

Abstract
Nicotine is reported to increase arousal and attention and to elevate mood, effects that are most often associated with changes in the function of monoaminergic neuromodulatory systems (Feldman et al., 1997). Recent studies have shown a nicotinic receptor-mediated presynaptic enhancement of fast glutamatergic (McGehee et al., 1995;Gray et al., 1996) and GABAergic (Léna and Changeux, 1997) transmission. However, the mechanism of nicotinic effects on metabotropic-mediated transmission in general, and on monoaminergic transmission in particular, is less well understood. We have examined nicotinic effects on dorsal raphe neurons of rats using whole-cell current and voltage-clamp recording techniques in vitro. In the majority of these neurons, activation of presynaptic nicotinic receptors induced a depolarization mediated by norepinephrine acting on α1 receptors. Blockade of this response revealed a hyperpolarization mediated by serotonin acting on 5-HT1A receptors. Because the norepinephrine effect was sensitive to methyllycaconitine (100 nm), it is concluded that nicotinic receptors with an α7 subunit can facilitate release of norepinephrine to activate metabotropic receptors. In contrast, methyllycaconitine-insensitive nicotinic receptors can induce 5-HT release in the dorsal raphe nucleus.
Keywords: norepinephrine, serotonin, acetylcholine, methyllycaconitine, dorsal raphe, electrophysiology
An interaction between brainstem cholinergic and serotonergic neurons mediated by cholinergic projections to the dorsal raphe (DR) nucleus has been implicated in the control of the desynchronized sleep–slow wave sleep cycle (McCarley and Hobson, 1975; Steriade and McCarley, 1990; McCarley et al., 1995). Elsewhere in the brain, there is also considerable overlap in the afferent targets of cholinergic (ACh), noradrenergic (NE), and serotonergic (5-HT) neurons that provides an anatomical basis for additional interactions between these neuromodulatory systems (Foote et al., 1983; Semba and Fibiger, 1989; Jacobs and Azmitia, 1992). Furthermore, biochemical studies have demonstrated a cholinergic enhancement of monoaminergic transmitter release in cortical tissue (Summers and Giacobini, 1995; Clarke and Reuben, 1996; Role and Berg, 1996; Wonnacott, 1997). However, the consequences of this enhanced monoamine release in target nuclei have not been examined at the cellular level.
The DR nucleus, which contains most of the rostrally projecting serotonergic neurons in the CNS (Tork, 1990), receives afferent input from both brainstem ACh and NE neurons (Jones and Moore, 1977). The NE input, via activation of postsynaptic α1 receptors (Yoshimura et al., 1985; Pan et al., 1994), acts as an excitatory drive onto DR neurons. This excitatory drive is likely to contribute to DR activity during waking (Baraban and Aghajanian, 1980) when NE neurons of the locus coeruleus have high tonic activity (Hobson et al., 1975; Aston-Jones and Bloom, 1981). In addition, serotonergic neurons of the DR can inhibit their own activity via the activation of 5-HT1Aautoreceptors (Aghajanian et al., 1972; Yoshimura and Higashi, 1985) that cause an increase in an inwardly rectifying potassium conductance (Williams et al., 1988; Penington et al., 1993). In contrast, the electrophysiological effects of cholinergic input onto DR neurons are less well characterized. These cholinergic effects are likely to be important because the majority of brainstem cholinergic neurons have their highest activity during waking and it is in this behavioral state that the highest activities of both NE and 5-HT neurons are observed (McCarley and Hobson, 1975; Steriade and McCarley, 1990; McCarley et al., 1995).
MATERIALS AND METHODS
Transverse brainstem slices were prepared from anesthetized 18- to 28-d-old hooded Long–Evans rats with procedures similar to those used in the preparation of slices containing the laterodorsal tegmental and pedunculopontine tegmental nuclei (LDT/PPT) nuclei (Luebke et al., 1993), except they were obtained from a more rostral region of the pontine–mesencephalic brainstem that contained the dorsal raphe nucleus. The whole-cell recording “blind-patch” technique ofBlanton et al. (1989) was used in conjunction with the use of a submerged-slice recording chamber with 500 μl of dead space and a rapid change drug application system (see below). Borosilicate glass patch electrodes, with a range of resistance from 4 to 6 MΩ, were filled with (in mm): K methylsulfate 130, CaCl21, MgCl2 3, HEPES 10, EGTA 3, MgATP 2, and NaGTP 0.2, 290–300 mOsm, pH 7.34. Current-clamp recordings were made with an Axoclamp 2A amplifier and pClamp 6.1 software (Axon Instruments, Burlingame, CA). Voltage-clamp recordings were obtained either with an Axoclamp 2A in discontinuous voltage-clamp mode or with an Axopatch 2D using series resistance compensation. The slices were continuously perfused with artificial CSF consisting of (in mm): NaCl 125, MgCl2 1.3, CaCl2 2.5, KH2PO4 3, glucose 10, and NaHCO326, pH 7.35, at 30°C. Drugs were applied by addition to the perfusion medium (flow rate of 1.5 ml/min) using multiple perfusion lines that were funneled into a single outlet near the recording area. The particular barrel to be used as a source of the medium could then be remotely selected for rapid media exchange (<1 sec in most cases) because of the small volume of dead space. Averaged data are reported as the mean ± SEM.
RESULTS
Nicotinic receptors mediate cholinergic hyperpolarization and depolarization in the dorsal raphe
Application of ACh to DR neurons of adult rats in vitroinduced a depolarization in seven neurons (6.0 ± 1.5 mV), a hyperpolarization in four neurons (4.0 ± 1.1 mV), and no effect on membrane potential in two neurons (<0.5 mV) (Fig.1A). Because the ACh effects might be mediated by either nicotinic or muscarinic receptors, the application of the muscarinic agonist methacholine was examined. At a concentration of 1 μm, no effects on membrane properties were observed in 9 of 11 neurons tested. In the remaining two neurons, methacholine evoked a small depolarization that was sensitive to the muscarinic antagonist atropine (5.0 μm).
Fig. 1.
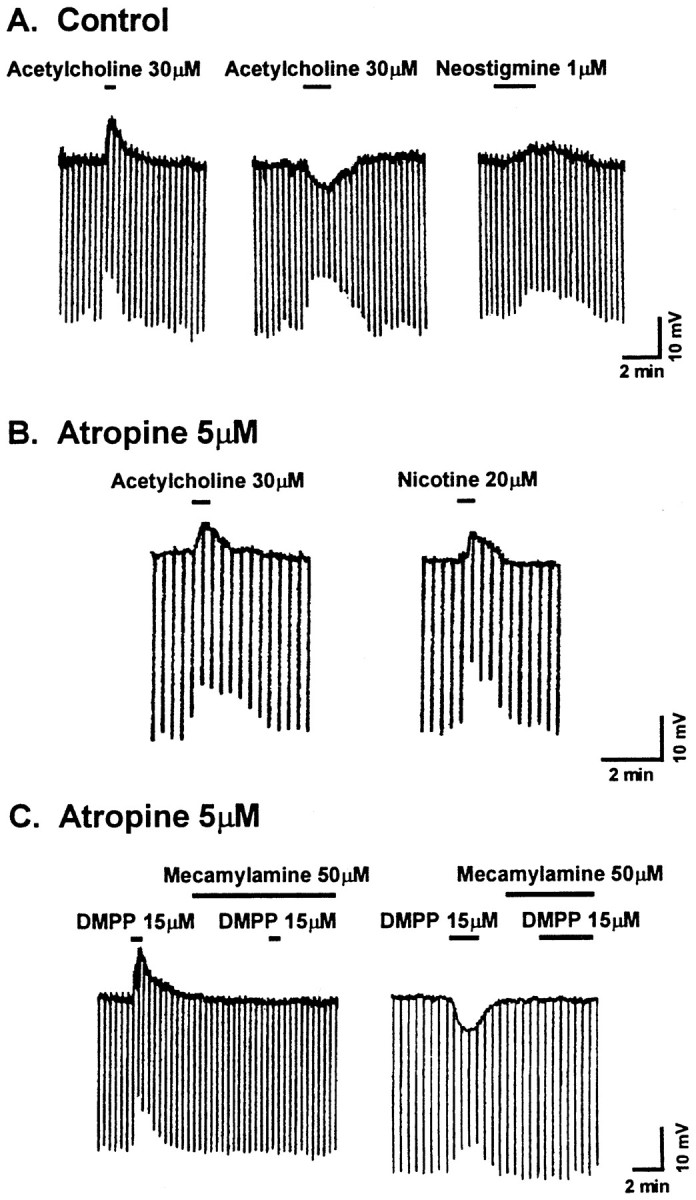
Acetylcholine activates nicotinic responses in dorsal raphe neurons. A, Three voltagetraces show typical depolarizing and hyperpolarizing membrane potential responses to acetylcholine and a depolarizing response to neostigmine from three different neurons. All responses are associated with a decrease in input resistance, measured with intracellular current injection (600 msec in duration; 50 pA in amplitude; downward deflections in all traces).B, In the presence of the muscarinic antagonist atropine, acetylcholine and nicotine both induce a membrane depolarization, suggesting activation of nicotinic receptors.C, The role of nicotinic receptors in the cholinergic responses is supported further by blockade of both depolarizing and hyperpolarizing responses to the nicotinic agonist DMPP by the nicotinic antagonist mecamylamine in two different neurons in the presence of atropine. The depolarizing response recovers by >50% after a 10 min wash in control medium, and the hyperpolarizing response recovers by >90% after a 6 min wash.
In subsequent experiments, atropine (5.0 μm) was applied to limit cholinergic effects to the activation of the nicotinic receptor subtypes only. Under these conditions, ACh evoked both depolarizing and hyperpolarizing responses in DR neurons, with the majority (60%) being depolarizing responses (n = 8; Fig. 1B). Both polarities of response were associated with a decrease in the membrane input resistance (Rm). Two additional neurons showed no alteration in the membrane potential but showed a decrease of 30 and 40% in their Rm values. Similar responses were also obtained with nicotine (20 μm;n = 7; Fig. 1B) or with the synthetic nicotinic receptor agonist 1,1-dimethyl-4-phenylpiperazinium (DMPP; 15 μm; n = 38; Fig. 1C), suggesting that these responses were mediated by nicotinic receptor activation. However, nicotine and another nicotinic agonist, lobeline, can inhibit [3H]MK-801 (dizocilpine maleate) binding in neural tissue, suggesting a potential action for these agonists at NMDA receptors (Aizenman et al., 1991). This did not seem to be the case because application of 2-amino-5-phosphonovalerate (APV), an NMDA antagonist (50 μm), did not affect the DMPP responses (n = 4).
Moreover, the nicotinic nature of these responses was supported further by the observation that they were blocked by a nicotinic receptor antagonist, mecamylamine (50 μm; n = 4; Fig. 1C), that was applied at a concentration unlikely to distinguish between different nicotinic receptor subtypes. Of two additional nicotinic receptor antagonists examined, one, dihydro-β-erythroidine (500 μm), was ineffective, and the other, hexamethonium, was only partially effective at high concentrations (1 mm). Interestingly, repeat application of DMPP resulted in a gradual reduction of the depolarizing response until only a hyperpolarizing response was elicited (data not shown).
The DR nucleus receives cholinergic afferent fibers that could activate the nicotinic receptors described above (Jones and Moore, 1977). In the experimental conditions of the in vitro slice, this activation might be subthreshold but could potentially be enhanced by blocking the major catabolic enzyme for cholinergic transmission, acetylcholinesterase. The addition of the cholinesterase inhibitor neostigmine (1 μm; n = 11) to the perfusion medium induced a decrease in input resistance in all cells (n = 11) and a depolarization in six cells (3.1 ± 0.5 mV), a hyperpolarization in three cells (2.5 ± 0.4 mV), and no change in membrane potential but a decrease in input resistance in the remaining two neurons tested (Fig. 1A).
Selective sensitivity to TTX and low calcium
The possibility of either a pre- or postsynaptic locus of the DMPP-mediated responses was examined next. In current clamp, application of tetrodotoxin (TTX; 0.6 μm; Fig.2A,B) antagonized the DMPP-induced depolarization (n = 7) and revealed a hyperpolarizing response in four of seven neurons tested, no hyperpolarization but a decrease in Rm in two of seven neurons, and a markedly reduced depolarization in the remaining neuron. These data were consistent with a depolarization that was indirect and mediated by a presynaptic activation of nicotinic receptors that required sodium-dependent (TTX-sensitive) inward current to evoke the depolarizing synaptic transmission. In addition, in perfusate containing low Ca2+ (10 μm) and high Mg2+ (10 mm), the DMPP-induced depolarization was also blocked, and a hyperpolarization was unmasked in all neurons tested (n = 4; Fig. 2C). This latter effect also suggested a calcium dependence of the depolarization, and although the site of the antagonism of the calcium flux was not determined in this study, it might be either the voltage-sensitive calcium channels or the nicotinic-gated cation channels or both.
Fig. 2.

The two nicotinic responses are selectively affected by either TTX or low calcium and high magnesium in the perfusion medium. A, Two voltage tracesfrom the same neuron before and during exposure to TTX show a TTX-dependent blockade of the depolarizing response that reveals a TTX-insensitive hyperpolarizing response to the nicotinic agonist DMPP.B, A histogram of the average of the voltage amplitude of the nicotinic response from the same neurons before (control) and during exposure to TTX demonstrates the reversal of polarity of this response.C, Voltage traces show that the addition of low calcium and high magnesium to the perfusion medium also reverses the polarity of the nicotinic response. D, In the presence of prazosin (1 μm), the nicotinic response is hyperpolarizing, and the hyperpolarization is unaffected by low calcium and high magnesium.
In contrast to the depolarizing DMPP response, the hyperpolarization was insensitive to TTX and insensitive to low Ca2+(Fig. 2A–D). This initially suggested a postsynaptic site of action similar to that for a nicotinic response described previously in septal neurons (Wong and Gallagher, 1989).
Nicotinic depolarization mediated by α1 noradrenergic receptors
Two of the putative neurotransmitters identified as excitatory to DR neurons include glutamate and norepinephrine. Both a fast glutamate-mediated EPSP (Pan and Williams, 1989a) and an α1 adrenoceptor-mediated slow EPSP (Yoshimura et al., 1985) have been described in DR neurons in vitro. Recently, nicotine has been demonstrated to increase glutamate release from presynaptic terminals in the cortex (McGehee et al., 1995; Gray et al., 1996). However, bath application of the NMDA receptor antagonist APV (100 μm) and the AMPA receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX; 10 μm) did not reduce the DMPP-mediated depolarization (n = 4). In contrast, the α1 adrenoceptor antagonist prazosin (1 μm; n = 4) blocked the depolarizing response and unmasked a membrane hyperpolarization (Fig.3A). Moreover, in voltage clamp, DMPP induced a nonreversing, barium-insensitive, inward current that had a voltage sensitivity similar to that of the current induced by the α1 receptor agonist phenylephrine (n = 4; Fig.3B,C), reported previously as barium-insensitive (Pan et al., 1994).
Fig. 3.

Both pharmacological and electrophysiological evidence suggests that noradrenergic α1 receptors and serotonergic 5-HT1A receptors mediate the nicotinic depolarizing and hyperpolarizing responses, respectively. A, Two voltagetraces illustrate a blockade of the DMPP-induced depolarizing response that reveals a hyperpolarizing response by exposure to the α1 antagonist prazosin added to the bath medium.B, A graph of current versus membrane potential shows the change in the neuronalI–V relationship when the α1 agonist phenylephrine is applied to the perfusion medium. C, Agraph similar to that described in B is shown except that DMPP was used in place of phenylephrine to show the similarity of the change induced in theI–V curves by these two agonists. (Barium was present in B and C to block the hyperpolarizing response.) D, Two voltagetraces taken during perfusion with prazosin (1 μm) show the DMPP-induced hyperpolarization that is blocked by the 5-HT1A antagonist pindobind-5-HT1A. E, F, Agraph of I–V curves shows the effect of 5-HT (E) and can be compared with the I–V curves showing the effect of the hyperpolarizing response to DMPP (F). In both cases, the agonists induce an inwardly rectifying current that reverses near the potassium equilibrium potential.
The DMPP and phenylephrine inward currents were both associated with a decrease in slope conductance that was more pronounced at the resting membrane potential than at more hyperpolarized potentials. However, under control conditions, the depolarizing response observed in most neurons was associated with a decrease in input resistance. This was likely to result from the induction by DMPP of both a voltage-sensitive inward current that decreased the whole-cell slope conductance (Fig.3C) and an increased inwardly rectifying conductance (Figs.3F,4A,B) that was small near the resting membrane potential and more pronounced at hyperpolarized potentials. This latter conductance was likely responsible for the hyperpolarization induced by nicotinic agonists when the depolarizing response was blocked. Under control conditions, nicotinic receptor activation induced both conductances; the result was a depolarization observed in conjunction with an apparent decrease in resistance. The apparent decrease in resistance likely reflected the measurement of the resistance by hyperpolarizing current pulses, because hyperpolarization would activate the inwardly rectifying current and thereby shunt the hyperpolarizing current pulse.
Fig. 4.

The inwardly rectifying current induced by DMPP is completely blocked by barium (100 μm). A, A graph of the whole-cell current [prazosin (1 μm) was present at all times] versus the membrane potential at which it was measured is shown with two curves. Thecontrol curve was obtained before exposure to DMPP (15 μm), and the curve labeled DMPP was obtained during the exposure. B, The curve shown was calculated by subtraction of the control curve from theDMPP curve to give the current generated by DMPP. An inward rectification of the current similar to that evoked by 5-HT1A agonists is apparent. C,D, The two graphs are similar to those inA and B except that barium (100 μm) was present. The current induced by DMPP was completely blocked.
Nicotinic hyperpolarization mediated by 5-HT1A receptors
The finding that prazosin blocked the DMPP-induced depolarization and left the DMPP-induced hyperpolarization intact enabled an examination of the hyperpolarizing response in isolation. In contrast to our initial supposition of a direct nicotinic action, the prazosin-insensitive hyperpolarization was blocked by the 5-HT1A antagonist pindobind-5-HT1A (1 μm; n = 6; Fig. 3D). In addition, the pindobind-sensitive current had a number of similarities to the 5-HT-induced inwardly rectifying potassium current, including voltage sensitivity, a similar reversal potential near the potassium equilibrium potential (Fig.3E,F), and a sensitivity to Ba2+ ions (Williams et al., 1988; Penington et al., 1993) (Fig. 4A–D).
Nicotinic-induced release of monoamines and reuptake antagonism
The DR nucleus has well characterized reuptake systems for both NE (Donnan et al., 1991; Tejani-Butt, 1992; Ordway et al., 1997) and 5-HT (Cortes et al., 1988; Hrdina et al., 1990; Stockmeier et al., 1996). Moreover, each uptake system has been shown to regulate the decay rate of synaptic responses mediated by the release of these two monoamines (Pan and Williams, 1989b). Consequently, any alteration of monoamine reuptake should affect the DMPP-mediated response of DR neurons if, indeed, the alteration results from presynaptic nicotinic receptor-mediated regulation of monoamine release. In the presence of barium (100 μm) and the NE reuptake inhibitor nisoxetine (50 nm), the time constant of decay for the DMPP-mediated depolarizing response (n = 5) was increased by 150 ± 10% (p ≤ 0.03, Walsh test; Fig.5A). Similarly, in the presence of prazosin and the 5-HT reuptake inhibitor fluoxetine (20 μm), the decay rate of the hyperpolarizing response to DMPP (n = 6) was increased by 220 ± 20% (p ≤ 0.02, Walsh test; Fig. 5B). These observations were consistent with the hypothesis of a DMPP-mediated action on presynaptic monoaminergic release sites. Thus, nicotinic receptor activation seemed to evoke the release of both NE and 5-HT and thereby activate both α1 and 5-HT1Areceptors on DR neurons, and furthermore, under control conditions, the depolarizing effect of α1 receptor activation predominated.
Fig. 5.

Antagonism of the noradrenaline and serotonin transporters prolongs the depolarizing and hyperpolarizing nicotinic responses, respectively. A, Two voltagetraces of the depolarizing DMPP response, in the presence of barium to block the hyperpolarizing response, show a decreased decay rate when the noradrenaline transport inhibitor nisoxetine is added to the perfusion medium. B, In the presence of prazosin to block the DMPP depolarizing response, the decay rate of the hyperpolarizing response is slowed by exposure to the serotonin transport inhibitor fluoxetine.
Methyllycaconitine selectively antagonizes the NE depolarizing response
The two nicotinic responses differed in regard to their sensitivity to one of the most selective of the nicotinic antagonists currently available. Methyllycaconitine (MLA), an antagonist of nicotinic receptors that possess an α7 subunit (Alkondon et al., 1992; Alkondon and Albuquerque, 1993), at a concentration of either 100 nm (n = 4; Fig.6) or 1.0 μm(n = 5) selectively blocked the DMPP-induced depolarization but did not affect the hyperpolarizing response. When the depolarization response was isolated by selective blockade of the hyperpolarizing response with barium (100 μm;n = 3; Fig. 6B) or with pindobind-5-HT1A (1 μm; n = 2), the MLA blockade of the depolarizing response was complete at 100 nm concentration. At the same concentration, MLA had no effect on the isolated (prazosin at 1 μm;n = 4; Fig. 6C) hyperpolarizing response. Mecamylamine (30 μm; n = 5) blocked both the DMPP-induced inward and outward currents, but the antagonism of the hyperpolarizing response was not complete, whereas the antagonism of the depolarizing response was. When the hyperpolarizing response was isolated (prazosin at 1 μm), 30 μmmecamylamine reduced it by 61 ± 11% (n = 5), whereas at 50 μm (n = 4), the isolated outward DMPP-induced current was completely blocked (Fig.1C).
Fig. 6.
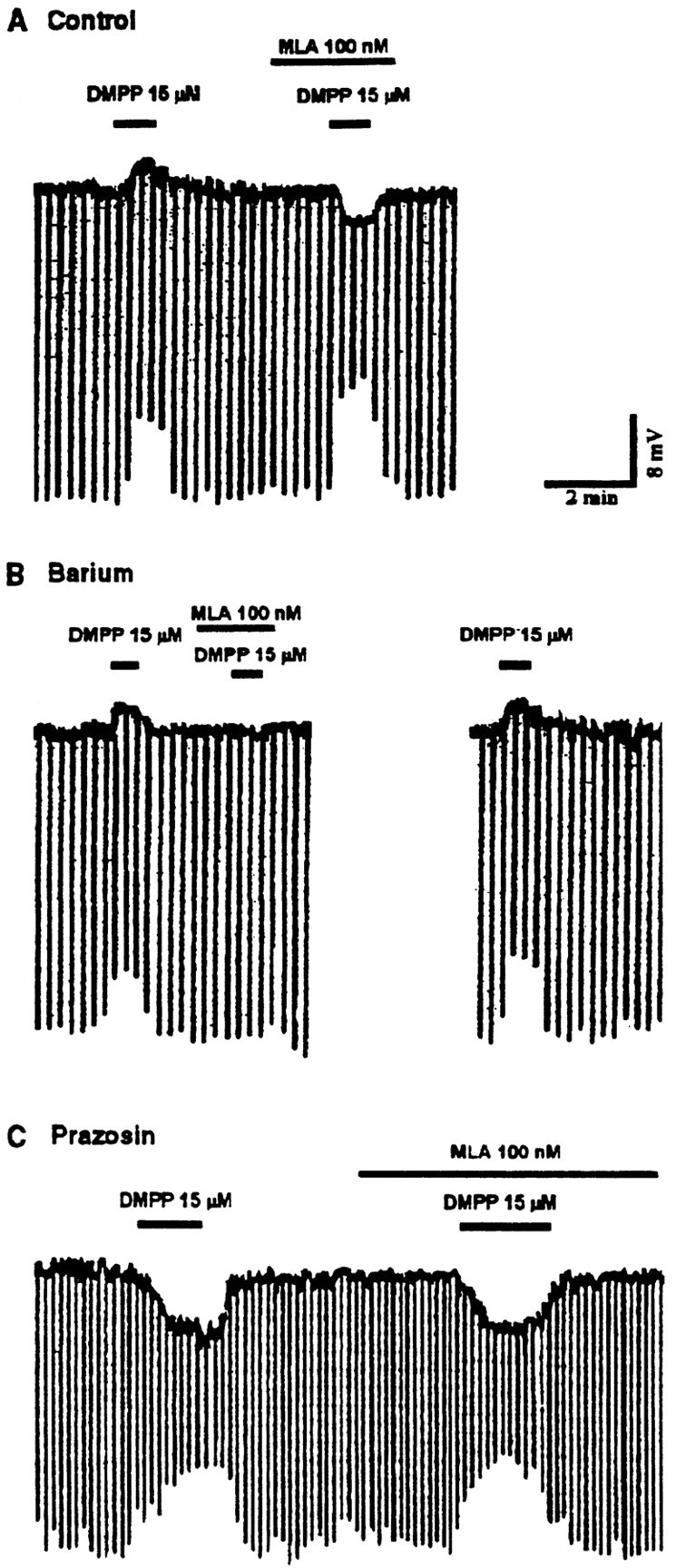
Methyllycaconitine selectively blocks the NE depolarizing response. A, A voltage traceshowing the blockade of the depolarizing response to DMPP by MLA reveals a hyperpolarizing response to DMPP. B, In the presence of barium (100 μm), the hyperpolarizing response to DMPP is blocked, and the depolarizing response can be seen in isolation. Under these conditions, the complete blockade of the response by MLA is confirmed. C, In the presence of prazosin (1 μm), the hyperpolarizing response to DMPP can be seen in isolation. Under these conditions, the response is unaffected by MLA.
DISCUSSION
In the majority of DR neurons, nicotinic agonists elicited a depolarization that was mimicked by the α1 agonist phenylephrine, blocked by the α1 antagonist prazosin, and prolonged by the NE uptake inhibitor nisoxetine. Furthermore, in control conditions the depolarizing response predominated and was selectively blocked by low concentrations of MLA. As seen with nicotinic stimulation of glutamate release (McGehee et al., 1995; Gray et al., 1996) and GABA release (Léna and Changeux, 1997), the depolarization was blocked by TTX, suggesting that the depolarizing effects of DMPP were caused by a presynaptic nicotinic receptor-mediated release of NE that acted on postsynaptic α1 receptors (Fig. 7). Spontaneous miniature postsynaptic monoaminergic currents could not be resolved in DR neurons, and this precluded an analysis of nicotinic effects on the frequency or amplitude of spontaneous miniature events. The dependence of the nicotinic-induced release of NE on voltage-sensitive sodium channels suggested a susceptibility of this effect to presynaptic inhibition, especially if the mechanism of inhibition was attributable to a shunting of depolarizing currents in the terminals. This stands in contrast to the TTX-insensitive, nicotinic-stimulated release of 5-HT discussed below.
Fig. 7.

Schematic of postulated mechanism of nicotinic receptor activation in the dorsal raphe nucleus. Cholinergic fibers from the cholinergic neurons of the laterodorsal and pedunculopontine nuclei may activate presynaptic nicotinic receptors located on both noradrenergic neurons from the locus coeruleus and serotonergic dorsal raphe neurons to facilitate release of noradrenaline and serotonin onto dorsal raphe neurons. The release of serotonin (*) may be from either dendritic or axonal sources or both.
When the α1 response was blocked, DMPP elicited a hyperpolarization, which was mimicked by 5-HT, antagonized by the 5-HT1Aantagonist pindobind, and prolonged by the 5-HT uptake inhibitor fluoxetine. This suggested that the hyperpolarizing effect of DMPP was caused by the release of 5-HT acting on postsynaptic 5-HT1Aautoreceptors (Fig. 7). Nicotinic-stimulated release of 5-HT, but not NE, was observed when either sodium-dependent action potentials or Ca2+ flux across the membrane were antagonized.
There are several possibilities that might account for the nicotinic-evoked release of 5-HT in the low Ca2+ and high Mg2+ solution. For example, transmitter release can be evoked in the absence of calcium flux across the membrane, as reported recently for insulin stimulation of neuropeptide secretion (Jonas et al., 1997), although in this case the receptor effector mechanism was metabotropic, unlike the ionotropic nicotinic receptors currently characterized in the CNS. In peripheral tissue, observations suggesting nicotinic-evoked neuropeptide secretion in the presence of calcium current antagonists are not unprecedented. Frog pituitary melanotrophs display a nicotinic-evoked release of α-MSH under these conditions that seemed to be mediated by a phosphoinositol-dependent increase of intracellular calcium (Garnier et al., 1994). However, this effect was not sensitive to other classical nicotinic agonists or antagonists, including acetylcholine. In mouse C2C12 myotubes, Grassi et al. (1993) reported a nicotinic receptor-induced increase in inositol phospholipid turnover that was sensitive to acetylcholine. Thus, metabotropic responses associated with transmitter release can be evoked by nicotinic agonists, although the molecular relationship of the receptors responsible for these effects to other nicotinic receptors remains to be determined.
An alternative, but not mutually exclusive, possibility is that 5-HT release from DR neurons requires less calcium than does the better-characterized fast amino acid transmitter systems. An inward current that would accompany an inward calcium flux was not induced by nicotinic agonists in this preparation. This suggests either an electrotonically distal location for this putative influx (for example, in axon terminals) and/or an amplitude of <5 pA of influx current. It should be noted that the low Ca2+ and high Mg2+ solution used in this study reduced but did not completely abolish Ca2+ flux or miniature synaptic currents. Accordingly, even in the presence of a partial blockade of calcium flux, as seen with the low Ca2+ and high Mg2+ medium used in this study, nicotinic activation may still provide sufficient calcium influx for 5-HT release of the same magnitude as that observed in control medium (Fig.2D). In this regard, a little-characterized aspect of 5-HT release in the dorsal raphe nucleus is that the presynaptic source of the 5-HT may be either recurrent axon terminals and/or dendodendritic sites. The possibility of the latter site is raised by observations of 5-HT-containing vesicles in dendrites of 5-HT neurons in the DR (Chazel and Ralston, 1987). However, it is not known whether 5-HT can be released from these vesicles nor what the relationship to intracellular calcium concentration might be.
Finally, it is conceivable that nonvesicular release of 5-HT occurs that may be altered by nicotinic activation. However, it seems unlikely that the 5-HT transporter is involved. The 5-HT transporter inhibitor fluoxetine did not abolish the nicotinic-induced hyperpolarization. Rather, this antagonist increased the time course of the decay of the hyperpolarization.
Numerous subunits of the nicotinic receptor have been described that can combine to form multiple functional receptors, suggesting a wide diversity of nicotinic acetylcholine receptors in vivo. One of the few pharmacological tools currently available with specificity for receptors containing the α7 subunit, MLA, was used in this study to block selectively the nicotinic depolarizing response that was caused by release of NE. Although the α8 and α9 subunits have some similarity with the α7 subunit and thus might bind MLA, there is no evidence for the existence of either of these two subunits in the brainstem tegmentum. This would suggest that the noradrenergic cells of the locus coeruleus can encode for α7 subunits and that nicotinic receptors capable of inducing the release of transmitter from their terminals also contain these α7 subunits.
In apparent contrast to the findings of the present study, evidence for a relative insensitivity of nicotinic-induced [3H]NE release in the hippocampus (as compared with nicotinic-induced dopamine release) to methyllycaconitine (Clarke and Reuben, 1996) and α-bungarotoxin (Sershen et al., 1997) has been reported. In the former study, the nicotinic-induced release of [3H]NE was not significantly affected by TTX, whereas in the latter, it was reduced but not eliminated, unlike the situation in the dorsal raphe. At the same concentration of nicotine used to assess electrophysiological effects in the dorsal raphe (20 μm, near maximal effect), the increase over the basal level of [3H]NE release was <25% of maximum in the hippocampus. Thus, both the mechanism for the nicotinic-evoked increase in [3H]NE release and the receptor mediating the effect in the hippocampus are probably different than that for the nicotinic-evoked NE synaptic potential observed in the dorsal raphe. The possibility of an electrophysiologically relevant nicotinic-induced NE response in the hippocampus that involves α7 subunits cannot yet be excluded.
At present, there is little evidence to suggest the nature of the nicotinic receptor(s) mediating the 5-HT response, except a lack of sensitivity for methyllycaconitine. Some selectivity of antagonism was also noted for mecamylamine, but this was only partial. In the hippocampus, mecamylamine showed little selectivity in blocking either methyllycaconitine-sensitive or -insensitive responses (Clarke and Reuben, 1996).
Implications for behavioral state control
During the behavioral state of waking, cholinergic, noradrenergic, and serotonergic activity is high. Under these conditions, nicotinic receptor activation may have a predominately excitatory effect on DR neurons, consistent with our results in control conditions in vitro (Fig. 1). This excitation would most likely have the greatest amplitude when both cholinergic and noradrenergic neurons are active. It could thus impart greater specificity of effect to divergent NE neuronal projections not only in DR target sites but in other CNS areas as well, because the NE terminals receiving coincidently active, cholinergic input could have the greatest influence.
Activation of 5-HT1A autoreceptors in the DR is sufficient to generate and maintain increases in REM sleep (Portas et al., 1996). In the transition from slow wave sleep to REM sleep, cholinergic activity increases, and monoaminergic activity ceases. Inhibition in the DR increases as indicated by the increased release of GABA in this region during REM (Nitz and Siegel, 1997). If the GABAergic inhibition during REM includes the noradrenergic terminals, then the possibility of a transition in polarity of cholinergic influence on DR neurons from an excitatory to an inhibitory effect exists. This is because nicotinic receptor stimulation of 5-HT release that is inhibitory may be resistant to GABAergic presynaptic inhibition. Nicotinic activation of the DR during REM sleep may thus increase a 5-HT1A-dependent inhibition.
Footnotes
This work was supported by the Department of Veterans Affairs and MH39, 683. We acknowledge and thank Kevin McLaughlin and Melissa Madrick for technical assistance.
Correspondence should be addressed to Dr. Robert W. Greene, Neuroscience Laboratory 151-C, Veterans Administration Medical Center, 940 Belmont Street, Brockton, MA 02401.
REFERENCES
- 1.Aghajanian GK, Haigler HJ, Bloom FE. Lysergic acid diethylamide and serotonin: direct actions on serotonin-containing neurons in rat brains. Life Sci [I] 1972;11:615–622. doi: 10.1016/0024-3205(72)90153-1. [DOI] [PubMed] [Google Scholar]
- 2.Aizenman E, Tang L-H, Reynolds IJ. Effects of nicotinic agonists on the NMDA receptor. Brain Res. 1991;551:355–357. doi: 10.1016/0006-8993(91)90958-x. [DOI] [PubMed] [Google Scholar]
- 3.Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- 4.Alkondon M, Pereira EF, Wonnacott S, Albuquerque EX. Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol. 1992;41:802–808. [PubMed] [Google Scholar]
- 5.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep–waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baraban JM, Aghajanian GK. Suppression of fringe activity of 5-HT neurons in the dorsal raphe by alpha-adrenoceptor antagonists. Neuropharmacology. 1980;19:355–363. doi: 10.1016/0028-3908(80)90187-2. [DOI] [PubMed] [Google Scholar]
- 7.Blanton MG, LoTurco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 8.Chazel G, Ralston HJ., III Serotonin-containing structures in the nucleus raphe dorsalis of the cat: an ultrastructural analysis of dendrites, presynaptic dendrites, and axon terminals. J Comp Neurol. 1987;259:317–329. doi: 10.1002/cne.902590302. [DOI] [PubMed] [Google Scholar]
- 9.Clarke PBS, Reuben M. Release of [3H]noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]dopamine release. Br J Pharmacol. 1996;117:595–606. doi: 10.1111/j.1476-5381.1996.tb15232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortes R, Soriano E, Pazos A, Probst A, Palacios JM. Autoradiography of antidepressant binding sites in the human brain: localization using [3H]imipramine and [3H]paroxetine. Neuroscience. 1988;27:473–496. doi: 10.1016/0306-4522(88)90282-5. [DOI] [PubMed] [Google Scholar]
- 11.Donnan GA, Kaczmarczyk SJ, Paxinos G, Chilco PJ, Kalnins RM, Woodhouse DG, Mendelsohn FA. Distribution of catecholamine uptake sites in human brain as determined by quantitative [3H]mazindol autoradiography. J Comp Neurol. 1991;304:419–434. doi: 10.1002/cne.903040307. [DOI] [PubMed] [Google Scholar]
- 12.Feldman RS, Meyer JS, Quenzer LF. Neuropsychopharmacology, pp 593–596. Sinauer; Sunderland, MA: 1997. Acute behavioral and physiological effects of nicotine. [Google Scholar]
- 13.Foote SL, Bloom FE, Aston-Jones G. Nucleus locus coeruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- 14.Garnier M, Lamacz M, Tonon MC, Vaudry H. Functional characterization of a nonclassical nicotine receptor associated with inositolphospholipid breakdown and mobilization of intracellular calcium pools. Proc Natl Acad Sci USA. 1994;91:11743–11747. doi: 10.1073/pnas.91.24.11743. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Grassi F, Giovannelli A, Fucile S, Eusebi F. Activation of the nicotinic acetylcholine receptor mobilizes calcium from caffeine-insensitive stores in C2C12 mouse myotubes. Pflügers Arch. 1993;422:591–598. doi: 10.1007/BF00374007. [DOI] [PubMed] [Google Scholar]
- 16.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 17.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brain stem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 18.Hrdina PD, Foy B, Hepner A, Summers RJ. Antidepressant binding sites in brain: autoradiographic comparison of [3H]paroxetine and [3H]imipramine localization and relationship to serotonin transporter. J Pharmacol Exp Ther. 1990;252:410–418. [PubMed] [Google Scholar]
- 19.Jacobs BL, Azmitia EC. Structure and function of brain serotonin system. Physiol Rev. 1992;72:165–229. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- 20.Jonas EA, Knox RJ, Smith TC, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of a unique neuronal Ca2+ pool and of neuropeptide secretion. Nature. 1997;385:343–346. doi: 10.1038/385343a0. [DOI] [PubMed] [Google Scholar]
- 21.Jones BE, Moore RY. Ascending projections of the locus coeruleus in the rat. II. Autoradiographic study. Brain Res. 1977;127:25–53. [PubMed] [Google Scholar]
- 22.Léna C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luebke JI, McCarley RW, Greene RW. Inhibitory action of muscarinic agonists on neurons in the rat laterodorsal tegmental nucleus in vitro. J Neurophysiol. 1993;70:2128–2134. doi: 10.1152/jn.1993.70.5.2128. [DOI] [PubMed] [Google Scholar]
- 24.McCarley RW, Hobson JA. Neuronal excitability modulation over the sleep cycle: a structural and mathematical model. Science. 1975;189:58–60. doi: 10.1126/science.1135627. [DOI] [PubMed] [Google Scholar]
- 25.McCarley RW, Greene RW, Rainnie D, Portas CM. Brain stem neuromodulation and REM sleep. Semin Neurosci. 1995;7:341–354. [Google Scholar]
- 26.McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 27.Nitz D, Siegel J. GABA release in the dorsal raphe nucleus: role in the control of REM sleep. Am J Physiol. 1997;273:R451–R455. doi: 10.1152/ajpregu.1997.273.1.R451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ordway GA, Stockmeier CA, Cason GW, Klimek V. Pharmacology and distribution of norepinephrine transporters in the human locus coeruleus and raphe nuclei. J Neurosci. 1997;17:1710–1719. doi: 10.1523/JNEUROSCI.17-05-01710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan ZZ, Williams JT. GABA- and glutamate-mediated synaptic potentials in rat dorsal raphe neurons in vitro. J Neurophysiol. 1989a;61:719–726. doi: 10.1152/jn.1989.61.4.719. [DOI] [PubMed] [Google Scholar]
- 30.Pan ZZ, Williams JT. Differentiated actions of cocaine and amphetamine on dorsal raphe neurons in vitro. J Pharmacol Exp Ther. 1989b;251:56–62. [PubMed] [Google Scholar]
- 31.Pan ZZ, Grudt TJ, Williams JT. Alpha 1-adrenoceptors in rat dorsal raphe neurons: regulation of two potassium conductances. J Physiol (Lond) 1994;478:437–447. doi: 10.1113/jphysiol.1994.sp020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penington NJ, Kelly JS, Fox AP. Whole-cell recordings of inwardly rectifying K+ currents activated by 5-HT1A receptors on dorsal raphe neurons of the adult rat. J Physiol (Lond) 1993;469:387–405. doi: 10.1113/jphysiol.1993.sp019819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portas CM, Thakkar M, Rainnie D, McCarley RW. Microdialysis perfusion of 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) in the dorsal raphe nucleus decreases serotonin release and increases rapid eye movement sleep in the freely moving cat. J Neurosci. 1996;16:2820–2828. doi: 10.1523/JNEUROSCI.16-08-02820.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 35.Semba K, Fibiger HC. Organization of central cholinergic systems. Prog Brain Res. 1989;79:37–63. doi: 10.1016/s0079-6123(08)62464-4. [DOI] [PubMed] [Google Scholar]
- 36.Sershen H, Balla A, Lajtha A, Vizi ES. Characterization of nicotinic receptors involved in the release of noradrenaline from the hippocampus. Neuroscience. 1997;77:121–130. doi: 10.1016/s0306-4522(96)00425-3. [DOI] [PubMed] [Google Scholar]
- 37.Steriade M, McCarley RW. Brainstem control of wakefulness and sleep. Plenum; New York: 1990. [Google Scholar]
- 38.Stockmeier CA, Shapiro LA, Haycock JW, Thompson PA, Lowy MT. Quantitative subregional distribution of serotonin 1A receptors and serotonin transporters in the human dorsal raphe. Brain Res. 1996;727:1–12. doi: 10.1016/0006-8993(96)00239-9. [DOI] [PubMed] [Google Scholar]
- 39.Summers KL, Giacobini E. Effects of located and repeated systemic administration of nicotine on extracellular levels of acetylcholine, norepinephrine, dopamine, and serotonin in rat cortex. Neurochem Res. 1995;20:753–759. doi: 10.1007/BF01705545. [DOI] [PubMed] [Google Scholar]
- 40.Tejani-Butt SM. [3H]Nisoxetine: a radioligand for quantitation of norepinephrine uptake sites by autoradiography or by homogenate binding. J Pharmacol Exp Ther. 1992;260:427–436. [PubMed] [Google Scholar]
- 41.Tork I. Anatomy of the serotonergic system. Ann NY Acad Sci. 1990;600:9–34. doi: 10.1111/j.1749-6632.1990.tb16870.x. [DOI] [PubMed] [Google Scholar]
- 42.Williams JT, Colmers WF, Pan ZZ. Voltage- and ligand-activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci. 1988;8:3499–3506. doi: 10.1523/JNEUROSCI.08-09-03499.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong LA, Gallagher JP. A direct nicotinic receptor-mediated inhibition recorded intracellularly in vitro. Nature. 1989;341:439–442. doi: 10.1038/341439a0. [DOI] [PubMed] [Google Scholar]
- 44.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 45.Yoshimura M, Higashi H. 5-Hydroxytryptamine mediates inhibitory postsynaptic potentials in rat dorsal raphe neurons. Neurosci Lett. 1985;53:69–74. doi: 10.1016/0304-3940(85)90099-0. [DOI] [PubMed] [Google Scholar]
- 46.Yoshimura M, Higashi H, Nishi S. Noradrenaline mediates slow excitatory synaptic potentials in rat dorsal raphe neurons in vitro. Neurosci Lett. 1985;61:305–310. doi: 10.1016/0304-3940(85)90481-1. [DOI] [PubMed] [Google Scholar]