinase assays were performed, using 100 μg of extract/immunoprecipitation. The resulting autoradiograph was scanned on a densitometer to determine the relative levels of Jun kinase activity at different times. The level of Jun kinase activity at time0 was set as 100%. The results shown are the average values for three independent experiments. Error bars indicate SE.
We then measured Jun kinase activity in extracts prepared from undifferentiated PC12 cells or PC12 cells that had been treated with NGF for 7 d and had acquired a neuronal phenotype. A representative result is shown in Figure 2B. On average (seven experiments), NGF treatment caused a fourfold increase in Jun kinase activity. This increase was not a rapid response to NGF and started ∼3 d after NGF addition (A. Eilers, unpublished observations). Furthermore, Jun kinase activity did not increase in PC12 cells that, instead of being treated with NGF, were simply left to proliferate for 7 d (data not shown). This result suggests that increases in the basal level of Jun kinase activity may be associated with neuronal differentiation. Finally, when NGF-dependent, differentiated PC12 cells were deprived of NGF, Jun kinase activity increased further, with a maximum induction of 4.5-fold at 16 hr after NGF withdrawal (Fig. 2C), in agreement with the observations of Xia et al. (1995).
We next measured Jun kinase activity in extracts prepared from sympathetic neurons that had been isolated from neonatal rats and cultured for 7 d in vitro. In the presence of NGF, sympathetic neurons contained very high levels of Jun kinase activity, which were comparable to those seen in UV-treated HeLa cells (data not shown). NGF withdrawal led to a twofold increase in Jun kinase activity after 4 hr (Fig. 3A). This increase was not the result of disturbing the cells or attributable to the presence of fresh serum in the −NGF medium, because Jun kinase was not activated in cells fed with fresh NGF-containing medium (Fig. 3, +NGF). In NGF-deprived cells, Jun kinase activity was also elevated at 8 hr but had returned to the basal level by 16 hr. Interestingly, the time at which JNK/SAPK activity started to increase (4 hr after NGF withdrawal) slightly precedes the time at which c-Jun levels and c-Jun phosphorylation start to increase (4–8 hr after NGF withdrawal; Ham et al., 1995). The activation of Jun kinase in sympathetic neurons caused by NGF withdrawal could be blocked by the addition of the survival agents CPTcAMP (at 100 μm) orN-acetylcysteine (at 30 mm) (Fig.3B).
Fig. 3.

Jun kinase is activated in sympathetic neurons after NGF withdrawal. A, Jun kinase assays were performed with extracts prepared from sympathetic neurons that had been withdrawn from NGF for 4, 8, or 16 hr (−NGF) or that had been refed with fresh NGF-containing medium (4 hr, +NGF), as described in Materials and Methods. Fifty micrograms of extract were used per immunoprecipitation. Relative Jun kinase activity was determined by scanning autoradiographs on a densitometer. The level of Jun kinase activity at time 0was set as 100%. The results shown are the average of three independent experiments. Error bars indicate SE. B,Survival agents prevent the NGF withdrawal-induced activation of Jun kinase in sympathetic neurons. Sympathetic neurons were refed with fresh NGF-containing medium (+NGF) or were deprived of NGF in the absence (−NGF) or presence of 100 μm CPTcAMP (−NGF +100 μm CPTcAMP) or 30 mmN-acetylcysteine (−NGF + 30 mm NAC). Four hours later, extracts were prepared, and immune complex kinase assays were performed using 50 μg of extract/immunoprecipitation. The results shown are the average of three independent experiments. Error bars indicate SE.
Jun kinase activation would be predicted to cause increased phosphorylation of the c-Jun N terminus at serines 63 and 73. To demonstrate directly that phosphorylation of serine 63 occurred in sympathetic neurons after NGF withdrawal, we made use of an antibody that only recognizes c-Jun when it is phosphorylated at this site. In immunoblotting experiments it has been shown that the phospho-c-Jun antibody does not recognize unphosphorylated c-Jun or phosphorylated Jun B or Jun D (Watson et al., 1998) (Lallemand and Yaniv, unpublished observations). The specificity of the phospho-c-Jun antibody was confirmed in the peptide competition experiment shown in Figure4A. Extracts from untreated (Fig. 4A, −UV) and UV-irradiated (Fig. 4A, +UV) Rat 1 cells were electrophoresed on an SDS-polyacrylamide gel, and immunoblotting with the phospho-c-Jun antibody was performed. Bands corresponding in size to phosphorylated forms of c-Jun were induced by UV treatment. These were competed away if the antibody was preincubated with the phosphopeptide used to generate it but not if the corresponding nonphosphorylated peptide was used. We then used the phospho-c-Jun antibody in immunofluorescence experiments with sympathetic neurons that had either been deprived of NGF or maintained in NGF-containing medium for 24 hr (Fig. 4B). NGF withdrawal led to the appearance of phospho-c-Jun staining in the nucleus of the majority of neurons (Fig. 4B, −NGF). This was not seen in neurons maintained in NGF-containing medium (Fig. 4B, +NGF). Phospho-c-Jun staining could be observed as early as 4 or 8 hr after the removal of NGF and was suppressed by survival agents such as 100 μm CPT-cAMP (data not shown). Thus, NGF withdrawal led to Jun kinase activation (Fig. 3A) and phosphorylation of c-Jun at serine 63 (Fig. 4B), a site known to be phosphorylated by Jun kinase.
Fig. 4.

c-Jun is phosphorylated on serine 63 when sympathetic neurons are deprived of NGF. A, The specificity of the phospho-c-Jun monoclonal antibody was verified in an immunoblotting experiment by performing peptide competition. Extracts were from untreated (−UV) or UV-irradiated (+UV) Rat 1 cells. The position of the phosphorylated forms of c-Jun (ph-c-Jun) is indicated. As predicted, UV treatment caused an increase in c-Jun phosphorylation. This signal was competed away by preincubating the antibody with the phosphopeptide used to generate it (phos) but not by an equal amount of the corresponding nonphosphorylated peptide (non-phos).B, Sympathetic neurons were isolated from neonatal rats and cultured for 7 d in vitro in the presence of NGF. The cells were then refed with medium lacking NGF, which had been supplemented with neutralizing anti-NGF antibody (−NGF), or with fresh NGF-containing medium (+NGF). Twenty-four hours later, the cells were fixed and stained with the phospho-c-Jun antibody and Hoechst dye as described in Materials and Methods. In the presence of NGF, weak background staining of the cell body was observed with the phospho-c-Jun antibody. After NGF withdrawal, nuclear phospho-c-Jun staining was apparent, indicating that the c-Jun protein in these cells was phosphorylated on serine 63. Phospho-c-Jun staining could be seen as early as 4 or 8 hr after NGF withdrawal (data not shown). Representative cells are shown.
p38 kinase is activated in differentiated PC12 cells but not in sympathetic neurons after NGF withdrawal
Although p38 kinase does not phosphorylate c-Jun in vitro, it can increase the rate of transcription of the c-jun gene by phosphorylating ATF-2 (Raingeaud et al., 1995;Raingeaud et al., 1996). Furthermore, it has been reported that p38 kinase is activated in differentiated PC12 cells after NGF withdrawal (Xia et al., 1995). We therefore performed immune complex kinase assays using an antibody raised against the Xenopus mpk2/p38 protein, which also recognizes mammalian p38 kinase (Rouse et al., 1994), and GSTATF-2[1–96] as substrate (Fig.5). As previously demonstrated by Xia et al. (1995), we found that NGF deprivation led to a threefold increase in p38 kinase activity in differentiated PC12 cells (Fig.5A). The kinetics of p38 activation in NGF-deprived PC12 cells were similar to those of Jun kinase (Fig. 2C). In contrast, p38 kinase was not activated in NGF-deprived sympathetic neurons at 4 or 8 hr after NGF withdrawal (Fig. 5B) or at later time points (data not shown). As an alternative approach, we also performed immunoblotting experiments with a p38 antibody and a phospho-p38 antibody that only recognizes the activated form of p38 phosphorylated on tyrosine 182 (Fig. 5C). Extracts from NGF-deprived sympathetic neurons or, as a control, PC12 cells that had been treated with anisomycin or UV radiation (known activators of p38 kinase) were tested. After immunoblotting, the resulting films were scanned on a densitometer to determine the levels of p38 and phospho-p38. The level of p38 protein was ∼10-fold lower in sympathetic neuron extracts compared with PC12 extracts. p38 protein levels were not significantly affected by NGF withdrawal or anisomycin or UV treatment. p38 phosphorylated on tyrosine 182 was readily detected in PC12 cells (30 sec ECL exposure), and anisomycin or UV treatment caused a sixfold to sevenfold increase in the level of phospho-p38 (Fig. 5C). In contrast, phospho-p38 could only be detected in the sympathetic neuron extracts after prolonged exposure of the film (a 30 min ECL exposure), and this low level of p38 phosphorylation did not increase after NGF withdrawal. In conclusion, the results of our kinase assays and immunoblotting experiments suggest that sympathetic neurons contain less p38 protein than PC12 cells and that this p38 kinase is not activated by NGF withdrawal. On the other hand, in PC12 cells p38 kinase is activated by treatment with anisomycin or UV radiation or after NGF deprivation, as reported previously (Xia et al., 1995).
Fig. 5.

p38 kinase is activated after NGF withdrawal in differentiated PC12 cells but not sympathetic neurons.A, Differentiated PC12 cells were withdrawn from NGF, and extracts were prepared at the times indicated. p38 kinase was immunoprecipitated from extracts using a Xenopusmpk2/p38 antibody that recognizes mammalian p38 (Rouse et al., 1994), and kinase assays were performed using GSTATF-2[1–96] as substrate. Relative p38 kinase activity was determined by scanning autoradiographs on a densitometer. The results of a representative experiment are shown. B, p38 kinase is not activated in NGF-deprived sympathetic neurons. p38 kinase assays were performed with extracts prepared from sympathetic neurons (SCG) or differentiated PC12 cells that had been deprived of NGF for the times indicated. Fifty micrograms of extract were used for each immunoprecipitation. A typical result is shown. The position of phosphorylated GSTATF2[1–96] is shown. C, p38 and phospho-p38 immunoblots were performed as described in Materials and Methods. Extracts were from sympathetic neurons (SCG) that had been deprived of NGF for 0, 4, 8, or 16 hr or were from control PC12 cells (−) or PC12 cells that had been exposed to anisomycin (A) or UV radiation (UV). p38 protein levels were 10-fold lower in SCG extracts compared with PC12 extracts. Activated p38 phosphorylated on tyrosine 182 (ph-p38) was readily detected in PC12 cell extracts (a 30 sec ECL exposure) but not in SCG extracts. Anisomycin or UV treatment increased phospho-p38 levels sixfold to sevenfold. Prolonged exposure of the same blot (a 30 min ECL exposure) revealed a low level of p38 phosphorylation in sympathetic neurons, which did not increase after NGF withdrawal.
Activated MEKK1 increases c-Jun levels and c-Jun phosphorylation and induces apoptosis in sympathetic neurons in the presence of NGF
To determine whether activation of the Jun kinase pathway was sufficient to induce apoptosis in sympathetic neurons, we tested the effect of microinjecting the cells with an expression vector for MEKK1. MEKK1 activates SEK1, which in turn activates Jun kinase (Sanchez et al., 1994; Yan et al., 1994). The MEKK1 expression vector we used has previously been shown to be a strong activator of the Jun kinase pathway and encodes a truncated, constitutively active form of MEKK1 with an N-terminal Myc epitope tag (Olson et al., 1995). We confirmed in immunofluorescence experiments with the Myc epitope-specific 9E10 antibody that neurons injected with the MEKK1 vector expressed the Myc-tagged protein. When injected at 0.4 mg/ml ∼40% of the cells clearly expressed MEKK1. To determine whether expression of the constitutively active MEKK1 protein in sympathetic neurons affected neuronal survival, we microinjected the MEKK1 vector or an empty expression vector (pSG5) together with a fluorescent marker (Mr 70,000 Texas Red–dextran). The injected cells were maintained in NGF-containing medium and were examined on an inverted fluorescence microscope at different times after injection. Four days after injection, 80% of the cells injected with the control vector pSG5 were still viable (Fig.6B). These cells had a normal morphology on phase, and Texas Red–dextran injected into the nucleus was retained within the nuclear envelope (Fig.6A). In contrast, most of the cells injected with MEKK1 had an apoptotic phenotype, with shrunken cell bodies and nuclei and Texas Red–dextran distributed throughout the cell body (Fig.6A). Only 20% of the cells injected with the MEKK1 vector were viable at 4 d (Fig. 6B). In addition, MEKK1 expression induced chromatin condensation, detected by Hoechst staining, and nuclear DNA fragmentation, detected by terminal transferase-mediated biotinylated UTP nick end-labeling (TUNEL) analysis (Ham, unpublished observations).
Fig. 6.

Overexpression of a constitutively active form of MEKK1 in sympathetic neurons increases c-Jun protein levels and induces apoptosis. A, Morphology of neurons 4 d after injection with pSG5 or MEKK1. Sympathetic neurons were microinjected with the empty vector pSG5 or an MEKK1 expression vector together with Texas Red–dextran (5 mg/ml) to mark the injected cells. The injected cells were left for 4 d in NGF-containing medium and then were examined on an inverted fluorescence microscope. The majority of cells injected with pSG5 had a normal morphology on phase, and the injected Texas Red–dextran was still retained within the nucleus. A representative cell is shown. In contrast, the majority of cells injected with MEKK1 had a clearly apoptotic morphology on phase (two cells injected with MEKK1 are shown), and the dextran marker was no longer retained within the nucleus. B, Kinetics of cell death after microinjection of MEKK1. Sympathetic neurons were microinjected with the MEKK1 vector or the control vector, pSG5, each at 0.1 mg/ml, together with Texas Red–dextran (5 mg/ml). The percentage of viable injected cells that remained at different times after injection was determined as described in Materials and Methods. The results shown are the average for four independent experiments. Error bars indicate SE. In each experiment, 200 neurons were injected per construct. C, Co-expression of SEKALprevents cell death induced by MEKK1 overexpression. Sympathetic neurons were microinjected with expression vectors for MEKK1 (0.1 mg/ml), SEKAL (0.4 mg/ml), or the control vector pSG5 (0.1–0.5 mg/ml) in the combinations indicated. Where necessary, the total DNA concentration was adjusted to 0.5 mg/ml by addition of pSG5. Texas Red–dextran was included in the injection mixes at 5 mg/ml. Several hours after injection, the number of Texas Red-containing cells was determined, and the cells were left in NGF-containing medium. Three days later, the number of viable injected cells that remained was determined. The results shown are the average of three independent experiments. Error bars indicate SE. D, Overexpression of MEKK1 increases c-Jun protein levels. Sympathetic neurons were injected with the vectors shown at the concentrations described inC. Forty-eight hours after injection, the percentage of surviving injected cells that expressed c-Jun protein was determined as described in Materials and Methods. The results shown are the average of seven independent experiments. Error bars indicate SE.
To determine whether the neuronal cell death induced by MEKK1 expression required SEK1 activity, we investigated the effect of co-injecting an expression vector for SEKAL together with the MEKK1 vector. MEKK1 activates SEK1 by phosphorylating serine 220 and threonine 224 (Yan et al., 1994). In SEKAL these residues have been mutated to alanine and leucine, respectively, so that the protein cannot be phosphorylated and remains inactive. SEKAL acts as a dominant negative mutant and prevents MEKK1 from activating Jun kinase (Yan et al., 1994). The SEKALprotein was tagged with the HA epitope and could be detected in the majority of neurons injected with the SEKAL expression vector in immunofluorescence experiments with an anti-HA antibody (data not shown). Like the control vector, injection of SEKALalone at 0.4 mg/ml had only a minor effect on neuronal viability in the presence of NGF (Fig. 6C). Co-injection of SEKAL(0.4 mg/ml) together with MEKK1 (0.1 mg/ml) prevented most of the cell death that would have been induced by MEKK1 at 3 d (Fig.6C). This result suggests that MEKK1 induces apoptosis in sympathetic neurons by activating SEK1 or a related kinase.
Finally, we investigated whether overexpression of MEKK1 could increase c-Jun protein levels in the presence of NGF. Sympathetic neurons were injected with an expression vector for MEKK1 or with the MEKK1 vector together with the SEKAL vector or a control vector. 48 hr later, the percentage of injected cells expressing c-Jun was determined. Overexpression of MEKK1 caused a fivefold increase in c-Jun levels compared with the control vector (Fig.6D). This increase was prevented by co-expression of SEKAL. Similar results were obtained when the injected cells were stained with the phospho-c-Jun-specific antibody. Forty-eight hours after injection, 25.5 ± 5.9% (average of three independent experiments ± SE) of the cells injected with the MEKK1 vector stained with the phospho-c-Jun antibody, whereas only 4.4 ± 0.9% of the cells injected with the control vector stained for phospho-c-Jun. Co-injection of SEKAL with MEKK1 reduced the percentage of phospho-c-Jun-positive cells from 25.5 to 5.4 ± 2.2%. It is notable that in these experiments, although all of the cells had been microinjected with the MEKK1 vector (at 0.1 mg/ml), only 25% of the cells remaining at 48 hr expressed c-Jun or phospho-c-Jun (Fig. 6D). This may be because only that percentage of cells contained high enough levels of MEKK1 to induce the expression of c-Jun. However, induction of c-Jun expression did correlate with induction of apoptosis, because between 48 and 72 hr after injection, approximately the percentage of cells that expressed c-Jun at 48 hr died (Fig. 6, compare B,D).
Expression of SEKAL does not prevent induction of c-Jun or apoptosis after NGF withdrawal
Because SEKAL is an efficient inhibitor of the Jun kinase pathway, we investigated whether expression of SEKAL could protect sympathetic neurons against NGF withdrawal-induced death. Sympathetic neurons were injected with pcDNA1 or pCDBcl-2 (a Bcl-2 expression vector), pSG5, or the SEKALvector together with Texas Red–dextran as a marker. The SEKAL vector was injected at the same concentration at which it had inhibited MEKK1-induced apoptosis (0.4 mg/ml). After injection, the cells were deprived of NGF and counted. Three days after NGF withdrawal, the percentage of viable injected neurons that remained was determined. The results are shown in Figure7A. As demonstrated previously (Ham et al., 1995), expression of Bcl-2 increased neuronal survival in the absence of NGF. In contrast, expression of SEKAL did not block NGF withdrawal-induced death and even appeared to decrease the percentage of viable neurons compared with pSG5. Furthermore, injection of the SEKAL expression vector at lower or higher concentrations (0.05 and 0.8 mg/ml) was not protective (J. Whitfield, unpublished observations).
Because expression of SEKAL did not prevent NGF withdrawal-induced death, we investigated whether its expression could prevent the increase in c-Jun protein levels that normally occurs after NGF withdrawal. Sympathetic neurons were injected with pCDFLAGΔ169, the SEKAL vector, or the empty pSG5 vector. The injected cells were deprived of NGF for 24 hr and then were fixed and stained with c-Jun antibody, and the percentage of injected cells expressing c-Jun was determined (Fig. 7B). As expected, the c-Jun dominant negative mutant FLAGΔ169 blocked expression of c-Jun. In contrast, expression of SEKAL did not significantly reduce the percentage of cells expressing c-Jun when compared with the corresponding empty vector pSG5. This result suggests that c-Jun expression is induced via a SEK1-independent pathway after NGF deprivation. We also found that, compared with the empty vector pSG5, microinjection of the SEKAL expression vector did not cause a significant decrease in the percentage of neurons that stained with the phospho-c-Jun antibody after NGF withdrawal (Whitfield, unpublished observations). In conclusion, the results of the injection experiments with SEKAL suggest that in sympathetic neurons the signaling pathway by which NGF withdrawal causes an increase in c-Jun protein levels, increased c-Jun phosphorylation, and apoptosis does not require the activity of SEK1.
DISCUSSION
In sympathetic neurons, the level of the transcription factor c-Jun increases after NGF withdrawal, AP-1 activity is required for cell death, and overexpression of c-Jun can induce apoptosis in the presence of NGF (Estus et al., 1994; Ham et al., 1995). Here we have studied the relationship between the Jun kinase and p38 kinase pathways and c-Jun induction and apoptosis in sympathetic neurons. In kinase assays we found that the basal level of Jun kinase activity in sympathetic neurons was very high. This was also the case in differentiated PC12 cells. In fact, Jun kinase activity increased fourfold when undifferentiated PC12 cells were treated with NGF, suggesting that this increase may be a feature of neuronal differentiation. After NGF withdrawal, Jun kinase activity increased twofold in sympathetic neurons, and this could be prevented by the addition of agents such as CPTcAMP or N-acetylcysteine, which promote neuronal survival in the absence of NGF (Rukenstein et al., 1991; Ferrari et al., 1995). Jun kinase activity was highest at 4 and 8 hr after NGF withdrawal, the time at which we previously found that c-Jun protein levels and N-terminal phosphorylation started to increase (Ham et al., 1995). Consistent with Jun kinase activation, we found in immunofluorescence experiments with a phospho-c-Jun-specific antibody that, after NGF withdrawal, c-Jun was phosphorylated on serine 63, one of the Jun kinase phosphorylation sites in the transactivation domain. In confirmation of the results of Xia et al. (1995), we found that in differentiated PC12 cells both Jun kinase and p38 kinase activity increased after NGF withdrawal. However, in the case of sympathetic neurons we did not observe activation of p38, suggesting that there are differences in the signaling pathways that are activated by NGF withdrawal in the two cell types. Consistent with this idea, we have found that neither expression of MKK3(Ala), a dominant negative mutant of MKK3 (MAP kinase kinase 3, an activator of p38 kinase) nor expression of MKK3(Glu), an activated form of MKK3, had any effect on c-Jun levels or NGF withdrawal-induced death in sympathetic neurons (Whitfield, unpublished observations).
Because c-Jun can activate transcription of the c-jungene (Angel et al., 1988), we investigated whether expression of the c-Jun dominant negative mutant FLAGΔ169 could inhibit the increase in c-Jun protein levels. We found that this was the case, suggesting that AP-1 activity is required for expression of c-Jun after NGF withdrawal. Furthermore, we demonstrated that the c-jun promoter was activated after NGF deprivation and that this required the jun1 and jun2 TRE elements. A hypothetical model consistent with these observations is presented in Figure 8. We propose that NGF withdrawal activates a kinase cascade that culminates in JNK/SAPK activation. Activated Jun kinases would then phosphorylate c-Jun and other transcription factors, such as ATF-2, which in turn would activate the transcription of target genes, including c-jun itself. One or more of the target genes would encode proteins that promote cell death.
Fig. 8.

Hypothetical model illustrating the relationship between the Jun kinase pathway and c-Jun expression and apoptosis in sympathetic neurons. After NGF withdrawal, Jun kinase activity, c-Jun levels, and c-Jun phosphorylation increase. Expression of c-Jun protein and apoptosis are blocked by the c-Jun dominant negative mutant FLAGΔ169, which inhibits AP-1 activity. NGF withdrawal-induced death is also blocked by injection of antibodies against c-Jun but not Jun B or Jun D (Estus et al., 1994). These results suggest that c-Jun activates target genes that promote apoptosis, as well as the c-jun gene itself. In other systems, JNK/SAPKs are activated by SEK1, which in turn is activated by MEKK1. Overexpression of MEKK1 in sympathetic neurons increases the level of c-Jun protein and induces apoptotic cell death, suggesting that activation of the Jun kinase pathway is sufficient to trigger apoptosis. The induction of c-Jun expression and apoptosis by MEKK1 is blocked by co-expression of the SEK1 dominant negative mutant SEKAL. However, expression of SEKAL does not block the expression of c-Jun induced by NGF withdrawal or NGF withdrawal-induced death. This might be because NGF withdrawal activates a JNKKK and JNKK that cannot be inhibited by SEKAL (a pathway parallel to MEKK1 and SEK1 that would activate JNK/SAPKs) or because the expression of c-Jun and apoptosis do not require Jun kinase activity and are induced by a different signaling pathway that is not affected by SEKAL (? leading to the c-jun gene). This would not be the p38 kinase pathway, because p38 is not activated in sympathetic neurons after NGF withdrawal.
Two predictions of our hypothetical model are (1) activation of the Jun kinase pathway in sympathetic neurons in the presence of NGF might induce apoptosis; and (2) inhibition of the pathway might protect sympathetic neurons against NGF-withdrawal-induced death. To test these ideas we performed microinjection experiments with expression vectors for a constitutively active form of MEKK1 that can activate the Jun kinase pathway strongly (Olson et al., 1995) and SEKAL, a dominant negative mutant of SEK1 (a downstream target of MEKK1 that activates Jun kinase) (Yan et al., 1994). We found that microinjection of the MEKK1 vector was sufficient to induce apoptosis in the presence of NGF, and in immunofluorescence experiments we found that expression of MEKK1 increased c-Jun levels and phospho-c-Jun staining. All of these effects could be blocked by co-expression of SEKAL. These results suggest that microinjection of the MEKK1 vector activated the Jun kinase pathway and induced apoptosis in an SEK1-dependent manner.
It is now clear that the pathway leading to Jun kinase activation is of great complexity, because at each level in the cascade a variety of different isoforms have been identified. There are three JNK genes, which through differential splicing, give rise to 10 different isoforms (Kyriakis et al., 1994; Gupta et al., 1996). In addition to SEK1, there is another Jun kinase kinase, MKK7 (Tournier et al., 1997), and others may exist (Moriguchi et al., 1995). Finally, as well as MEKK1, a number of other Jun kinase kinase kinases (JNKKKs) have been identified that can activate the JNK/SAPK pathway. These include MEKK2, MEKK3, MEKK4, and members of the mixed lineage kinase family (for review, seeKyriakis and Avruch, 1996; Gerwins et al., 1997). To determine whether inhibition of the Jun kinase pathway would block the NGF withdrawal-induced death of sympathetic neurons, we investigated whether expression of SEKAL could increase neuronal survival after NGF deprivation. Surprisingly, although injection of the SEKAL expression vector blocked MEKK1-induced apoptosis (Fig. 6C), it did not prevent NGF deprivation-induced death or expression of c-Jun after NGF withdrawal (Fig. 7). This contrasts with the results of Xia et al. (1995), who reported that expression of an SEK1 dominant negative mutant protected differentiated PC12 cells against NGF withdrawal-induced death. The reasons for this discrepancy are unclear. The mechanism by which SEKAL inhibits activation of Jun kinase is unknown. It might function by sequestering JNKKKs, such as MEKK1, that activate SEK1. Alternatively, SEKAL might bind to JNK/SAPKs and thereby prevent SEK1 from interacting with its substrate. If SEKAL functions exclusively by sequestering upstream JNKKKs, it might not inhibit c-Jun expression or NGF withdrawal-induced death if the Jun kinases in sympathetic neurons were activated by a JNKKK and JNKK that could not be inhibited by SEKAL (Fig. 8). If, on the other hand, SEKAL sequesters JNKs, an alternative interpretation of our results might be that induction of c-Jun and apoptosis after NGF withdrawal do not require Jun kinase activity, although AP-1 activity is required. Alternatively, it is possible that SEKAL may not be able to bind to all JNK isoforms. Experiments in which SEK1 dominant negative mutants have been used or in which SEK genes have been inactivated have sometimes given unexpected results. InDrosophila, the hep gene encodes a Jun kinase activator homologous to SEK1 (Sluss et al., 1996). Surprisingly, thehep1 loss-of-function mutation was found to have increased Jun kinase activity when extracts fromhep1 and wild-type embryos were compared (Riesgo-Escovar et al., 1996). Furthermore, in mammalian cells activation of Jun kinase by the tumor promoter arsenite was not inhibited by an SEK1 dominant negative mutant, which could in the same cells inhibit activation of Jun kinase by MEKK1 (Cavigelli et al., 1996). Finally, in experiments with cells isolated fromsek1−/− knock-out mice, activation of Jun kinase by anisomycin or heat shock was inhibited, whereas activation by sorbitol or UV radiation was unaffected (Nishina et al., 1997). Taken together, these results suggest that some signals can activate Jun kinase by SEK1-independent pathways, which therefore may not be inhibited by SEK1 dominant negative mutants.
What molecules might be responsible for activating the Jun kinase pathway and increasing c-Jun protein levels after NGF withdrawal? In some systems it has been reported that Jun kinase activation is a consequence of caspase activation (Frisch et al., 1996). However, in the case of sympathetic neurons it has been shown that a broad spectrum caspase inhibitor, Boc-aspartyl(OMe)-fluoromethylketone, which protects sympathetic neurons against apoptosis and which inhibited PARP cleavage and ICE homolog-1 (ICH-1) activation, did not prevent the induction of c-jun RNA after NGF withdrawal (Deshmukh et al., 1996). This suggests that the induction of c-jun RNA after NGF withdrawal does not require caspase activity and that the signaling pathway that regulates c-Jun expression is upstream of caspase activation. Similarly, caspase inhibitors prevented apoptosis, but not Jun kinase activation, when differentiated PC12 cells were deprived of NGF (Park et al., 1996). In other systems it has been demonstrated that the small GTPases Cdc42 and Rac1 can specifically activate the Jun kinase pathway (Coso et al., 1995; Minden et al., 1995; Olson et al., 1995). Interestingly, expression of dominant negative mutants of Cdc42 or Rac1 in sympathetic neurons can inhibit the expression of c-Jun protein after NGF withdrawal and can prevent NGF withdrawal-induced death (C. Bazenet, M. Mota, and L. L. Rubin, unpublished observations), suggesting that activation of Cdc42, Rac1, or related proteins may trigger the signaling pathway that leads to c-Jun induction.
In conclusion, we have demonstrated that in sympathetic neurons NGF withdrawal leads to Jun kinase activation and increased phosphorylation of c-Jun. Activation of the Jun kinase pathway by expression of MEKK1 was sufficient to induce apoptosis in the presence of NGF. However, expression of a dominant negative mutant of SEK1 did not block expression of c-Jun or apoptosis after NGF withdrawal, suggesting that c-Jun expression may be regulated by an SEK1-independent pathway (Fig. 8). Future work will be directed toward identifying the components of the kinase cascade that lead to Jun kinase activation in sympathetic neurons after NGF deprivation and to establishing whether Jun kinase is activated in other models of neuronal cell death.
Note added in proof
The data presented here are consistent with the recent results ofVirdee et al. (1997) who also observed that Jun kinase activity and c-Jun phosphorylation increase in NGF-deprived sympathetic neurons.
Footnotes
This work was supported by the Eisai Company of Japan. We thank Chantal Bazenet and Jim Staddon for helpful discussions and critical reading of this manuscript. We are also grateful to Alan Ashworth and Mike Olson for providing the expression vector for MEKK1, Leonard Zon for providing the SEKAL vector, Peter Angel for the c-jun CAT reporter genes, and Dominique Lallemand, John Kyriakis, and Angel Nebreda for providing the phospho-c-Jun, Jun kinase, and p38 antibodies. We also thank Howard Desmond for peptide synthesis.
A.E. and J.W. made equal contributions to this work.
Correspondence should be addressed to Dr. Jonathan Ham, Eisai London Research Laboratories, Bernard Katz Building, University College London, Gower Street, London WC1E 6BT, UK.
REFERENCES
- 1.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 2.Cavigelli M, Li WW, Lin A, Su B, Yoshioka K, Karin M. The tumour promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. EMBO J. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- 3.Coso OA, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 4.Dérijard B, Raingeaud J, Barrett T, Wu I-H, Han J, Ulevitch RJ, Davis RJ. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 5.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doherty P, Mann DA, Walsh FS. Comparison of the effects of NGF, activators of protein kinase C, and a calcium ionophore on the expression of Thy-1 and N-CAM in PC12 cell cultures. J Cell Biol. 1988;107:333–340. doi: 10.1083/jcb.107.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari G, Yan CYI, Greene LA. N-Acetylcysteine (D- and L-stereoisomers) prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frisch SM, Vuori K, Kelaita D, Sicks S. A role of Jun-N-terminal kinase in anoikis: suppression by bcl-2 and crmA. J Cell Biol. 1996;135:1377–1382. doi: 10.1083/jcb.135.5.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerwins P, Blank JL, Johnson GL. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J Biol Chem. 1997;272:8288–8295. doi: 10.1074/jbc.272.13.8288. [DOI] [PubMed] [Google Scholar]
- 11.Gupta S, Campbell D, Dérijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 12.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 13.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 14.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 15.Hirai S-I, Ryseck R-P, Mechta F, Bravo R, Yaniv M. Characterization of jun D: a new member of the jun proto-oncogene family. EMBO J. 1989;8:1433–1439. doi: 10.1002/j.1460-2075.1989.tb03525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 17.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 18.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 19.Lallemand D, Spyrou G, Yaniv M, Pfarr CM. Variations in Jun and Fos protein expression and AP-1 activity in cycling, resting and stimulated fibroblasts. Oncogene. 1997;14:819–830. doi: 10.1038/sj.onc.1200901. [DOI] [PubMed] [Google Scholar]
- 20.Lin A, Minden A, Martinetto H, Claret F-X, Lange-Carter C, Mercurio F, Johnson GL, Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290. doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- 21.Livingstone C, Patel G, Jones N. ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM. Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minden A, Lin A, Claret F-X, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 24.Moriguchi T, Kawasaki H, Matsuda S, Gotoh Y, Nishida E. Evidence for multiple activators for stress-activated protein kinases/c-Jun amino-terminal kinases. J Biol Chem. 1995;270:12969–12972. doi: 10.1074/jbc.270.22.12969. [DOI] [PubMed] [Google Scholar]
- 25.Nishina H, Fischer KD, Radvanyi L, Shahinian A, Hakem R, Rubie EA, Bernstein A, Mak TW, Woodgett JR, Penninger JM. Stress-signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature. 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- 26.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 27.Papavassiliou AG, Treier M, Bohmann D. Intramolecular signal transduction in c-Jun. EMBO J. 1995;14:2014–2019. doi: 10.1002/j.1460-2075.1995.tb07193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway. J Biol Chem. 1996;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 29.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 30.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 31.Raingeaud J, Whitmarsh AJ, Barrett T, Dérijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 1996;10:2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- 33.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 34.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 36.Sluss HK, Han Z, Barrett T, Davis RJ, Ip Y-T. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996;10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 37.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusion proteins with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 38.Suda K, Barde Y-A, Thoenen H. Nerve growth factor in mouse and rat serum: correlation between bioassay and radioimmunoassay determinations. Proc Natl Acad Sci USA. 1978;75:4042–4046. doi: 10.1073/pnas.75.8.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ. Mitogen-activated kinase kinase 7 is an activator of the c-Jun amino-terminal kinase. Proc Natl Acad Sci USA. 1997;94:7337–7342. doi: 10.1073/pnas.94.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P. ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 1995;14:1798–1811. doi: 10.1002/j.1460-2075.1995.tb07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Virdee K, Bannister AJ, Hunt SP, Tolkovsky AM. Comparison between the timing of JNK activation, c-Jun phosphorylation, and onset of death commitment in sympathetic neurons. J Neurochem. 1997;69:550–561. doi: 10.1046/j.1471-4159.1997.69020550.x. [DOI] [PubMed] [Google Scholar]
- 42.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 44.Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]