

Abstract
Bcl-2 is a cytoplasmic protein that blocks apoptosis in a wide variety of cell types. Here we report a novel role for Bcl-2 in the early stages of neuronal development. Shortly after differentiating from progenitor cells, sensory neurons undergo a distinct morphological change; initially they have small, spindle-shaped, phase-dark cell bodies that become large, spherical, and phase-bright. Early sensory neurons cultured from the trigeminal ganglia ofbcl-2−/− embryos at embryonic day 11 (E11) and E12 underwent this change more slowly than trigeminal neurons of wild-type embryos of the same ages. The delay was not attributable to the well documented role of Bcl-2 in preventing apoptosis, because Bcl-2-deficient early sensory neurons survived as well as wild-type neurons. Accordingly, there was a significantly smaller number of the more mature type of neuron in the early trigeminal ganglia of bcl-2−/−embryos, yet the number of neurons in the trigeminal ganglia ofbcl-2−/− and wild-type embryos was similar. The absence of Bcl-2 did not cause a uniform delay in the developmental program of sensory neurons, because the time course of nerve growth factor receptor expression (both trkA and p75) was unaffected in the trigeminal neurons ofbcl-2−/− embryos. These findings indicate that Bcl-2 expression is required for the normal progression of a particular early maturational change in embryonic sensory neurons.
Keywords: Bcl-2, neuronal differentiation, apoptosis, neurotrophin, sensory neuron, gene-targeted mice
Bcl-2 is a widely expressed cytoplasmic protein that plays a key role in regulating cell survival in the immune system and nervous system. Mice with targeted null mutations in the bcl-2 gene have markedly reduced numbers of B and T cells attributable to increased apoptosis (Nakayama et al., 1993, 1994; Veis et al., 1993), whereas mice carrying a transgene causing high levels of Bcl-2 expression in the immune system show extended survival of B and T cells (McDonnell et al., 1989, 1990). Overexpression of Bcl-2 in cultured neurons prevents their death after neurotrophin deprivation (Garcia et al., 1992; Allsopp et al., 1993b), and mice expressing a bcl-2 transgene under the control of a neuron-specific enolase promoter have increased numbers of neurons in several regions (Martinou et al., 1994). The survival response of cultured cranial sensory neurons to neurotrophins during the phase of naturally occurring neuronal death is markedly reduced by antisensebcl-2 RNA (Allsopp et al., 1995). Likewise, cranial sensory neurons from bcl-2−/− embryos do not survive as well in vitro with neurotrophins as wild-type embryos during the peak period of naturally occurring neuronal death and are lost to a greater extent than wild-type neurons during this period of development in vivo (Piñón et al., 1997). Sympathetic neurons from postnatalbcl-2−/− mice die more rapidly after nerve growth factor (NGF) deprivation in vitrothan wild-type neurons (Greenlund et al., 1995), and postnatalbcl-2−/− mice have significantly fewer sensory, autonomic, and motor neurons than wild-type mice (Michaelidis et al., 1996). Furthermore, Bcl-2 is structurally and functionally similar to the ced-9 gene product of Caenorhabditis elegans that also prevents programmed cell death (Hengartner and Horvitz, 1994).
In addition to the wealth of data indicating that Bcl-2 plays a key role in regulating cell survival, work on several cell lines has raised the possibility that Bcl-2 influences neuronal differentiation. Bcl-2 overexpression in a neural crest-derived line enhances neurite outgrowth and increases the expression of neuron-specific enolase (Zhang et al., 1996). Midbrain-derived dopaminergic lines stably expressing Bcl-2 extended longer neurites than control-transfected cells (Oh et al., 1996), and Bcl-2 enhances the differentiation of the PC12 cells grown in serum-free conditions (Batistatou et al., 1993).
Because the developmental significance of overexpressing Bcl-2 in cell lines is difficult to interpret, we compared the early development of newly differentiated sensory neurons ofbcl-2−/− and wild-type mouse embryos. Shortly after sensory neurons differentiate from progenitor cells, they undergo a distinctive and clearly recognized morphological change; initially they have small, spindle-shaped, phase-dark cell bodies that subsequently enlarge and become spherical and phase-bright (Wright et al., 1992). We show that Bcl-2-deficient neurons retain a more immature morphology longer than wild-type neurons both in vitro andin vivo, and that this delay in early neuronal maturation is not a consequence of the antiapoptotic function of Bcl-2.
MATERIALS AND METHODS
Experimental animals. Bcl-2 null mutant mice were a gift of Dennis Loh, Nippon Roche Research Center, Kamakura, Japan (Nakayama et al., 1993, 1994). Embryos were obtained from overnight matings of bcl-2+/− mice. Pregnant females were killed at the required stage of gestation, and the genotypes of the embryos were determined by a PCR-based technique using DNA isolated from embryonic tissues. There were no consistent differences in the stage of development of wild-type and Bcl-2-deficient embryos in each litter using the staging criteria ofTheiler (1972).
Neuronal cultures. Separate dissociated cultures of trigeminal ganglion neurons were established from each embryo in E11 and E12 litters resulting from matings ofbcl-2+/− mice. The trigeminal neurons of each embryo were cultured in triplicate in 60-mm-diameter poly-d-ornithine- and laminin-coated plastic tissue culture dishes at a density of 500–2000 neurons per dish in 5 ml of serum-free medium (Davies et al., 1993). Because many trigeminal neurons switch survival dependence from brain-derived neurotrophic factor (BDNF) to NGF between E11 and E12 (Buchman and Davies, 1993; Paul and Davies, 1995), E11 neurons were grown with BDNF (5 ng/ml), and E12 neurons were grown with NGF (5 ng/ml).
Cohorts of morphologically immature neurons were defined in these cultures 18 hr after plating, and the fate of each neuron in these cohorts was followed for up to 3 d in culture. This was done by recording the location 18 hr after plating of all immature neurons within a 9 × 9 mm grid inscribed on the undersurface of the culture dishes (Davies, 1989) and by monitoring at intervals if each of these neurons was still immature, had assumed a more mature morphology, or had degenerated.
Quantification of neurons in the trigeminal ganglion. E12, E13, and E14 mouse embryos in litters resulting from overnight matings of bcl-2+/− mice were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.3, and then embedded, serially sectioned at 8 μm, and stained with cresyl fast violet, and the total number of morphologically recognizable neurons in both trigeminal ganglia was quantified as described previously (Piñón et al., 1996). Neurons were identified by virtue of Nissl substance and large, round, pale-stained nuclei. Because of their small size, immature neurons cannot be distinguished from progenitor cells and glial cells in sections stained with cresyl fast violet and are therefore not included in these counts.
To identify all neurons (immature and mature) in the trigeminal ganglia of younger embryos, neurofilament staining was used. The heads of E12 embryos were fixed in Carnoy’s fluid (60% ethanol, 30% chloroform, and 10% acetic acid) for 20 min before dehydration and wax embedding. Serial sections of the heads were cut at 8 μm, mounted on poly-l-lysine-coated slides, cleared in xylene, and dehydrated before quenching (10% methanol and 3% hydrogen peroxide in PBS). Nonspecific interactions were blocked using 10% horse serum in PBS with 0.4% Triton X-100. The sections were incubated in primary antibody (monoclonal anti-neurofilament 160; Sigma, St. Louis, MO) (used at 1:500) for 1 hr and then labeled using biotinylated secondary antibody (1:200), avidin, and biotinylated horseradish peroxidase macromolecular complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA). The substrate used for the peroxidase reaction was 1 mg/ml diaminobenzidine tetrachloride (Sigma). After staining, the sections were washed in tap water before rehydration and mounting. Estimation of neuronal number was performed as for cresyl fast violet-stained sections (Piñón et al., 1996). Here, neurons were identified as neurofilament-positive cells.
Measurement of specific mRNA levels. A quantitative reverse transcription-PCR (RT-PCR) technique was used to measure the levels of trkA and p75 mRNAs in total RNA extracted from pairs of trigeminal ganglia dissected from wild-type andbcl-2−/− mice (Wyatt and Davies, 1993). The reverse transcription and PCR reactions were calibrated by the inclusion of known amounts of cRNA competitor templates for each of the mRNAs in the reverse transcription reaction. The cRNA competitor templates were synthesized in vitro from cDNA competitor constructs. Details of the competitors, primers, and reaction conditions are provided elsewhere (Wyatt and Davies, 1993, 1995; Wyatt et al., 1997). Previous detailed comparisons of this method with quantitative Northern blotting have demonstrated its accuracy and reproducibility over a wide range of mRNA concentrations (Wyatt and Davies, 1993).
RESULTS
Neuronal maturation in vitro
Sensory neurons undergo a clearly recognized morphological change at an early stage in their development. After differentiating from progenitor cells, sensory neurons initially have small, spindle-shaped, phase-dark cell bodies. Subsequently, the cell bodies enlarge to become spherical and phase-bright (Wright et al., 1992). We refer to developing sensory neurons exhibiting these two morphologically distinct appearances as immature and mature early sensory neurons, respectively (Fig. 1). In the current study, we have focused on this morphological change in the embryonic mouse trigeminal ganglion, a population of sensory neurons in which development is extensively characterized (Davies and Lumsden, 1984,1986; Davies, 1987; Piñón et al., 1996; Wilkinson et al., 1996). The majority of neurons in this ganglion differentiate from proliferating progenitor cells between E9.5 and E13.5, and the first axons first emerge from the trigeminal ganglion at E9.5. The number of neurons peaks between E12 and E14 and decreases by 50% to reach a stable number by birth as a result of neuronal apoptosis that peaks at E14. We therefore studied the development of trigeminal neurons during the key early stages between E11 and E14 when most neurons are differentiating from progenitor cells and extending axons to their targets.
Fig. 1.
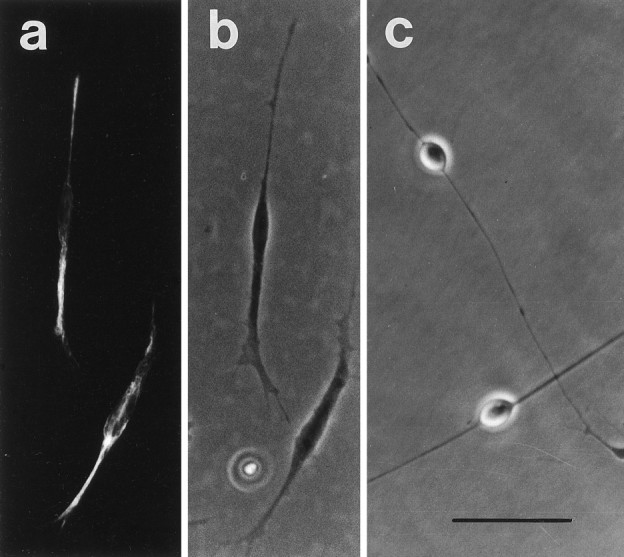
Photomicrographs of E11 trigeminal ganglion cultures showing the typical morphology of immature (a,b) and mature (c) early sensory neurons. The paired immunofluorescence (a) and phase-contrast (b) photomicrographs demonstrate that cells identified as immature neurons are stained with anti-neurofilament antiserum (Wright et al., 1992). Scale bar, 50 μm.
We observed that the proportion of immature neurons in dissociated cultures of early trigeminal ganglia frombcl-2−/− embryos was consistently higher than the proportion of immature neurons in cultures established from wild-type embryos of the same age (Fig.2). In cultures established from trigeminal ganglia at E11 and E12 when many neurons are differentiating from progenitor cells and extending axons toward their targets in vivo (Davies and Lumsden, 1984; Wilkinson et al., 1996), the proportion of immature neurons was highest in cultures established frombcl-2−/− embryos at all time intervals examined (24, 48, and 72 hr). Furthermore, the proportion of immature neurons in cultures established frombcl-2+/− embryos was intermediate between that in cultures established frombcl-2−/− and wild-type embryos, suggesting a gene dosage effect. Similar results were obtained in E11 and E12 cultures of nodose ganglion neurons (data not shown).
Fig. 2.

Bar charts showing the percentage of neurons that are immature in dissociated cultures of E11 and E12 trigeminal ganglia from bcl-2−/− and wild-type embryos after 24, 48, and 72 hr. The means + SE of the results of cultures established from 13 bcl-2−/−, 26bcl-2+/−, and 14bcl-2+/+ embryos at E11 and 17bcl-2−/−, 29bcl-2+/−, and 18bcl-2+/+ embryos at E12 are shown.
An elevated number of immature neurons in dissociated cultures of early sensory ganglia from bcl-2−/− embryos could be attributable to a delay in the maturation of Bcl-2-deficient neurons, to differences in progenitor cell differentiation, or to differences in neuronal survival in these cultures. To directly address the question of whether Bcl-2-deficient early sensory neurons mature more slowly than wild-type neurons, we defined cohorts of immature neurons in cultures of E11 and E12 trigeminal neurons 18 hr after plating and followed the fate of each neuron in these cohorts for up to 3 d in culture (Fig. 3). These studies showed that Bcl-2-deficient neurons retain an immature morphology much longer than wild-type neurons; the maturation of Bcl-2-deficient neurons was delayed by 18–24 hr compared with wild-type neurons. These marked differences in the rate of neuronal maturation cannot be attributed to differences in the survival of Bcl-2-deficient and wild-type neurons, because very few neurons died in these cohorts throughout the period studied (Fig. 3). Because all data were obtained before the genotypes were ascertained, these data were not influenced by observer bias.
Fig. 3.

Graphs summarizing the results of neuron cohort experiments comparing the maturation and survival of trigeminal neurons from wild-type and bcl-2−/− embryos at E11 and E12. Cohorts of immature neurons were defined 18 hr after plating, and the number of immature neurons remaining in these cohorts (top graphs) and the number of neurons surviving in these cohorts (bottom graphs) are expressed as percentages of the initial cohort size at intervals up to 72 hr. The means ± SE of the results of cultures established from 24bcl-2−/− and 23bcl-2+/+ embryos at E11 and 22bcl-2−/− and 17bcl-2+/+ embryos at E12 are shown. The initial cohort size in each culture ranged from 20 to 60 neurons.
Development of early trigeminal neurons in vivo
To determine whether neuronal maturation is delayed in the trigeminal ganglia of bcl-2−/− embryosin vivo, we counted the morphologically recognizable mature neurons in the trigeminal ganglia of wild-type andbcl-2−/− embryos at several developmental stages. Neurogenesis in the mouse trigeminal ganglion and the recruitment of axons to the trigeminal nerve occur between E9.5 and E13.5 (Davies and Lumsden, 1984; Wilkinson et al., 1996). We began comparing the trigeminal ganglia of wild-type andbcl-2−/− embryos at E12 because large numbers of neurons become distinguishable by morphological criteria from other cell types at this stage (Davies and Lumsden, 1984). Because of their small size, immature neurons cannot be distinguished from progenitor cells and glial cells in cresyl fast violet-stained sections and are therefore not included in these counts. Figure4 shows that there are significantly greater numbers of morphologically distinct neurons in the trigeminal ganglia of wild-type embryos at both E12 (p = 0.0008; n = 17, t test) and E13 (p < 0.006; n = 18,t test). By E14, however, the number of neurons of the trigeminal ganglia of bcl-2−/− embryos and wild-type embryos was not significantly different (p > 0.1; n = 22, ttest). Because all data were obtained before the genotypes were ascertained, these data are not influenced by observer bias.
Fig. 4.

Graph of the numbers of morphologically recognizable neurons in cresyl violet-stained sections of the trigeminal ganglia of wild-type andbcl-2−/− embryos at E12, E13, and E14 in the trigeminal ganglia of wild-type andbcl-2−/− embryos at E12, and bar chart of the numbers of neurons positively identified by neurofilament staining in the trigeminal ganglia of wild-type andbcl-2−/− embryos at E12. The means ± SE are shown (n = 53 embryos for mature neurons; n = 11 embryos for neurofilament-positive cells).
To ascertain whether the reduced number of morphologically identifiable neurons in the early trigeminal ganglia ofbcl-2−/− embryos was attributable to a reduction in the number of neurons in the ganglion, we positively identified all neurons (mature and immature) in the trigeminal ganglion at E12 using neurofilament staining (Fig.5). Estimates of neurofilament-positive cells in the ganglia of wild-type andbcl-2−/− embryos revealed no significant difference at this stage (Fig. 4). These observations suggest that the reduction in morphologically recognizable neurons in the trigeminal ganglia of bcl-2−/−embryos during the early stages of ganglion formation is not attributable to a reduction in neurons in the ganglion. These in vivo observations are consistent with the results of our in vitro studies that suggest that Bcl-2-deficient neurons retain an immature morphology longer in vivo than wild-type neurons.
Fig. 5.
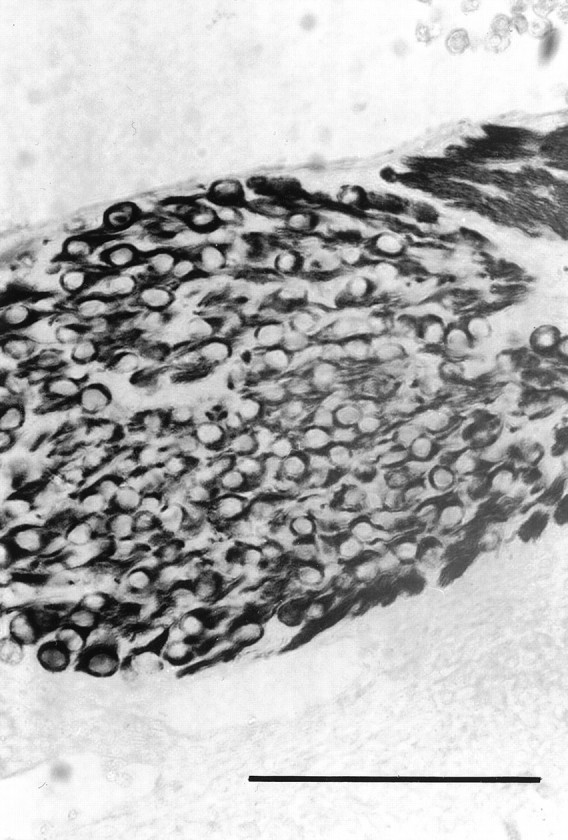
Photomicrograph of a section of the trigeminal of an E12 bcl-2−/− embryo stained for neurofilament protein. Scale bar, 100 μm.
Quantification of trkA and p75 mRNAs
To determine whether the delayed morphological maturation of early sensory neurons in bcl-2−/− embryos is a feature of a general delay in the program of development of these neurons, we used competitive RT-PCR to quantify the level of mRNAs encoding the NGF receptors trkA and p75. TrkA is a receptor tyrosine kinase that is essential for the survival response of neurons to NGF (Kaplan et al., 1991; Klein et al., 1991; Allsopp et al., 1993a), whereas p75 has several functions (Bothwell, 1995; Davies, 1997) including enhancing the survival response of trigeminal neurons to NGF (Davies et al., 1993). The levels of these mRNAs are known to increase rapidly during the early stages of trigeminal ganglion development and are expressed predominantly if not exclusively in the neurons of the ganglion in the embryo (Wyatt et al., 1990; Wyatt and Davies, 1993;Davies et al., 1995). Figure 6 shows that the levels of both mRNAs increased to the same extent in the trigeminal ganglia of wild-type and bcl-2−/−embryos between E12 and E14.
Fig. 6.

Graphs of the levels of trkA and p75 mRNA in the trigeminal ganglia of wild-type andbcl-2−/− embryos at E12, E13, and E14. The means ± SE are shown (n = 42 embryos).
DISCUSSION
We have obtained compelling evidence for a novel role for Bcl-2 during the early stages of neuronal development that is distinct from its well recognized antiapoptotic function. Newly differentiated sensory neurons initially have small, spindle-shaped, phase-dark cell bodies that subsequently enlarge, becoming spherical and phase-bright. By tracking the morphology of individual neurons at intervals in cultures established from the early trigeminal ganglia ofbcl-2−/− and wild-type embryos, we have demonstrated that this clearly recognizable morphological change is markedly delayed in Bcl-2-deficient neurons. This delay is not a consequence of a decrease in the viability of Bcl-2-deficient neurons, because early trigeminal neurons ofbcl-2−/− and wild-type embryos survive equally well in vitro. Thus, the absence of Bcl-2 has a marked effect on the in vitro development of early sensory neurons that is unrelated to its role in regulating cell survival. Interestingly, later in development during the peak period of naturally occurring neuronal death, Bcl-2-deficient trigeminal neurons do die more rapidly than wild-type neurons in NGF-supplemented medium. This decrease in the in vitro viability of older embryonic Bcl-2-deficient trigeminal neurons is correlated with an increase in the number of dying neurons in the trigeminal ganglion at this stagein vivo and a reduction in the number of neurons in the ganglion before birth (Piñón et al., 1997). Reductions in the number of neurons in dorsal root and sympathetic ganglia and in motoneuron populations have also been reported inbcl-2−/− mice postnatally (Michaelidis et al., 1996).
Our in vitro observations of the delay in the morphological maturation of early Bcl-2-deficient sensory neurons were supported by an in vivo analysis of the trigeminal ganglia ofbcl-2−/− and wild-type embryos. In cresyl fast violet-stained sections, there were fewer morphologically recognizable neurons in the trigeminal ganglia ofbcl-2−/− embryos compared with wild-type embryos at E12 and E13, the stage when many neurons are differentiating from progenitor cells and starting to extend axons to their targets (Davies and Lumsden, 1984; Davies, 1987). Because of their small size, immature neurons are not distinguishable from progenitor cells and other non-neuronal cells in this histological analysis. The total neuronal complement of the trigeminal ganglion ofbcl-2−/− embryos was, however, normal during this period of development, as revealed by counting the neurofilament-positive cells in the trigeminal ganglia ofbcl-2−/− and wild-type embryos. The most parsimonious explanation for these findings is that as in vitro, the morphological maturation of early sensory neurons is delayed in vivo in the absence of Bcl-2.
Of particular interest is our demonstration that the delay in the morphological maturation of early Bcl-2-deficient neurons is not a consequence of a general retardation of the development ofbcl-2−/− embryos or of sensory neurons within these embryos. There was no apparent difference in the stage-specific external features (Theiler, 1972) ofbcl-2−/− and wild-type embryos in each litter, and the time course and level of expression of p75 and trkA mRNAs were virtually identical in embryos of both genotypes. Because these mRNAs are expressed predominantly if not exclusively in the neurons of the embryonic trigeminal ganglion, and their levels increase markedly with the acquisition of the NGF survival response (Wyatt et al., 1990; Wyatt and Davies, 1993), these findings also reinforce our evidence based on estimates of the number of neurofilament-positive cells that the neuronal complement of the early trigeminal ganglion is similar in bcl-2−/− and wild-type embryos.
Several studies of the consequences of manipulating the expression of Bcl-2 in various cell lines and neurons have raised the possibility that Bcl-2 may have functions in addition to its well known antiapoptotic role. The differentiation of the PC12 pheochromocytoma cell line into neurons is enhanced by overexpressing Bcl-2. However, this effect is only observed when the cells are grown in serum-free medium (Batistatou et al., 1993). Midbrain-derived dopaminergic cell lines stably expressing Bcl-2 extend longer neurites than control-transfected cells but do not have increased expression of many neuron-specific proteins (Oh et al., 1996). Bcl-2 overexpression in a neural crest-derived cell line enhances the outgrowth of neurite-like processes and also increases the expression of neuron-specific enolase (Zhang et al., 1996). Several observations indicate that the level of Bcl-2 expression in mouse retinal ganglion cells (RGCs) influences the ability of their axons to regenerate into co-cultured tectal explants (Chen et al., 1997). At early fetal stages, RGCs normally express high levels of Bcl-2 and are able to extend axons into co-cultured tectal tissue of the same age, whereas late fetal RGCs express low levels of Bcl-2 and are not able to grow axons into tectal tissue. The ability of early fetal RGCs to regenerate into tectal tissue is substantially reduced in retinal explants obtained frombcl-2−/− embryos, whereas late fetal and adult RGCs from transgenic mice overexpressing Bcl-2 in neurons are able to regenerate into tectal tissue. Recently, Bcl-2 expression has been shown to influence axonal growth rate during development. Early sensory neurons from bcl-2−/− mouse embryos extend axons more slowly in vitro than neurons from wild-type embryos of the same age (Hilton et al., 1997).
In summary, we have demonstrated a novel role for endogenously expressed Bcl-2 in sensory neurons shortly after they differentiate from progenitor cells. At this stage of development, Bcl-2 promotes an early morphological change in the neurons but does not influence certain other aspects of their development, such as the time course of NGF receptor expression. This action of Bcl-2 is unrelated to its well characterized antiapoptotic role and may provide an explanation for the widespread expression of Bcl-2 in embryonic nervous system during the earliest stages of neuronal development (Abe et al., 1993; Merry et al., 1994), whereas abnormal degeneration of neurons in Bcl-2-deficient mice is not observed until the late fetal and postnatal periods (Michaelidis et al., 1996; Piñón et al., 1997).
Footnotes
This work was supported by grants from the Cancer Research Campaign and the Wellcome Trust. We thank Dennis Loh for the bcl-2mutant mice, Debbie Hughes for assistance with genotyping, and Gene Burton and John Winslow for the purified recombinant BDNF and NGF.
Correspondence should be addressed to Dr. Alun Davies at the above address.
REFERENCES
- 1.Abe DS, Harada N, Yamada K, Tanaka R. Bcl-2 gene is highly expressed during neurogenesis in the central nervous system. Biochem Biophys Res Commun. 1993;191:915–921. doi: 10.1006/bbrc.1993.1304. [DOI] [PubMed] [Google Scholar]
- 2.Allsopp TE, Robinson M, Wyatt S, Davies AM. Ectopic trkA expression mediates a NGF survival response in NGF-independent sensory neurons but not in parasympathetic neurons. J Cell Biol. 1993a;123:1555–1566. doi: 10.1083/jcb.123.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993b;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 4.Allsopp TE, Kiselev S, Wyatt S, Davies AM. Role of Bcl-2 expression in the BDNF survival response. Eur J Neurosci. 1995;7:1266–1272. doi: 10.1111/j.1460-9568.1995.tb01116.x. [DOI] [PubMed] [Google Scholar]
- 5.Batistatou A, Merry DE, Korsmeyer SJ, Greene LA. Bcl-2 affects survival but not neuronal differentiation of PC12 cells. J Neurosci. 1993;13:4422–4428. doi: 10.1523/JNEUROSCI.13-10-04422.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 7.Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development. 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- 8.Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- 9.Davies AM. The growth rate of sensory nerve fibres in the mammalian embryo. Development. 1987;100:307–311. doi: 10.1242/dev.100.2.307. [DOI] [PubMed] [Google Scholar]
- 10.Davies AM. Intrinsic differences in the growth rate of early nerve fibres related to target distance. Nature. 1989;337:553–555. doi: 10.1038/337553a0. [DOI] [PubMed] [Google Scholar]
- 11.Davies AM. The yin and yang of nerve growth factor. Curr Biol. 1997;7:38–40. doi: 10.1016/s0960-9822(06)00016-9. [DOI] [PubMed] [Google Scholar]
- 12.Davies AM, Lumsden AG. Relation of target encounter and neuronal death to nerve growth factor responsiveness in the developing mouse trigeminal ganglion. J Comp Neurol. 1984;223:124–137. doi: 10.1002/cne.902230110. [DOI] [PubMed] [Google Scholar]
- 13.Davies AM, Lumsden AG. Fasciculation in the early mouse trigeminal nerve is not ordered in relation to the emerging pattern of whisker follicles. J Comp Neurol. 1986;253:13–24. doi: 10.1002/cne.902530103. [DOI] [PubMed] [Google Scholar]
- 14.Davies AM, Lee KF, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- 15.Davies AM, Wyatt S, Nishimura M, Phillips H. NGF receptor expression in sensory neurons develops normally in embryos lacking NGF. Dev Biol. 1995;171:434–438. doi: 10.1006/dbio.1995.1293. [DOI] [PubMed] [Google Scholar]
- 16.Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 17.Greenlund LJ, Korsmeyer SJ, Johnson EM. Role of BCL-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 18.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 19.Hilton M, Middleton G, Davies AM. Bcl-2 influences axonal growth rate in embryonic sensory neurons. Curr Biol. 1997;7:798–800. doi: 10.1016/s0960-9822(06)00339-3. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan DR, Hempstead BL, Martin ZD, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- 21.Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 1991;65:189–197. doi: 10.1016/0092-8674(91)90419-y. [DOI] [PubMed] [Google Scholar]
- 22.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowsky H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischaemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 23.McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ. Bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 24.McDonnell TJ, Nunez G, Platt FM, Hockenberry D, London L, McKearn JP, Korsmeyer SJ. Deregulated Bcl-2-immunoglobulin transgene expands a resting but responsive immunoglobulin M- and D-expressing B-cell population. Mol Cell Biol. 1990;10:1901–1907. doi: 10.1128/mcb.10.5.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. Bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- 26.Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic neurons and sensory neurons during the early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama K, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, Fields LE, Lucas PJ, Stewart V, Alt FW, Loh DY. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 28.Nakayama K, Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh YJ, Swarzenski BC, O’Malley KL. Overexpression of Bcl-2 in a murine dopaminergic neuronal cell line leads to neurite outgrowth. Neurosci Lett. 1996;202:161–164. doi: 10.1016/0304-3940(95)12235-4. [DOI] [PubMed] [Google Scholar]
- 30.Paul G, Davies AM. Trigeminal sensory neurons require extrinsic signals to switch neurotrophin dependence during the early stages of target field innervation. Dev Biol. 1995;171:590–605. doi: 10.1006/dbio.1995.1307. [DOI] [PubMed] [Google Scholar]
- 31.Piñón LGP, Minichiello L, Klein R, Davies AM. Timing of neuronal death in trkA, trkB and trkC mutant embryos reveals developmental changes in sensory neuron dependence on Trk signalling. Development. 1996;122:3255–3261. doi: 10.1242/dev.122.10.3255. [DOI] [PubMed] [Google Scholar]
- 32.Piñón LGP, Middleton G, Davies AM. Bcl-2 is required for cranial sensory neuron survival at defined stages of embryonic development. Development. 1997;124:4173–4178. doi: 10.1242/dev.124.20.4173. [DOI] [PubMed] [Google Scholar]
- 33.Theiler K. The house mouse (development and normal stages from fertilisation to 4 weeks). Springer; Berlin: 1972. [Google Scholar]
- 34.Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 35.Wilkinson GA, Fariñas I, Backus C, Yoshida CK, Reichardt LF. Neurotrophin-3 is a survival factor in vivo for early mouse trigeminal neurons. J Neurosci. 1996;16:7661–7669. doi: 10.1523/JNEUROSCI.16-23-07661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wright EM, Vogel KS, Davies AM. Neurotrophic factors promote the maturation of developing sensory neurons before they become dependent on these factors for survival. Neuron. 1992;9:139–150. doi: 10.1016/0896-6273(92)90229-7. [DOI] [PubMed] [Google Scholar]
- 37.Wyatt S, Davies AM. Regulation of expression of mRNAs encoding the nerve growth factor receptors p75 and trkA in developing sensory neurons. Development. 1993;119:635–648. doi: 10.1242/dev.119.3.635. [DOI] [PubMed] [Google Scholar]
- 38.Wyatt S, Davies AM. Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol. 1995;130:1435–1446. doi: 10.1083/jcb.130.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wyatt S, Shooter EM, Davies AM. Expression of the NGF receptor gene in sensory neurons and their cutaneous targets prior to and during innervation. Neuron. 1990;4:421–427. doi: 10.1016/0896-6273(90)90054-j. [DOI] [PubMed] [Google Scholar]
- 40.Wyatt S, Piñón LGP, Ernfors P, Davies AM. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 1997;16:3115–3123. doi: 10.1093/emboj/16.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang KZ, Westberg JA, Holtta E, Andersson LC. Bcl-2 regulates neural differentiation. Proc Natl Acad Sci USA. 1996;93:4504–4508. doi: 10.1073/pnas.93.9.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]