

Abstract
Brain acetylcholinesterase (AChE) forms stable complexes with amyloid-β peptide (Aβ) during its assembly into filaments, in agreement with its colocalization with the Aβ deposits of Alzheimer’s brain. The association of the enzyme with nascent Aβ aggregates occurs as early as after 30 min of incubation. Analysis of the catalytic activity of the AChE incorporated into these complexes shows an anomalous behavior reminiscent of the AChE associated with senile plaques, which includes a resistance to low pH, high substrate concentrations, and lower sensitivity to AChE inhibitors. Furthermore, the toxicity of the AChE–amyloid complexes is higher than that of the Aβ aggregates alone. Thus, in addition to its possible role as a heterogeneous nucleator during amyloid formation, AChE, by forming such stable complexes, may increase the neurotoxicity of Aβ fibrils and thus may determine the selective neuronal loss observed in Alzheimer’s brain.
Keywords: AChE, Aβ-amyloid fibrils, AChE-Aβ-amyloid fibril complexes, amyloid formation, Alzheimer’s disease, neurotoxicity
Alzheimer’s disease (AD), the most common form of dementia in adults, is a neurodegenerative disorder characterized by selective neuronal loss and the presence of two different types of fibril deposits: amyloid plaques and neurofibrillary tangles (Soto et al., 1994; Selkoe, 1996). Recent advances in the molecular genetics of AD indicate that it is a complex disorder with mutations in many genes, and the apolipoprotein E genotype is a risk factor (Selkoe, 1997). However, at least 60% of AD patients do not have a family history of the disease. Therefore, there is a need to search for the mechanisms responsible for the progressive cognitive decline observed in most of the sporadic cases, which correspond to the great majority of Alzheimer’s patients (Katzman and Kawas, 1994;Inestrosa et al., 1996a; Van Leeuwen et al., 1998). The fact that neurodegenerative changes occur around senile plaques (Mann and Esiri, 1989), and that the neurotoxicity of Aβ depends on its aggregation into amyloid fibrils (Lorenzo and Yankner, 1994), indicates that amyloid fibrils are the primary pathogenic mechanism in AD (Yankner, 1996). It has also been suggested that endogenous factors that modulate Aβ fibrillogenesis and deposition could play a significant role in the pathogenesis of the disease (Inestrosa et al., 1996b; Harper and Lansbury, 1997). In this context, several studies have revealed numerous other proteins associated with the amyloid plaque deposits. These proteins include apolipoprotein E (apoE) (Namba et al., 1991), α1-anti-chymotrypsin (Abraham et al., 1988), heparan sulfate proteoglycans (Snow et al., 1988), and acetylcholinesterase (AChE) (Ulrich et al., 1990). AChE plays a key role in cholinergic transmission in the CNS of mammals (Inestrosa and Perelman, 1989, 1990;Taylor, 1991) and may also participate in noncholinergic mechanisms (Massoulié et al., 1993; Small et al., 1996). Most of the cortical AChE activity present in Alzheimer’s brain is predominantly associated with the amyloid core of senile plaques rather than with the neuritic component found at the periphery (Ulrich et al., 1990;Gómez-Ramos et al., 1992; Morán et al., 1993). Histochemical studies have demonstrated that the AChE associated with senile plaques differs enzymatically from the AChE associated with normal fibers and neurons with respect to optimum pH, inhibitor sensitivity, and inhibition by excess substrate (Mesulam et al., 1987;Geula and Mesulam, 1989; Schätz et al., 1990; Wright et al., 1993). AChE directly promotes the assembly of Aβ peptide into amyloid fibrils (Alvarez et al., 1995; Inestrosa et al., 1996b), and recent studies from our laboratory have shown that AChE forms complexes with small Aβ peptide fragments (Alvarez et al., 1997). It was therefore our aim to investigate whether the characteristics of AChE observed in the amyloid plaques of Alzheimer’s brain could be reproduced underin vitro conditions, using complexes obtained with whole Aβ1–40 peptide. We report here that the incorporation of AChE into Alzheimer’s amyloid aggregates results in the formation of stable complexes that change the biochemical and pharmacological properties of the enzyme and cause an increase in the neurotoxicity of the amyloid-β fibrils, suggesting that AChE could play a pathogenic role in AD by influencing the process leading to amyloid toxicity and the appearance of AD.
MATERIALS AND METHODS
Materials. Synthetic peptide Aβ1–40, corresponding to residues 1–40 of the human Aβ sequence, was purchased from Chiron Corporation (Emeryville CA). The following inhibitors were purchased from Sigma (St. Louis, MO): edrophonium chloride, tetrahydro-aminoacridine hydrochloride (Tacrine), and propidium iodide. Fasciculin was purified as described previously (Dajas et al., 1987; Karlsson et al., 1984), and the monoclonal antibody 25B1 directed against fetal bovine serum AChE was obtained as described by Gentry et al. (1995).
Purification of brain AChE. The tetrameric G4AChE form (sedimentation coefficient, 10.7 S) was purified from bovine caudate nucleus, using acridine affinity chromatography, as described previously (Inestrosa et al., 1987). Both specific activities (6000 U/mg protein), and staining intensities after SDS-PAGE (Laemmli, 1970) (a single band of 66 kDa) were used to verify purity.
AChE assay. AChE activity was determined by the method ofEllman et al. (1961).
Preparation of 125I-Tyr derivatives of AChE and Aβ1–40. For the iodination of AChE and the Aβ peptide, the iodo-bead method was used (Markwell, 1982); 100 μg of AChE was labeled with 500 μCi 125I-Na, and 100 μg of Aβ1–40 peptide was labeled with 1000 μCi125I-Na (Comisión de Energía Nuclear de Chile). A single bead was used in each case and incubated in 200 μl of 0.1 m phosphate buffer, pH 7.0, for 15 min. The iodinated enzyme was separated from the unreacted 125I on a Sephadex G-25 column and submitted to 12% SDS-PAGE (Laemmli, 1970). Autoradiography showed a main band (at 66 kDa) coincident with the electrophoretic migration of native AChE. The specific activity of the standard 125I-AChE used in our experiments was estimated on the order of 2.4 mCi/mmol. For the iodinated Aβ peptide, a Sepharose Bio-Gel P-2 column was used to separate the peptide from free125I, and the labeled peptide fraction was submitted to Tris-Tricine 16% SDS-PAGE (Schagger and von Jagow, 1987). Autoradiography showed a single band (at 4 kDa) coincident with the electrophoretic pattern of the native Aβ1–40 peptide. The specific activity of the standard 125I-Aβ peptide used in our experiments was estimated on the order of 0.6 mCi/mmol, and the peptide was used immediately after radioiodination.
Generation of AChE–Aβ complexes in vitro. To obtain AChE–Aβ complexes, Aβ1–40 peptide was incubated with or without AChE in a stirred aggregation assay performed as described previously (Jarrett et al., 1993; Evans et al., 1995). Briefly, stock solutions were prepared by dissolving lyophilized aliquots of the Aβ peptide in dimethyl sulfoxide (DMSO) at 15 mg/ml (3.5 mm). Aliquots of peptide stock solution (70 nmol in ≃20 μl of DMSO) were added to PBS, pH 7.4, for a final volume of 725 μl. For the aggregation experiments performed with AChE, peptide stock (70 nmol in DMSO) was added to 680 μl of PBS buffer containing AChE (100 nm; 25 μg of the enzyme in ∼25 μl of buffer: 20 mm sodium phosphate, 1 mm EDTA, 0.1% Triton X-100, and 10 mm caproic acid, pH 7.4). The solutions were stirred continuously (210 rpm), and aggregation was measured versus buffer blank by turbidity at 405 nm. After 6 hr of incubation with stirring, the mixtures were incubated without stirring for 4–5 d after which the aggregates formed were analyzed.
Congo red (CR) assay. CR binding to amyloid aggregates was used as described previously (Klunk et al., 1989). Aliquots (40 μl) of the aggregation mixtures were added to 960 μl of a solution of 25 μm CR, 100 mm phosphate buffer, pH 7.4, and 150 mm NaCl and incubated for 30 min. Absorbance was measured at 480 and 540 nm. The CR binding was estimated by CR(M) = (A540/25,295) − (A480/46,306).
AChE activity in amyloid fibrils. Four hundred-microliter aliquots of the Aβ1–40 fibrils formed in the presence of AChE in aggregation assays (5 d of incubation) were centrifuged at 14,000 rpm for 30 min. The supernatants were removed, and the AChE–Aβ complexes were resuspended in 400 μl of PBS. To remove noncomplexed AChE, the aggregates were washed three times by centrifugation, and resuspension in 400 μl of PBS was accomplished by vortex stirring. The final pellet was resuspended in 100 μl of PBS. To quantify the fraction of AChE present in the AChE–Aβ complexes, the enzymatic activity in the final pellet and supernatants was determined by the assay of Ellman et al. (1961). In addition,125I-AChE was used to form AChE–Aβ complexes, and the radioactivity in the same fractions was also quantified. Furthermore, AChE activity associated with the amyloid fibrils in vitrowas detected by histochemical staining of the washed aggregates (Karnovsky and Roots, 1964). The amyloid character of the Aβ1–40 fibrils was evidenced by the binding of thioflavine-T (LeVine, 1993) and detected under a fluorescence microscope, as described previously (Alvarez et al., 1997). Control samples in which Aβ was incubated alone were subjected to the same procedures.
Dissociation studies of the AChE–Aβ complexes. A 10 μl aliquot of the 125I-AChE–Aβ complexes (final pellet) was added to 290 μl of each of the agents listed below. The complexes were incubated in polypropylene tubes with each agent for 1 hr with gentle stirring. The aggregates were then centrifuged at 14,000 rpm for 30 min, and both the AChE activity and radioactivity present in the pellet and the supernatant were determined. The agents used were 1% SDS, 6 m guanidine-HCl, 6 m guanidine isothiocyanate, 8 m urea, PBS-1% Tween 20, PBS-1% Triton X-100, 1 m NaCl, and 2 m MgCl2.
Velocity sedimentation analysis. AChE (G4 form) alone or the AChE–125I-Aβ complexes produced in the stirred kinetic assays were submitted to velocity sedimentation analysis in 5–20% sucrose gradients, containing a 70% sucrose cushion at the bottom of the centrifuge tube. A Combi-Sorvall ultracentrifuge was used. Fractions were collected from the bottom of the gradient as described previously (Inestrosa et al., 1996b).
Immunogold labeling of the AChE–Aβ complexes. For immunogold staining, AChE–Aβ complexes were adsorbed onto nickel grids covered by a carbon-stabilized Formvar film and air-dried. After washing in buffer (0.05 m Tris-HCl, pH 7.4) for 2 min, nonspecific binding was blocked by incubation in 0.05 mTris-HCl, pH 7.4, with 1% ovalbumin for 1 hr. The grids were then placed on a droplet of anti-AChE polyclonal antibody diluted 1:50 in 0.05 m Tris-HCl, pH 7.4, with 1% ovalbumin and incubated at 4°C overnight. These were passed under five droplets of washing solution (0.05 m Tris-HCl, pH 7.4, with 0.05% Tween 20) for 5 min each time, placed on a droplet of anti-rabbit IgG conjugated to 10 nm colloidal gold particles for 1 hr (Sigma; diluted 1:20 in 0.05m Tris-HCl, pH 7.4, with 1% ovalbumin), and passed under another five droplets of washing solution (Naslund et al., 1995). Before the final examination under a JEOL 100-B electron microscope, the specimens were negatively stained with 2% uranyl acetate in water.
Determination of kinetic parameters. The kinetics of AChE activity were determined using appropriate dilutions of acetylthiocholine in the activity assay; the concentrations used were in the range of 0.03–1.0 mm, and separate blanks were used for each substrate concentration. The reaction was started by the addition of enzyme or AChE–Aβ complex. The Kmand Vmax values for acetylthiocholine were calculated by regression analysis of the linear portions of the Lineweaver–Burk plots (1/V vs 1/S). The substrate inhibition constants (Kss) were determined from a plot of 1/V versus [S] for [S] > 1.0 mm (Radic et al., 1991).
Inhibition of AChE activity. The IC50 values for edrophonium, Tacrine, and propidium (where IC50 is the inhibitor concentration required for 50% inhibition of AChE activity) were determined by incubating AChE and AChE–Aβ complexes with varying concentrations of each inhibitor. The reaction was started by the addition of the substrate mixture, with the acetylthiocholine concentration fixed at 0.75 mm, and incubated for 30 min at 37°C. IC50 values were calculated by logarithmic plot analysis. Inhibition coefficient values (Ki) were calculated from the equationKi = IC50/1 + (S/Km), as described byHobbiger and Peck (1969) where S represents the concentration of acetylthiocholine (0.75 mm), and Km represents the Michaelis–Menten constant.
pH dependence. To study the pH dependence of the enzymatic activity of AChE and the AChE–Aβ complexes, activity assays were performed at different pH values. The optimal pH was determined over a range of 4.0–9.0 in PBS buffer. pH values were measured with a pH meter standardized at 25°C to ±0.01 pH unit. The buffer was titrated to the required pH by the addition of HCl for pH values <5.0 and by NaOH for pH values >8.0.
Cytotoxicity assays in PC12 cells. Rat pheochromocytoma (PC12) cells (Greene and Tischler, 1976; Inestrosa et al., 1981) were cultured in DMEM, 10% fetal calf serum (FCS), 5% horse serum, 100 U/ml penicillin/streptomycin, and 2 mml-Gln. For the cytotoxicity assays, cells were seeded in 96-well plates in serum-free medium with 2 μm insulin at a density of 4 × 103 cells/100 μl per well (Solomon et al., 1997). AChE–Aβ complexes, amyloid fibrils, or PBS (control) were added to the wells at different concentrations in a final volume of 10 μl. The cells were incubated for 48 hr at 37°C, after which cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method (Mosmann, 1983). This involves determining the mitochondrial dehydrogenase activity in intact cells by incubating for 4 hr at 37°C with 1 mg/ml MTT. The reaction was stopped by the addition of cell lysis buffer (50% dimethylformamide and 20% SDS, pH 7.4), and the plates were incubated overnight, after which MTT reduction was determined in a Uniskan microplate spectrophotometer at 540 and 650 nm. Results are expressed as the percentage of control values.
Avian retina cell cultures. Primary cultures of retina cells were prepared following the protocol described previously (De Mello et al., 1976; Vogel and Nirenberg, 1976; Puro et al., 1977). Briefly, 8-d-old chick embryo retinas were dissected under sterile conditions, cleared from pigment epithelium, and placed in a Ca2+- and Mg2+-free salt-balanced medium (Sheffield and Moscona, 1970). Trypsin, to a final concentration of 0.05%, was added to the medium and incubated at 37°C for 8 min. The trypsinized tissue was then centrifuged at 500 × gfor 1 min, and the supernatant was discarded. The pellet was resuspended in 5 ml of Eagle’s basal medium plus 5% FCS and mechanically dissociated by pipetting the tissue 10 times with a 5 ml pipette (Paes de Carvalho and De Mello, 1982). The number of cells per 35 mm plastic dish was ∼30 × 106 in 2 ml of medium. The plates were then transferred to a humidified atmosphere of 95% air and 5% CO2 at 37°C. The medium was changed every other day.
Cytotoxicity assays in retina cells. For the cytotoxicity assays, 6 d retina cell cultures were treated with either AChE–Aβ complexes or β-amyloid fibrils, previously aged for 5 d and added to the wells at a final Aβ peptide concentration of 2.5 μm and incubated for 24 hr at 37°C (Campos et al., 1997). Then retina cells were analyzed by phase-contrast microscopy (20× magnification) using an Olympus inverted microscope. Phase-contrast images were photographed with Kodak (Rochester, NY) Ektachrome 64T 35 mm film.
RESULTS
AChE Forms a stable complex with the amyloid fibrils
Because we have previously shown that AChE is able to promote the aggregation of the Aβ peptide (Inestrosa et al., 1996b), in the present work we study the characteristics of the amyloid fibrils formed in the presence of AChE. To obtain amyloid fibrils, we performed kinetic stirred aggregation assays, following Aβ aggregation by turbidity (Jarrett et al., 1993; Evans et al., 1995), in the presence or absence of AChE. After incubation for 5 hr, Aβ1–40peptide without AChE aggregated to a lesser extent (twofold less) than the peptide incubated with AChE (Fig.1A). Fibrils formed in the presence of AChE were separated from soluble enzyme by velocity sedimentation in 5–20% sucrose gradients (Fig. 1B), using a 70% sucrose cushion at the bottom of the tubes to isolate the high molecular weight amyloid–AChE complexes. Figure1B1 shows the sedimentation profile of native brain AChE (10.7 S), and Figure 1B2 shows a high molecular weight AChE aggregate at the 20–70% sucrose interface. Using125I-Aβ peptide these AChE aggregates were shown to co-sediment with a labeled high molecular weight amyloid aggregate (Fig. 1B3). The radioactivity detected in the low-density side of the gradient probably reflects the distribution of soluble Aβ or small Aβ aggregates. At least 58% of the AChE activity present in these gradients appeared associated with the Aβ fibrils, suggesting that AChE is able to bind and interact with the amyloid aggregates forming a macromolecular complex. The fraction of AChE incorporated into the AChE–Aβ complexes was established by extensive washing to remove noncomplexed AChE. Table1 shows that ∼78% of AChE activity remains associated with the complexes. When 125I-AChE was used to estimate the amount of enzyme incorporated into the fibrils, at least 50% of the initial radioactivity was found associated with the complexes. These results indicate that AChE forms a complex with the Aβ fibrils in vitro, with an average of 60% of the AChE becoming incorporated into the amyloid fibrils. The stoichiometric ratio of AChE/Aβ in the final complexes was ∼1:1000.
Fig. 1.

The AChE is associated with Aβ amyloid fibrils.A, Aβ peptide aggregation assay. Aβ peptide (97 μm in PBS, pH 7.4) was incubated in a kinetic stirred assay, with (•) or without (○) AChE (0.1 μm). Aβ aggregation was followed by turbidity at 405 nm. B, Sedimentation velocity analysis. Native brain AChE (10.7 S) as control and 125I-Aβ amyloid aggregates produced in a stirred kinetic assay with AChE were submitted to velocity sedimentation analysis in 5–20% sucrose gradients containing a 70% sucrose cushion at the bottom of the centrifuge tube. A Combi-Sorval ultracentrifuge was used. Fractions were collected from the bottom of the gradient as described previously (Inestrosa et al., 1994). AChE activity and radioactivity in the corresponding fractions were determined.1, Control G4 AChE; 2, AChE–Aβ aggregates; 3, 125I-Aβ amyloid aggregates.
Table 1.
AChE activity is associated with Aβ1–40aggregates
| Fraction | AChE activity ± SD (U) | % of the initial | AChE (cpm ± SD) | % of the initial |
|---|---|---|---|---|
| Initial | 1.941 ± 0.480 | 100.00 | 686.5 ± 131.6 | 100.00 |
| SN | 0.228 ± 0.106 | 10.78 | 329.9 ± 98.6 | 48.05 |
| W1 | 0.023 ± 0.002 | 1.18 | 21.0 ± 1.9 | 3.05 |
| W2 | 0.005 ± 0.004 | 0.26 | 3.0 ± 0.3 | 0.43 |
| W3 | 0.001 ± 0.002 | 0.05 | ||
| Aβ1–40aggregates | 1.553 ± 0.611 | 78.00 | 341.7 ± 70.9 | 49.77 |
A 400 μl aliquot of the incubation mixture (Aβ1–40 with AChE) of the turbidity assays (after 5 d of incubation) was centrifuged (SN), and Aβ1–40aggregates were resuspended in 40 μl and washed three times with 10 volumes of the PBS (W1, W2, and W3). AChE associated with and removed from aggregates was determined by enzymatic activity by the method ofEllman et al. (1961). The results are the mean ± SD of four or five experiments.
The stability of the complexes was analyzed by performing incubations in the presence of strong dissociating agents, some of which had previously been shown to dissociate Aβ polymers obtained from AD brain (Masters et al., 1985). AChE was followed by enzymatic activity or by following 125I-AChE radioactivity. The interaction of the enzyme with the Aβ fibrils was resistant to high ionic strength conditions, including 1 m NaCl and 2 mMgCl2 (Table 2), and was only partially affected by PBS-Tween 20 and PBS-Triton X-100. These detergent buffers were able to remove ∼10 and 20%, respectively, of the total AChE activity associated with the amyloid aggregates. Urea (8m) released ∼12% of the AChE activity from the Aβ fibrils, and the chaotropic agent 6 m guanidine-HCl released 57%. On the other hand, SDS and guanidine isothiocyanate released 84 and 82%, respectively, of the enzyme present in the complexes. These results are consistent with the notion that stable AChE–Aβ complexes are formed when AChE is used to accelerate the formation of amyloid aggregates.
Table 2.
Solubilization of AChE from Aβ1–40aggregates
| (cpm in the supernatant/cpm total × 100(% ± SD) | (activity in the supernatant/total activity × 100(%) | |
|---|---|---|
| PBS | 3.20 ± 0.65 | 2.19 |
| PBS-1% Tween 20 | 9.30 ± 3.96 | 12.01 |
| PBS-1% Triton X-100 | 15.42 ± 2.23 | 32.18 |
| 1 m NaCl | 1.82 ± 0.51 | 2.87 |
| 2 m MgCl2 | 1.73 ± 0.31 | 1.30 |
| 8 m urea | 12.5 ± 0.38 | ND |
| 1% SDS | 84.15 ± 8.48 | ND |
| 6 m guanidine-HCl | 57.45 ± 20.10 | ND |
| 6m guanidine isothiocyanate | 82.25 ± 14.87 | ND |
Amyloid aggregates formed in presence of 125I-AChE were washed with PBS three times. After washing with 10 volumes of each solution, the AChE solubilized was determined in the supernatant after centrifugation by counts per minute and activity. The results represent the mean ± SD of three or four experiments. ND, Not determined.
The association of AChE with the amyloid aggregates is an early event during fibril formation
To investigate how early during the Aβ–AChE complex formation the enzyme becomes associated with the fibrils, 40 μl aliquots were taken from turbidity assays at different incubation times, and the AChE activity present in the pellets was determined. Figure2 shows that the AChE activity present in the incubation mixture became rapidly associated with the Aβ aggregates. In fact, as early as 30 min after mixing, most of the AChE added was present in the precipitate fraction containing amyloid aggregates (Fig. 2, •). Concomitant with this, most of the free enzyme was removed from the soluble fraction (Fig. 2, ○). The amyloid character of the Aβ aggregates formed in the presence of AChE was determined by Congo red assays (Fig. 2, inset). This result indicates that the formation of AChE–Aβ complexes is an early event during the Aβ aggregation process.
Fig. 2.

Temporal dependence of AChE–Aβ fibril complex formation. Turbidimetric stirred assays of Aβ with AChE were incubated for 90 min. Then aliquots of 40 μl were taken at different incubation times, diluted in 400 μl of PBS, and centrifuged, and AChE activity was determined in the fibril pellet (•) and the supernatant fraction (○) by the assay of Ellman et al. (1961). The amyloid character of the aggregates formed in the presence of AChE was determined by Congo red assays (inset) in the pellet fraction.
Morphological examination of the AChE–Aβ complexes
The structure of the AChE–Aβ complexes was examined under fluorescence microscope after staining with thioflavine-T; the aggregates showed a strong green fluorescence, indicating their amyloid character (Fig. 3A,B) (Roher et al., 1986). When these aggregates were histochemically stained for AChE using the method of Karnovsky and Roots (1964), they showed the typical brown of this staining reaction (Fig. 3C,D), confirming the presence of AChE activity in the AChE–Aβ complexes. Amyloid aggregates generated in the absence of AChE did not show such histochemical staining (data not shown). Finally, we examined the amyloid filaments to establish whether the filaments were indeed complexed to AChE. Using immunogold electron microscopy with antibodies directed against AChE, small specks could be seen in the AChE–Aβ complexes (Fig. 4A) and AChE–Aβ fibers (Fig. 4B). Aβ complexes alone did not show specific labeling (Fig. 4C). The electron microscopic examination of the amyloid filaments demonstrated the existence of the AChE–Aβ complex.
Fig. 3.

Thioflavine-T fluorescence and AChE histochemical staining of the AChE–Aβ complexes. A, B, Amyloid aggregates formed in a kinetic stirred assay were washed three times with PBS and then incubated for 4 min in a solution containing an excess of thioflavine-T (5 mg/ml). Samples were examined under an Axioplan fluorescence microscope (D-7082; Zeiss, Oberkochen, Germany) at 400× magnification. C, D, Amyloid aggregates from a kinetic stirred assay were washed three times with PBS, after which the fibrils were stained for AChE with the histochemical staining reaction of Karnovsky and Roots (1964).
Fig. 4.
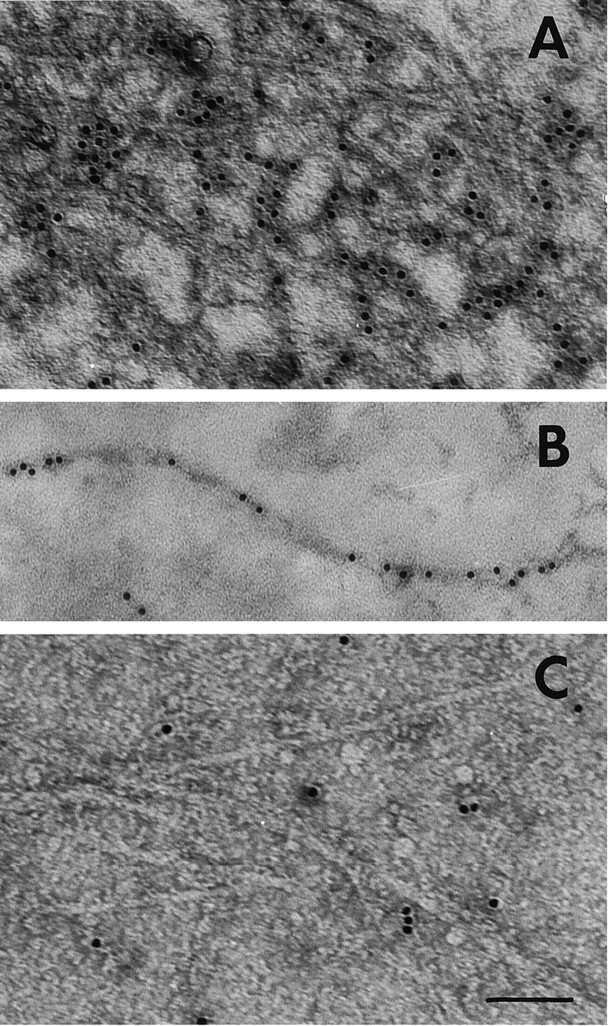
Immunogold labeling of the AChE–Aβ complexes.A, AChE–Aβ complexes were labeled with a polyclonal antibody raised against AChE, followed by an anti-IgG antibody conjugated to 10 nm colloidal gold particles, and were visualized by negative staining. B, Select example of an amyloid fiber labeled with anti-AChE conjugated to gold particles. C, Control sample of high molecular mass Aβ aggregates without AChE that were treated with the same specific antibody as above. In each case, 10 μl aliquots taken from an Aβ peptide kinetic stirred aggregation assay, performed either in the presence or absence of AChE, were adsorbed onto 300-mesh Formvar-coated grids, negative-stained with 2% uranyl acetate, and viewed for fibrils with a Philips electron microscope. Scale bar: A, C, 90 nm;B, 80 nm.
Enzymatic properties of the AChE present in the AChE–Aβ complexes
There are previous studies indicating that the AChE associated with the amyloid deposits of AD brains appears to have unique properties (Geula and Mesulam, 1989; Kalaria et al., 1992). Therefore, a thorough biochemical and pharmacological analysis of the amyloid-associated AChE was performed. Kinetic studies were performed, and the effects of different inhibitors, excess substrate, and low pH were evaluated. Figure 5Ashows the Lineweaver–Burk plot (1/V vs 1/S) obtained for soluble AChE and Aβ-complexed AChE. It is apparent that the kinetic parameters change, and that the Km andVmax values for the amyloid were higher than for the soluble AChE (Table 3). When the AChE–Aβ complexes were incubated under different pH conditions, it became clear that the enzyme associated with amyloid was more resistant to low-pH conditions than the free enzyme (Fig. 5B). When the incubations were performed in the presence of increasing substrate concentrations, the Aβ-associated AChE was more resistant to inhibition by excess acetylthiocholine (Fig. 5C). A shift toward the right is observed on the rising side of the curve (in the lower concentration range), whereas on the descending side of the curve (high concentrations), the data are most coincident (although not identical) for both AChE and AChE–Aβ. Moreover, the activity peak for AChE–Aβ is sharper and narrower than that for AChE alone. This behavior probably indicates that once the substrate has gained access (in the higher concentration range), the effects of excess substrate on both AChE and AChE–Aβ are very similar if not identical. However, at lower concentrations, it is more difficult for the substrate to bind complexed AChE than free AChE for reasons of physical hindrance.
Fig. 5.
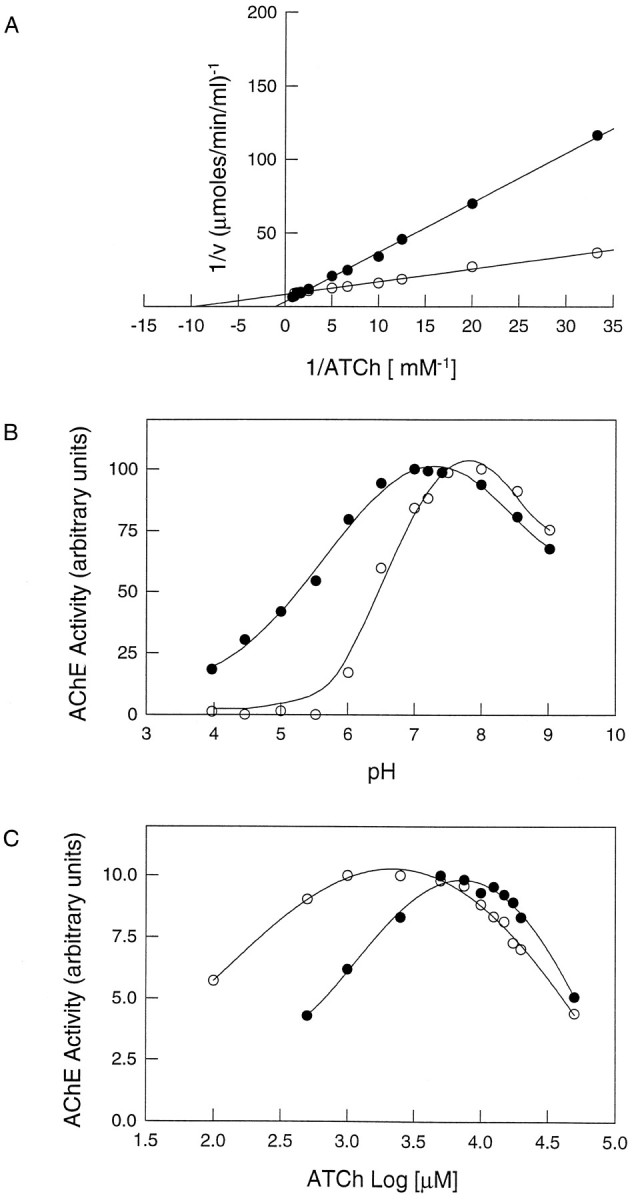
Biochemical characterization of the AChE activity associated with amyloid-β fibrils. AChE–Aβ complexes were washed exhaustively with PBS using four cycles of centrifugation and resuspension to remove noncomplexed AChE, and then amyloid-associated AChE activity was studied (•). Soluble native brain AChE was used as control (○). A, Lineweaver–Burk (double-reciprocal) plots. AChE activity, free and associated with amyloid-β fibrils, was measured over a range of substrate concentrations. B, pH dependence. Optimal pH for AChE and AChE–Aβ complex activity was determined over a pH range of 4.0–9.0 in phosphate buffer.C, Activity of AChE as a function of substrate concentration. The rate of hydrolysis is plotted as a log function of acetylthiocholine substrate concentration. The bell-shaped curves show that both free AChE and the AChE–Aβ complexes are inhibited by excess substrate. However, the AChE–Aβ complexes require higher acetylthiocholine concentrations for optimal activity than the free enzyme.
Table 3.
Comparison of the kinetic parameters of acetylthiocholine hydrolysis and effects of the specific inhibitors of AChE (IC50) on the free AChE and that associated with amyloid-β fibrils
| Parameter | AChE | AChE–Aβ complex |
|---|---|---|
| Kinetic parameters | ||
| Km(mm) | 0.097 ± 0.007 | 0.764 ± 0.025* |
| Vmax (μmol/min/ml) | 0.129 ± 0.015 | 0.222 ± 0.029* |
| Kss(mm) | 30.4 ± 1.1 | 48.1 ± 8.5** |
| Optimal pH | 7.5–8.0 | 7.0 |
| Activity at pH 5.0 (%) | 3.2 ± 0.8 | 41.1 ± 3.0* |
| IC50 values | ||
| Tacrine (nm) | 445.0 ± 17.6 | 1074.8 ± 73.3* |
| Edrophonium (μm) | 5.36 ± 0.48 | 17.6 ± 4.6*** |
| Propidium (μm) | 34.6 ± 1.20 | 72.0 ± 4.8* |
| Inhibition coefficients (Ki) | ||
| Tacrine (nm) | 51.0 | 542.4 |
| Edrophonium (μm) | 0.61 | 8.90 |
| Propidium (μm) | 3.96 | 36.3 |
The Km and Vmax was determined from Lineweaver–Burk plots (1/V vs 1/S), with 0.03–1.0 mm acetylthiocholine.Kss was determined by plotting of 1/Vas a function of substrate concentration (0.1–50 mm). For determination of IC50 values, samples were incubated with appropriate concentrations of the various inhibitors for 30 min at room temperature, and the AChE activity was determined colorimetrically.Ki values are determined as specified in Materials and Methods. Values represent means ± SD of four to six separate assays made in duplicate for AChE and three to five for AChE–Aβ complex.
*Significantly different from control with p < 0.001 by nonparied Student’s t test; ***p < 0.01; **p < 0.05.
Finally, the AChE associated with amyloid fibrils appeared more resistant to inhibition by anti-cholinesterase agents (Table 3). In particular, when fasciculin (a 61 amino acid peptide isolated from mamba venom (Rodriguez Ithurralde et al., 1983; Dajas et al., 1987) was used, a much higher concentration (micromolar) was required to inhibit the amyloid-associated AChE than the free soluble enzyme (nanomolar) (Fig. 6A). A similar trend was observed when monoclonal antibody 25B1 directed against AChE (Gentry et al., 1995), which also blocks the amyloid formation elicited by the enzyme, (Reyes et al., 1997) was used (Fig.6B). The Ki values obtained for the other anti-cholinesterase agents, Tacrine, edrophonium, and propidium, were 16-, 14-, and 9-fold higher, respectively, for the complexed AChE than for the soluble enzyme (Table 3). In all cases, a ∼5- to 10-fold increase in the substrate or inhibitor concentrations was required to reach the same level of AChE inhibition when AChE was associated with Aβ aggregates. That is to say, the effects were independent of the structure or size of the inhibitor molecules under consideration. As such, these results are consistent with a physical phenomenon, i.e., the location or occlusion of the enzyme enmeshed within a fibrillar environment. In a broad context, our results are consistent with the idea that the association of AChE with Aβ fibrils determines a change in its enzymatic properties, as occurs in the senile plaques of Alzheimer’s brain.
Fig. 6.
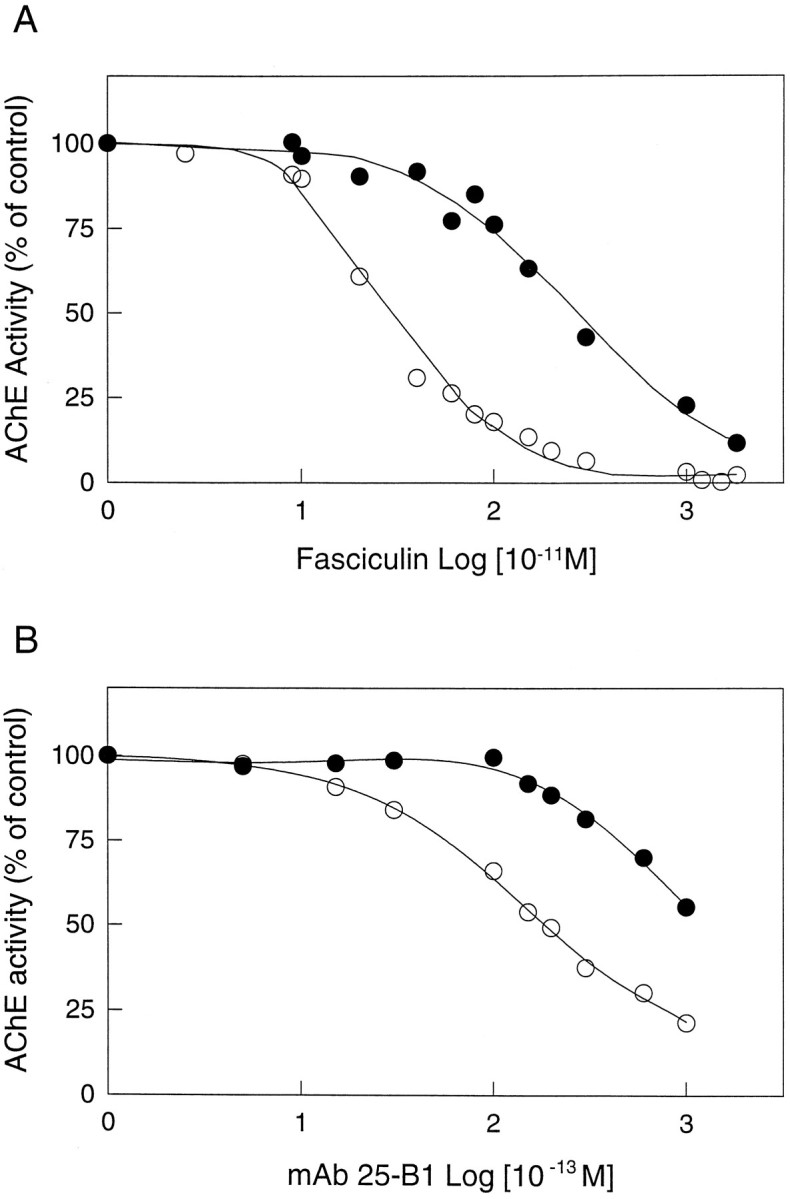
Inhibition of the AChE–Aβ complex by fasciculin and the monoclonal antibody 25-B1 (mAb 25-B1), two peripheral anionic binding site ligands. The activity is plotted as a function of fasciculin (A) and mAb 25-B1 (B) concentrations. Free AChE (○) and AChE–Aβ complexes (•) are both inhibited by fasciculin and mAb 25-B1. However, the AChE–Aβ complexes require higher concentrations of both compounds to reach the same level of inhibition as free AChE. The IC50 values for free AChE were 249 ± 15 pm for fasciculin and 19.0 ± 0.5 pm for mAb 25–B1. The corresponding values for the AChE–Aβ complexes were 2746 ± 28 pm for fasciculin and >100 pmfor mAb 25–B1. The IC50 values represent the mean ± SD of three identical samples run in separate experiments.
AChE–Aβ complexes exhibit increased neurotoxicity in relation to amyloid fibrils alone
Its has been demonstrated that amyloid fibrils are toxic to neuronal cells in culture (Lorenzo and Yankner, 1994; Yankner, 1996), and several studies have shown a strong correlation between Aβ neurotoxicity and the aggregation state of the Aβ peptide (Busciglio et al., 1992; Mattson et al., 1992; Pike et al., 1993). Hence we hypothesized that the fibrils of the AChE–Aβ complexes would be neurotoxic (Inestrosa et al., 1997), and to evaluate this point, we performed experiments using AChE–Aβ complexes versus Aβ aggregates in neuronal cell cultures. First, rat pheochromocytoma (PC12) cell cultures (Greene and Tischler, 1976) were treated with fibrillar Aβ and AChE–Aβ complexes. Cell viability was assessed by a decrease in their ability to reduce MTT (Mosmann, 1983). As shown in Figure7, Aβ neurotoxicity was dose-dependent in the range of 1–25 μm, and cell toxicity decreased to ∼27% of control cells measured in the absence of Aβ peptide. However, when the PC12 cells were treated with Aβ fibrils formed in the presence of AChE, a major decrease in MTT reduction was apparent in comparison with Aβ fibrils alone. This effect was dependent on the concentration of both the Aβ peptide and the AChE used in the complexes; the highest concentration of AChE added to the cells in complexed form was 25 nm, in comparison with 25 μm Aβ peptide. At this concentration, cell survival was minimal, and the cytotoxic effect of the fibrillar Aβ was enhanced almost twofold, whereas AChE added alone, at 25 nm, did not show a toxic effect (data not shown). Further experiments were performed using primary neuronal cultures to discard any possibility of a specific susceptibility of PC12 cells to the AChE–amyloid aggregates. The effect of the AChE–Aβ complexes was studied in chick primary cultures of retina cells (Vogel and Nirenberg, 1976; Puro et al., 1977; Paes de Carvalho and De Mello, 1982). Figure8A shows a control 6 d neuronal cell culture. These cells are known to display abundant neurite outgrowth and present several types of neurotransmitter receptors, including GABA, dopamine, and acetylcholine of both nicotinic and muscarinic sort (Enna and Snyder, 1976; Vogel et al., 1976; Sugiyama et al., 1977; Makman et al., 1980). After 24 hr of treatment with β-amyloid fibrils (2.5 μm), the retina cells showed a clear loss of neurites and a decrease in cell density (Fig. 8B). However, when the neuronal cultures were exposed to AChE–Aβ complexes, aged for 5 d, dramatic changes were observed. Only a minimal number of cells were seen to present neurites, and most cells were dead after 24 hr of culture (Fig.8C). Summarizing, studies with two very different cell culture systems indicate that the AChE–Aβ complex is more toxic than Aβ fibrils alone, suggesting that the incorporation of AChE into Alzheimer’s amyloid fibrils could lead to an increase in their neurotoxic properties. The AChE associated with the β-amyloid deposits present in Alzheimer’s brain may indeed be a relevant factor in triggering the neurodegeneration induced by senile plaques in specific regions of the brain.
Fig. 7.

AChE–Aβ complexes are more toxic than Aβ fibrils alone in PC12 cells. Rat pheochromocytoma PC12 cells were treated with Aβ amyloid fibrils as a control (white bars) or with AChE–Aβ amyloid fibrils (black bars) for 48 hr at different Aβ concentrations. The cell viability after treatment was measured by the MTT reduction assay. Significantly different from control: *p < 0.05; **p < 0.001 by nonpaired Student’st test; n.s., not significant.
Fig. 8.
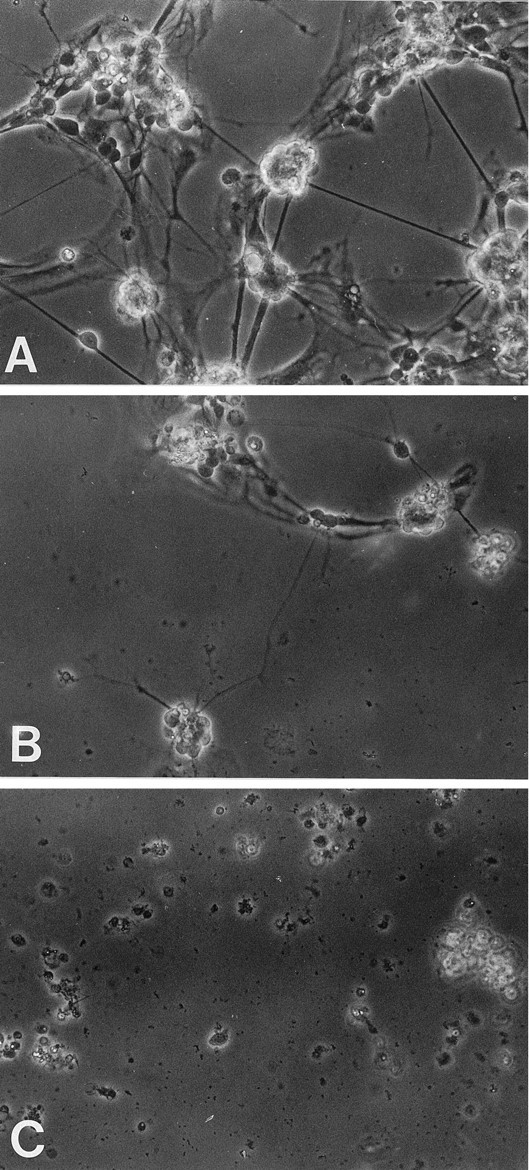
AChE–Aβ complexes are more toxic than Aβ fibrils alone in primary cultures of retina cells. Six-day chick retina cell cultures were incubated for 24 hr in a medium supplemented with PBS as a control (A), β-amyloid fibrils (2.5 μm) (B), and AChE–Aβ complexes (2.5 μm for the Aβ peptide) (C). The retina cell cultures were observed by phase contrast microscopy using 40× magnification under an Olympus inverted microscope. Representative photographs are shown.
DISCUSSION
The interaction of AChE with Aβ aggregates results in the formation of a stable AChE–Aβ complex
In the present study, the ability of purified AChE to form an AChE–Aβ1–40 complex was demonstrated. AChE was able to bind and interact with amyloid aggregates forming stable macromolecular complexes. In fact, such complexes were joined by strong intermolecular bonds because they were partially disrupted only by chaotropic agents (6 m guanidine-HCl and 6 m guanidine isothiocyanate), agents known to be able to solubilize amyloid cores from Alzheimer’s brain (Masters et al., 1985) and to disrupt amyloid fibrils (Naiki et al., 1991), and by SDS detergent, able to disassemble amyloid fibrils formed by the Aβ1–40 peptide (Soreghan et al., 1994). High ionic strength buffers were unable to dissociate the enzyme from these complexes. A very high percentage of the initial AChE present during fibrillogenesis remained bound to the Aβ fibrils, as estimated by the incorporation of 125I-AChE or by enzymatic activity measurements. Immunogold labeling confirmed that AChE was tightly bound to the amyloid fibrils.
It would appear that aggregation with AChE is not a promiscuous process but, rather, thermodynamically favored, because only a small amount of AChE is required to promote aggregation (Fig. 1). In fact, the stoichiometric ratio of AChE/Aβ in the final complexes was ∼1:1000, and it is possible that this ratio represents the successive incorporation of Aβ monomers onto an initial Aβ fibril containing an AChE–Aβ complex. A morphological examination of such complexes showed that their structural properties were typical of amyloid; i.e., they displayed fluorescent staining with thioflavine-T and a fine structure similar to that of amyloid Aβ fibrils, as determined by negative staining under the electron microscope. Finally, our biochemical data indicated that the incorporation of AChE into the amyloid complex was an early event during the polymerization and growth of the fibrils. This suggests a role for the AChE–Aβ complex at the beginning of the amyloidogenic process, whereby AChE may act as a heterogeneous nucleus, increasing the rate of fibrillogenesis and stabilizing the growing amyloid fibrils. Consistent with this possibility is the finding that when AChE was incubated with partially formed amyloid fibrils, only a small proportion of the enzyme (2%) was able to bind to the fibrils (data not shown) (R. Alarcón and N. C. Inestrosa, unpublished results). Such behavior can be expected from heteronucleators in a polymerization process, which act during the initial nucleation phase of filament formation (Ferrone et al., 1985).
The formation of an AChE–Aβ complex in vitro is reminiscent of the formation of apoE–Aβ complexes observed in vivo (Naslund et al., 1995; Castaño et al., 1995). In this case, the existence of the apoE–Aβ complex together with genetic evidence has been used to support a role for apoE as a risk factor in AD (Selkoe, 1996).
The AChE–Aβ complex changes the biochemical properties of the enzyme
The formation of an AChE–Aβ complex is entirely consistent with the fact that AChE and butyrylcholinesterase have been identified within Aβ deposits, such as those found in preamyloid diffuse and mature senile plaques and cerebral blood vessels (Morán et al., 1993; Geula and Mesulam, 1994), and emphasizes the potential relevance of AChE in AD. Most of the AChE found in senile plaques is associated with the amyloid cores, and only a small portion is associated with the neuritic components at the periphery of the plaques (Ulrich et al., 1990; Carson et al., 1991). Histochemical studies have demonstrated that the AChE associated with senile plaques differs from the enzyme found in normal fibers and neurons with respect to optimal pH, inhibition by excess substrate, and inhibitor sensitivity (Mesulam et al., 1987; Geula and Mesulam, 1989; Schätz et al., 1989; Wright et al., 1993). Furthermore, biochemical studies have indicated that senile plaque-associated AChE is only partially extracted using collagenase digestions (Nakamura et al., 1990), heparan sulfate extractions (Kalaria et al., 1992), or high-salt buffers plus detergent (Mimori et al., 1997). Interestingly, the AChE present in the AChE–Aβ complexes reported here showed unique properties similar to those of senile plaque-associated AChE. In fact, the complexed enzyme (1) presented Km and Vmaxvalues higher than those of the free enzyme and was more resistant to (2) incubation under low pH conditions, (3) inhibition by anti-cholinesterase agents, and (4) inhibition by excess acetylthiocholine. Regarding the latter, the shift toward the right seen in the lower concentration side of the curve (Fig. 5C) suggests that the fibrils establish a physical barrier that hinders access of the substrate to the enzyme active site. This would explain why higher concentrations of acetylthiocholine are required and would also account for the reactions obtained with small organic inhibitors as well as larger ones. This physical trapping of the enzyme is also consistent with our observation (Fig. 2) that formation of the AChE–Aβ complex is an early rather than late event in the fibrillogenic process. Maturation of the fibrils inhibits or precludes occlusion of the enzyme within Aβ, whereas fibrils still in their initial phases retain the freedom to trap the enzyme. In addition, as indicated in Table 2, such a mechanism is distinct from the hydrophobic interactions typical of globular proteins and allows for the observation that not all denaturants, certainly not high-salt media, are able to solubilize the fibril components. Physical trapping of the enzyme could also account for the general observation that butyrylcholinesterase, which has no peripheral anionic binding site and displays different kinetic properties to AChE, is also found amid amyloid fibrils. This theory is quite interesting, in that a structural gene defect in AChE, butyrylcholinesterase, or any other enzyme species caught midst the amyloid fibrils is not a requisite.
AChE–Aβ complexes increase the neurotoxicity of Alzheimer’s fibrils
The neurotoxicity of Aβ fibrils, together with the genetic evidence linking the amyloid precursor protein (APP) with AD, constitute the most relevant evidence to support a central role for the Aβ peptide in the pathogenesis of AD (Selkoe, 1996; Yankner, 1996). The observation, in AD brain studies, that neurodegenerative changes occur around senile plaques (Mann and Esiri, 1989) and the demonstration that Aβ together with various C-terminal fragments of APP containing the Aβ sequence is neurotoxic in cell cultures gave rise to the hypothesis that Aβ may be the primary cause of neuronal degeneration in AD (Yankner et al., 1989; Yankner, 1996). The neurotoxicity of Aβ is dependent on its aggregation state (Busciglio et al., 1992; Pike et al., 1993); it requires the assembly of Aβ into amyloid fibrils, whereas nonfibrillar, amorphous aggregates of Aβ are not neurotoxic (Lorenzo and Yankner, 1994; Busciglio et al., 1995;Howllett et al., 1995). Our studies showing that the AChE–Aβ complex presented a stronger cytotoxic effect than Aβ fibrils alone in both PC12 cells and primary retina cells indicate that the incorporation of AChE into amyloid fibrils changes their cytotoxic properties. This result is interesting because, although Aβ is a primary factor in the pathogenesis of AD (Selkoe, 1997), there are several reports suggesting that Aβ deposition is not the sole determinant of disease progression. For example, substantial numbers of amyloid plaques occasionally appear in the brain in nondemented individuals, suggesting that Aβ deposition does not invariably lead to AD (Dickson et al., 1991). It is possible that this may be in part attributable to differences in the protein composition and structure of the plaques in demented and nondemented individuals (Barcikowska et al., 1989; Cras et al., 1991; Mesulam and Geula, 1994). Besides the capacity of AChE to modulate the toxicity of amyloid fibrils in vitro, other macromolecules such as apoE and apoJ have also been shown to regulate the toxicity of these fibrils. However, both of these elements were found to decrease amyloid toxicity in culture (Oda et al., 1995; Boggs et al., 1996; Puttfarcken et al., 1997), suggesting that an adequate balance between toxic elements and the microenvironment defines the final toxicity of Aβ fibrils. In this context, it is interesting to mention that the potential role of AChE in AD is further supported by the fact that AChE systems, in particular those more vulnerable to AD such as the lightly stained neurons located in the entorhinal cortex, the CA1 and subiculum of the hippocampus, and the amygdala (Shen, 1994,Kasa et al., 1997), are the first to be affected in the pathological process of AD. Although the cellular mechanism of action of Aβ is not precisely understood, several processes have been proposed. In brief, Aβ has been shown to alter Ca2+ homeostasis (Mattson et al., 1992; Arispe et al., 1993), to enhance excitotoxic mechanisms (Mattson et al., 1992), and to generate free radicals (Hensley et al., 1994), although some controversy arises in the latter case (Sayre et al., 1997). Thus, AChE could affect the toxic signaling of Aβ at several points. An alternative possibility is that AChE per se activates neuronal cell death (Calderón et al., 1996,1998). In any case, further experiments are required to fully understand the mechanisms by which the AChE–Aβ complex is more toxic for neuronal cells in culture than Aβ fibrils alone.
Footnotes
This research was supported by Fondo Nacional de Desarrollo Científico y Technológico Grant 1971240 to N.C.I., Comisión Nacional de Investigación Científica y Technológica PhD Thesis Award 2960052 to A.A., and a predoctoral fellowship from Dirección de Investigación y Postgrado, Pontificia Universidad Católica de Chile to C.O. N.C.I. is a recipient of a Presidential Chair in Science from the Chilean Government. F.J.M. is on leave from Hospital La Princesa (Madrid, Spain).
Correspondence should be addressed to Dr. Nibaldo C. Inestrosa, Molecular Neurobiology Unit, Catholic University of Chile, P.O. Box 114-D, Santiago, Chile.
REFERENCES
- 1.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor α1-antichymotrypsin in the brain amyloid deposits of the Alzheimer’s disease. Cell. 1988;52:487–501. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez A, Bronfman F, Pérez CA, Vicente M, Garrido J, Inestrosa NC. Acetylcholinesterase, a senile plaque component, affects the fibrillogenesis of amyloid-β-peptides. Neurosci Lett. 1995;201:49–52. doi: 10.1016/0304-3940(94)12127-c. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez A, Opazo C, Alarcon R, Garrido J, Inestrosa NC. Acetylcholinesterase promotes the aggregation of amyloid-β-peptide fragments by forming a complex with the growing fibrils. J Mol Biol. 1997;272:348–361. doi: 10.1006/jmbi.1997.1245. [DOI] [PubMed] [Google Scholar]
- 4.Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid β-protein [AβP-(1–40)] in bilayer membrane. Proc Natl Acad Sci USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barcikowska M, Wisniewski HM, Bancher C, Grundke-Iqbal I. About the presence of paired helical filaments in dystrophic neurites participating in the plaque formation. Acta Neuropathol (Berl) 1989;78:225–231. doi: 10.1007/BF00687751. [DOI] [PubMed] [Google Scholar]
- 6.Boggs LN, Fuson KS, Baez M, Churgay L, Mcclure D, Becker G, May PC. Clusterin (ApoJ) protects against in vitro amyloid β(1–40) neurotoxicity. J Neurochem. 1996;67:1324–1327. doi: 10.1046/j.1471-4159.1996.67031324.x. [DOI] [PubMed] [Google Scholar]
- 7.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 8.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-Amyloid fibrils induce Tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 9. Calderón FH, De Ferrari GV, Luza SC, von Bernhardi R, Inestrosa NC. Toxic effects of acetylcholinesterase in neuronal cell cultures. Mol Biol Cell [Suppl] 7 1996. 652a, 3791. [Google Scholar]
- 10.Calderón FH, von Bernhardi R, De Ferrari GV, Luza S, Aldunate R, Inestrosa NC (1998) Toxic effects of acetylcholinesterase on neuronal and glial-like cells in vitro. Mol Psychiatry, in press. [DOI] [PubMed]
- 11.Campos EO, De Mello FG, Inestrosa NC (1997) Toxic effects of amyloid-β-peptide (Aβ), acetylcholinesterase (AChE) and AChE-Aβ complex on cultured cells of avian retina. Paper presented at XI Meeting of the Chilean Cell Biology Society, Termas de Cauquenes, Chile, September.
- 12.Carson KA, Geula C, Mesulam M-M. Electron microscopic localization of cholinesterase activity in Alzheimer brain. Brain Res. 1991;540:204–208. doi: 10.1016/0006-8993(91)90508-s. [DOI] [PubMed] [Google Scholar]
- 13.Castaño EM, Prelli F, Pras M, Frangione B. Apolipoprotein E carboxyl-terminal fragments are complexed to amyloid A and J. J Biol Chem. 1995;270:17610–17615. doi: 10.1074/jbc.270.29.17610. [DOI] [PubMed] [Google Scholar]
- 14.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci USA. 1991;88:7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dajas F, Bolioli B, Castello ME, Silveira R. Rat striatal acetylcholinesterase inhibition by fasciculin (a polypeptide from green mamba snake venom). Neurosci Lett. 1987;77:87–91. doi: 10.1016/0304-3940(87)90612-4. [DOI] [PubMed] [Google Scholar]
- 16.De Mello FG, Bachrach U, Nirenberg M. Ornithine and glutamic acid decarboxylase activities in the developing chick retina. J Neurochem. 1976;27:847–851. doi: 10.1111/j.1471-4159.1976.tb05145.x. [DOI] [PubMed] [Google Scholar]
- 17.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1991;13:179–189. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 18.Ellman GL, Courtney KD, Andres V, Featherstone RM. A new rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 19.Enna SJ, Snyder SH. Gamma-aminobutyric acid (GABA) receptor binding in mammalian retina. Brain Res. 1976;115:174–179. doi: 10.1016/0006-8993(76)90834-9. [DOI] [PubMed] [Google Scholar]
- 20.Evans KC, Berger EP, Cho C-G, Weisgraber KL, Lansbury PT. Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:763–767. doi: 10.1073/pnas.92.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J Mol Biol. 1985;183:611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 22.Gentry MK, Moorad DR, Hur RS, Saxena A, Ashani Y, Doctor BP. Characterization of monoclonal antibodies that inhibit the catalytic activity of acetylcholinesterases. J Neurochem. 1995;64:842–849. doi: 10.1046/j.1471-4159.1995.64020842.x. [DOI] [PubMed] [Google Scholar]
- 23.Geula C, Mesulam M-M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 1989;498:185–189. doi: 10.1016/0006-8993(89)90419-8. [DOI] [PubMed] [Google Scholar]
- 24.Geula C, Mesulam M-M. Cholinergic systems and related neuropathological predilection patterns in Alzheimer disease. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer disease. Raven; New York: 1994. pp. 263–291. [Google Scholar]
- 25.Gómez-Ramos P, Mufson EJ, Morán MA. Ultrastructural localization of acetylcholinesterase in neurofibrillary tangles, neuropil and senile plaques in aged and Alzheimer’s brain. Brain Res. 1992;569:229–237. doi: 10.1016/0006-8993(92)90634-l. [DOI] [PubMed] [Google Scholar]
- 26.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 28.Hensley K, Carrey JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer’s disease. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hobbiger F, Peck AW. Hydrolysis of suxamethonium by different types of plasma. Br J Pharmacol. 1969;37:258–271. doi: 10.1111/j.1476-5381.1969.tb09543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howllett DR, Jennings KH, Lee DC, Clarck MSG, Brown F, Wetzel R, Wood SJ, Camilleri D, Roberts GW. Aggregation state and neurotoxic properties of Alzheimer β-amyloid peptide. Neurodegeneration. 1995;4:23–32. doi: 10.1006/neur.1995.0003. [DOI] [PubMed] [Google Scholar]
- 31.Inestrosa NC, Perelman A. Distribution and anchoring of molecular forms of acetylcholinesterase. Trends Pharmacol Sci. 1989;10:325–329. doi: 10.1016/0165-6147(89)90067-9. [DOI] [PubMed] [Google Scholar]
- 32.Inestrosa NC, Perelman A. Association of acetylcholinesterase with the cell surface. J Membr Biol. 1990;118:1–9. doi: 10.1007/BF01872200. [DOI] [PubMed] [Google Scholar]
- 33.Inestrosa NC, Reiness CG, Reichardt LF, Hall ZW. Cellular localization of the molecular forms of acetylcholinesterase in rat pheochromocytoma PC12 cells treated with nerve growth factor. J Neurosci. 1981;1:1260–1267. doi: 10.1523/JNEUROSCI.01-11-01260.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inestrosa NC, Roberts WL, Marshall TL, Rosenberry TL. Acetylcholinesterase from bovine caudate nucleus is attached to membranes by a novel subunit distinct from those of acetylcholinesterase in other tissues. J Biol Chem. 1987;262:4441–4444. [PubMed] [Google Scholar]
- 35.Inestrosa NC, Alvarez A, Calderon FH. Acetylcholinesterase is a senile plaque component that promotes assembly of β-peptide into Alzheimer’s filaments. Mol Psychiatry. 1996a;1:359–361. [PubMed] [Google Scholar]
- 36.Inestrosa NC, Alvarez A, Pérez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996b;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 37.Inestrosa NC, Alvarez A, Garrido J, Calderón F, Bronfman FC, Dajas F, Gentry MK, Doctor BP. Acetylcholinesterase promotes Alzheimer’s β-amyloid fibril formation. In: Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, editors. Alzheimer’s disease: biology, diagnosis and therapeutics. Wiley; London: 1997. pp. 501–510. [Google Scholar]
- 38.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy-terminus of the β-amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 39.Kalaria RN, Kroon SN, Grahovac I, Perry G. Acetylcholinesterase and its association with heparan sulphate proteoglycans in cortical amyloid deposits of Alzheimer’s disease. Neuroscience. 1992;51:177–184. doi: 10.1016/0306-4522(92)90482-h. [DOI] [PubMed] [Google Scholar]
- 40.Karlsson E, Mbugua P, Rodriguez-Ithurralde D. Fasciculins, anticholinesterase toxins from the venom of the green mamba Dendroaspis angusticeps. J Physiol (Paris) 1984;79:232–240. [PubMed] [Google Scholar]
- 41.Karnovsky MJ, Roots L. A “direct-coloring” thiocholine method for cholinesterases. J Histochem Cytochem. 1964;12:219–221. doi: 10.1177/12.3.219. [DOI] [PubMed] [Google Scholar]
- 42.Kasa P, Rakonczay Z, Gulya K. The cholinergic system in Alzheimer’s disease. Prog Neurobiol. 1997;52:511–535. doi: 10.1016/s0301-0082(97)00028-2. [DOI] [PubMed] [Google Scholar]
- 43.Katzman R, Kawas CH. The epidemiology of the dementia and Alzheimer disease. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer disease. Raven; New York: 1994. pp. 105–122. [Google Scholar]
- 44.Klunk WE, Pettegrew JW, Abraham DJ. Two simple methods for quantifying low-affinity dye-substrate binding. J Histochem Cytochem. 1989;37:1293–1297. doi: 10.1177/37.8.2666512. [DOI] [PubMed] [Google Scholar]
- 45.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 46.LeVine H. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lorenzo A, Yankner BL. β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makman MH, Dvorkin B, Horowitz SG, Thal LJ. Properties of dopamine agonist and antagonist binding sites in mammalian retina. Brain Res. 1980;194:403–418. doi: 10.1016/0006-8993(80)91221-4. [DOI] [PubMed] [Google Scholar]
- 49.Mann DMA, Esiri MM. The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci. 1989;89:169–179. doi: 10.1016/0022-510x(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 50.Markwell MA. A new solid-state reagent to iodinate proteins. I. Conditions for the efficient labeling of antiserum. Anal Biochem. 1982;125:427–432. doi: 10.1016/0003-2697(82)90025-2. [DOI] [PubMed] [Google Scholar]
- 51.Massoulié J, Pezzementi L, Bon S, Krejci E, Vallette FM. Molecular and cellular biology of cholinesterases. Prog Neurobiol. 1993;41:31–91. doi: 10.1016/0301-0082(93)90040-y. [DOI] [PubMed] [Google Scholar]
- 52.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer’s disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel R. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mesulam M-M, Geula C. Butyrylcholinesterase reactivity differentiates the amyloid plaques of aging from those of dementia. Ann Neurol. 1994;36:722–724. doi: 10.1002/ana.410360506. [DOI] [PubMed] [Google Scholar]
- 55.Mesulam M-M, Geula C, Morán A. Anatomy of cholinesterase inhibition in Alzheimer’s disease: effect of physostigmine and tetrahydroaminoacridine on plaques and tangles. Ann Neurol. 1987;22:683–691. doi: 10.1002/ana.410220603. [DOI] [PubMed] [Google Scholar]
- 56.Mimori Y, Nakamura S, Yukawa M. Abnormalities of acetylcholinesterase in Alzheimer’s disease with special reference to effect of acetylcholinesterase inhibitors. Behav Brain Res. 1997;83:25–30. doi: 10.1016/s0166-4328(97)86041-x. [DOI] [PubMed] [Google Scholar]
- 57.Morán MA, Mufson EJ, Gómez-Ramos P. Colocalization of cholinesterases with β-amyloid protein in aged and Alzheimer’s brain. Acta Neuropathol (Berl) 1993;85:362–369. doi: 10.1007/BF00334445. [DOI] [PubMed] [Google Scholar]
- 58.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 59.Naiki H, Higuchi K, Nakakuki K, Takeda T. Kinetic analysis of amyloid fibril polymerization in vitro. Lab Invest. 1991;65:104–110. [PubMed] [Google Scholar]
- 60.Nakamura S, Kawashima S, Nakang S, Tsuji T, Araki W. Subcellular distribution of acetylcholinesterase in Alzheimer’s disease: abnormal localization and solubilization. J Neural Trans [Suppl] 1990;30:13–23. doi: 10.1007/978-3-7091-3345-3_2. [DOI] [PubMed] [Google Scholar]
- 61.Namba Y, Tomonaga M, Kawaski H, Otomon E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jacob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 62.Naslünd J, Thyberg J, Tjernberg LO, Wernstedt C, Karstrom AR, Bogdanovic N, Gandy SE, Lannfelt L, Terenius L, Nordstedt C. Characterization of stable complexes involving apolipoprotein E and the amyloid β peptide in Alzheimer’s disease brain. Neuron. 1995;15:219–228. doi: 10.1016/0896-6273(95)90079-9. [DOI] [PubMed] [Google Scholar]
- 63.Oda T, Wals P, Osterburg HH, Johnson SA, Pasinetti GM, Morgan TD, Rozovsky Y, Stine WB, Snyder SW, Holzman TF, Fratt GA, Finch CE. Clusterin (ApoJ) alters the aggregation of amyloid β-peptide (Aβ1–42) and forms slowly sedimenting Aβ complexes that cause oxidative stress. Exp Neurol. 1995;136:2–31. doi: 10.1006/exnr.1995.1080. [DOI] [PubMed] [Google Scholar]
- 64.Paes de Carvalho R, De Mello FG. Adenosine-elicited accumulation of adenosine 3′,5′-cyclic mono-phosphate in the chick embryo retina. J Neurochem. 1982;38:493–500. doi: 10.1111/j.1471-4159.1982.tb08655.x. [DOI] [PubMed] [Google Scholar]
- 65.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puro DG, De Mello FG, Nirenberg M. Synapse turnover: the formation and termination of transient synapses. Proc Natl Acad Sci USA. 1977;74:4977–4981. doi: 10.1073/pnas.74.11.4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puttfarcken PS, Manelli AM, Falduto MT, Godfrey SG, LaDu MJ. Effect of apolipoprotein E on neurite outgrowth and β-amyloid-induced toxicity in developing rat primary hippocampal cultures. J Neurochem. 1997;68:760–769. doi: 10.1046/j.1471-4159.1997.68020760.x. [DOI] [PubMed] [Google Scholar]
- 68.Radic Z, Reiner E, Taylor P. Role of the peripheral anionic site on acetylcholinesterase: inhibition by substrates and coumarin derivatives. Mol Pharmacol. 1991;39:98–104. [PubMed] [Google Scholar]
- 69.Reyes AE, Perez DR, Alvarez A, Garrido J, Gentry MK, Doctor BP, Inestrosa NC. A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem Biophys Res Commun. 1997;232:652–655. doi: 10.1006/bbrc.1997.6357. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez-Ithurralde D, Silveira R, Barbeito L, Dajas F. Fasciculin a powerful anticholinesterase polypeptide from Dendroaspis angusticeps. Neurochem Int. 1983;5:267–274. doi: 10.1016/0197-0186(83)90028-1. [DOI] [PubMed] [Google Scholar]
- 71.Roher A, Wolfe D, Palutke M, KuKuruga D. Purification, ultrastructure, and chemical analysis of Alzheimer disease amyloid plaque core protein. Proc Natl Acad Sci USA. 1986;83:2662–2666. doi: 10.1073/pnas.83.8.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sayre LM, Zagorski MG, Surewicz WK, Krafft GA, Perry G. Mechanisms of neurotoxicity associated with amyloid β deposition and the role of free radicals in the pathogenesis of Alzheimer’s disease: a critical appraisal. Chem Res Toxicol. 1997;10:518–526. doi: 10.1021/tx970009n. [DOI] [PubMed] [Google Scholar]
- 73.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 74.Schätz CR, Geula C, Mesulam M. Competitive substrate inhibition in the histochemistry of cholinesterase activity in Alzheimer’s disease. Neurosci Lett. 1990;117:56–61. doi: 10.1016/0304-3940(90)90119-t. [DOI] [PubMed] [Google Scholar]
- 75.Selkoe DJ. Amyloid β-protein and the genetics of Alzheimer’s disease. J Biol Chem. 1996;271:18295–18298. doi: 10.1074/jbc.271.31.18295. [DOI] [PubMed] [Google Scholar]
- 76.Selkoe DJ. Alzheimer’s disease: genotypes, phenotype, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 77.Sheffield JB, Moscona AA. Electron microscopic analysis of aggregation of embryonic cells: the structure and differentiation of aggregates of neural retina cells. Dev Biol. 1970;23:36–61. doi: 10.1016/s0012-1606(70)80006-9. [DOI] [PubMed] [Google Scholar]
- 78.Shen ZX. Acetylcholinesterase provides deeper insights into Alzheimer’s disease. Med Hypotheses. 1994;43:21–30. doi: 10.1016/0306-9877(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 79.Small DH, Michaelson S, Sberna G. Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem Int. 1996;28:453–483. doi: 10.1016/0197-0186(95)00099-2. [DOI] [PubMed] [Google Scholar]
- 80.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Susuki S, Hassell J, Wight TN. The presence of heparan sulfate proteoglycan in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 81.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soreghan B, Kosmoski J, Glabe C. Surfactant properties of Alzheimer’s Aβ peptides and the mechanism of amyloid aggregation. J Biol Chem. 1994;269:28551–28554. [PubMed] [Google Scholar]
- 83.Soto C, Brañes MC, Alvarez J, Inestrosa NC. Structural determinants of the Alzheimer’s amyloid β-peptide. J Neurochem. 1994;63:1191–1198. doi: 10.1046/j.1471-4159.1994.63041191.x. [DOI] [PubMed] [Google Scholar]
- 84.Sugiyama H, Daniels MP, Nirenberg M. Muscarinic acetylcholine receptors of the developing retina. Proc Natl Acad Sci USA. 1977;74:5524–5528. doi: 10.1073/pnas.74.12.5524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taylor P. The cholinesterases. J Biol Chem. 1991;266:4025–4028. [PubMed] [Google Scholar]
- 86.Ulrich J, Meier-Ruge W, Probst A, Meier E, Ipsen S. Senile plaques: staining for acetylcholinesterase and A4 protein. A comparative study in the hippocampus and entorhinal cortex. Acta Neuropathol (Berl) 1990;80:624–628. doi: 10.1007/BF00307630. [DOI] [PubMed] [Google Scholar]
- 87.Van Leeuwen FW, de Kleijn DPV, van den Hurk HH, Neubauer A, Sonnemans MAF, Sluijs JA, Koycu S, Ramdjielal RDJ, Salehi A, Martens GJM, Grosveld FG, Burbach JPH, Hol EM. Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer’s and down patients. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- 88.Vogel Z, Nirenberg M. Localization of acetylcholine receptors during synaptogenesis in retina. Proc Natl Acad Sci USA. 1976;73:1806–1810. doi: 10.1073/pnas.73.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vogel Z, Daniels MP, Nirenberg M. Synapse and acetylcholine receptor synthesis by neurons dissociated from retina. Proc Natl Acad Sci USA. 1976;73:2370–2374. doi: 10.1073/pnas.73.7.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wright CI, Geula C, Mesulam M-M. Protease inhibitors and indoleamines selectively inhibit cholinesterases in the histopathologic structures of Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:683–686. doi: 10.1073/pnas.90.2.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yankner BA. Mechanism of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 92.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]