

Abstract
Estrogen replacement therapy appears to delay the onset of Alzheimer’s disease (AD), but the mechanisms for this action are incompletely known. We show how the enhancement of synaptic sprouting by estradiol (E2) in response to an entorhinal cortex (EC) lesion model of AD may operate via an apolipoprotein E (apoE)-dependent mechanism. In wild-type (WT) mice, ovariectomy decreased commissural/associational sprouting to the inner molecular layer of the dentate gyrus, with synaptophysin (SYN) as a marker. E2 replacement returned SYN in the inner layer to levels of EC-lesioned, ovary-bearing controls and increased the area of compensatory synaptogenesis in the outer molecular layer. In EC-lesioned apoE-knock-out (KO) mice, however, E2 did not enhance sprouting. We also examined apoJ (clusterin) mRNA, which is implicated in AD by its presence in senile plaques, its transport of Aβ across the blood–brain barrier, and its induction by neurodegenerative lesioning. ApoJ mRNA levels were increased by E2 replacement in EC-lesioned WT mice but not in apoE-KO mice. These data suggest a mechanism for the protective effects of estrogens on AD and provide a link between two important risk factors in the etiology of AD, the apoE ε4 genotype and an estrogen-deficient state. This is also the first evidence that SYN, a presynaptic protein involved in neurotransmitter release, is regulated by E2 in the adult brain, and that apoE is necessary for the induction of apoJ mRNA by E2 in brain injury.
Keywords: Estrogen, apolipoprotein E, Alzheimer’s disease, sprouting, synaptophysin, brain lesion, apolipoprotein J
This study examines the interactions of estradiol (E2) and ovariectomy (OVX) with apolipoproteins E and J during reactive synaptogenesis in the hippocampus. Estrogen replacement therapy reduces the risk of Alzheimer’s disease (AD) by incompletely known mechanisms. In the CA1 neuronal field of the hippocampus, which is devastated in AD (Simic et al., 1997) E2 induces excitatory synapses and dendritic spines (Woolley and McEwen, 1992, 1993). E2 also enhances compensatory sprouting in response to entorhinal cortex lesioning (ECL), a model for the deafferenting aspect of AD (Geddes et al., 1985). In the response to ECL, the molecular layer of the dentate gyrus is reinnervated by sprouting from multiple pathways. The inner molecular layer receives an increase in commissural/associational (C/A) afferents, whereas the outer one-third receives afferents from the septohippocampal pathways, contralateral entorhinal cortex, and local interneurons (Steward and Loesche, 1977; Scheff, 1989). Ovariectomy reduces and E2 replacement reinstates compensatory sprouting by C/A neurons to the inner molecular layer (Morse et al., 1986). Effects of E2 on outer molecular layer sprouting have not been reported.
We also considered two apolipoproteins in neuronal sprouting. Apolipoprotein E (apoE) is a 37 kDa glycoprotein that mediates cholesterol transport in the CNS (Poirier et al., 1993a) and peripheral circulation (Koo et al., 1985). ApoE is involved in the response to neural injury (Boyles et al., 1990; Poirier et al., 1991; Poirier, 1994), maintenance of dendritic complexes (Masliah et al., 1995), and neuronal remodeling in vitro (Nathan et al., 1994; Fagan et al., 1996) and in AD (Arendt et al., 1997). Because apoE mRNA levels in the brain are induced by E2 (Srivastava et al., 1996; Stone et al., 1997), we hypothesized that E2 supports synaptic sprouting via increased apoE production, possibly for the transport of cholesterol and other hydrophobic membrane components. To test this hypothesis we manipulated E2 levels in wild-type (WT) and apoE-knock-out (KO) mice via OVX and E2 replacement and examined compensatory synaptic sprouting 2 weeks after entorhinal cortex lesioning.
ApoJ also mediates cholesterol transport (Jordan-Stark et al., 1994). Like apoE, apoJ mRNA is increased in experimental lesions and AD (May et al., 1990) and is under steroidal control (Day et al., 1990). The apoE ε4 allele, the most general risk factor for AD currently described, is associated with decreased apoE and increased apoJ in the AD brain (Bertrand et al., 1995). We hypothesized that apoJ mRNA levels would show a similar, brain-wide increase in apoE-KO mice, with apoJ compensating for the reduction in brain apolipoproteins caused by the removal of apoE.
MATERIALS AND METHODS
Surgery and estrogen replacement. Female mice (C57Bl/6J and apoE-KO; The Jackson Laboratory, Bar Harbor, ME) were maintained in a controlled light and temperature environment, with food and water ad libitum. ApoE-KO mice were developed, verified (by Southern blot and double immunodiffusion), and deposited at The Jackson Laboratory by Piedrahita et al. (1992). Animals underwent an OVX or sham OVX under 2,2,2-tribromoethanol anesthesia (0.4 gm/kg). After 1 week, mice were lesioned by perforant path transection with a stereotaxic retractable wire knife (Scouten wire knife; Kopf, Tujunga, CA) under the same anesthesia. The retracted assembly is inserted into the entorhinal cortex (0.5 mm anterior, 3.2 mm lateral of λ, and 1 mm ventral from dura). The extended blade was then lowered 2 mm ventrally twice at angles to avoid the hippocampus. For E2replacement, OVX mice were given 17β-estradiol at a concentration of 440 ng/ml in acidified drinking water, which produces serum E2 levels of 8 pg/ml during the day (approximately estrus or early diestrus levels) and 23 pg/ml at night (low proestrus levels) and was sufficient to suppress LH hypersecretion caused by OVX (Gordon et al., 1986). Thus E2 levels in E2-replaced mice did not exceed physiological levels. Control mice underwent anesthesia and sham ovariectomy or ECL. In sham ovariectomies skin and peritoneal wall incisions were made. In sham ECL scalps were incised, and holes were drilled in the skull with a hand-held dental drill; wire knife cuts were not made. Postoperative mortality was 60% higher in apoE-KO mice than in WT mice.
After 2 weeks the mice were anesthetized and perfusion-fixed in phosphate buffer, pH 7.4, containing 4% paraformaldehyde. Brains (n = 36, six per group) were immersion-fixed 1 d at 4°C in buffered paraformaldehyde, immersed in 30% sucrose for 3 d (4°C), and sectioned by cryostat (25 μm).
Immunohistochemistry. Sections were treated with 1% BSA and 1% normal goat serum in PBS to block nonspecific binding and then treated with primary antibody (rabbit anti-human synaptophysin at 1.5 μg/ml; Dako, Carpenteria, CA) for 90 min at room temperature. After washing in PBS, sections were exposed to biotinylated secondary antibody (goat anti-rabbit) for 1 hr and peroxidase-conjugated streptavidin for 30 min. (Vectastain; Vector Laboratories, Burlingame, CA). The reaction was completed with immunoperoxidase-DAB visualization.
In situ hybridization. Sections were washed in PBS and dehydrated in an ethyl alcohol series (30–100%). Sections were prehybridized for 1 hr at 55°C (prehybridization buffer: 0.75m NaCl, 50% formamide, 10% dextran sulfate, and 0.05m phosphate, pH 7.4) and hybridized with a35S-labeled cRNA probe. Sections were hybridized with35S-labeled cRNA for 3 hr at 55°C. Sense cRNA probes serve as controls for background signal. Slides were then covered with NTB2 emulsion (Eastman Kodak, Rochester, NY) and exposed for 10 d for cellular analysis. After development, slides were counter stained with cresyl violet.
Image analysis and statistics. Molecular layer width and optical density (OD) were measured with IPLab Spectrum image analysis software (Signal Analytics Corporation). To ensure that all measurements were objective, light and optical density threshold levels were established before measurement and were identical for each brain and treatment. Inner, middle, and outer molecular layers were distinguished as described previously (Masliah et al., 1991). Data were analyzed by two-way ANOVA on SuperANOVA statistical software (Abacus Concepts, Berkeley, CA). All statistical tests were run before data normalization.
RESULTS
Synaptophysin immunoreactivity
To estimate synaptic density in the dentate gyrus, we examined the immunoreactivity for the presynaptic protein synaptophysin (SYN) as described by Masliah et al. (1991). SYN immunoreactivity in the molecular layer of the normal dentate gyrus (unlesioned mice) did not differ between WT and apoE-KO mice (WT, 25.52 ± 2.1 OD units; KO, 29.6 ± 8.8 OD units, not significant). Fourteen days after EC lesioning, both WT and apoE-KO mice (with ovaries) displayed the expected laminated pattern of SYN immunoreactivity in the dentate gyrus (Masliah et al., 1991), in which immunostaining was increased in the inner and outer molecular layers but decreased in the middle molecular layer (Fig. 1A,D). Integrated density of the inner molecular layer was 15% lower in apoE-KO mice than in wild-type mice (Fig.2A).
Fig. 1.
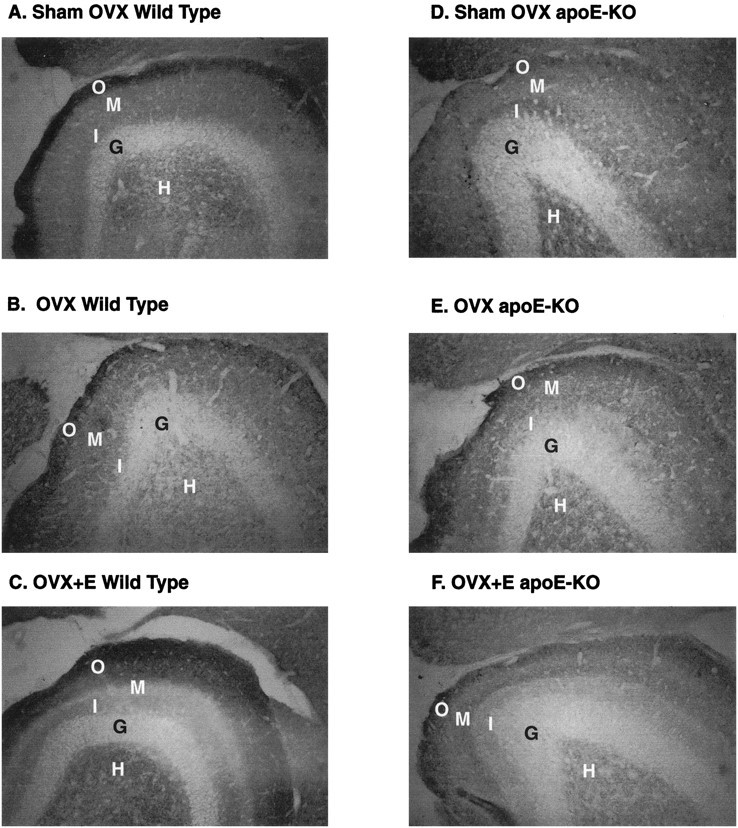
Synaptophysin immunoreactivity in the dentate gyrus of ECL wild-type and ApoE-KO mice. C57BL/6J mice (A–C) and apoE-KO mice (D–F) showed different responses to estradiol after ECL. Two weeks after surgery, sham-OVX mice (A, WT; D, apoE-KO) showed less immunoreactivity in the middle molecular layer than in the inner or outer layer of the dentate gyrus. In wild-type C57BL/6J mice, OVX causes a decrease in both the relative thickness and OD of the inner layer (B), whereas estradiol replacement brought inner layer thickness and OD back to sham-OVX levels (C). In apoE-KO mice, OVX (E) and estradiol replacement (F) made no significant changes in inner molecular layer thickness or OD. Width of the outer molecular layer was also increased in the WT E2-replaced mice (C). In apoE-KO mice, outer molecular layer width was unaffected by estrogen treatment (D–F).H, Hilus; G, granular layer;O, outer; M, middle; I, inner molecular layer.
Fig. 2.

Markers for sprouting in response to ECL show E2-dependent effects in wild-type but not ApoE-KO mice.A, The absolute width and OD of SYN immunoreactivity in the inner molecular layer were measured with an image-processing analysis system, and integrated density (thickness × optical density) was calculated. The y-axis gives integrated density as a percent of that for the WT, ovary-bearing (control) mice. There was a decrease in both thickness (15%; p < 0.05) and OD (35%; p < 0.05) of the inner layer of WT ovariectomized mice. E2 replacement returned inner layer width, OD, and integrated density to control levels. ApoE-KO mice did not show any significant changes in response to E2. Ovary-bearing apoE-KO mice showed a reduced integrated density (15%; NS) when compared with ovary-bearing controls. *Significantly different from control; p < 0.05. B, The OD of the outer molecular layer reached the threshold of image analysis software; however, the width of the outer layer was measured. They-axis gives the width of the outer molecular layer, expressed as a percent of the total molecular layer width. Although OVX did not cause a significant decrease in outer layer width in WT mice, E2-replaced mice showed a significant increase in outer layer width. In contrast, no E2 effect on outer molecular layer width was detected in aopE-KO mice. *Significantly different from inner layer width; p < 0.05.
OVX of WT mice decreased the C/A sprouting response to EC lesioning, as shown by the thickness and intensity of the SYN immunoreactivity of the inner molecular layer (Figs. 1B,2A). E2 replacement in OVX-WT mice restored expression of SYN to that of ovary-bearing, lesioned controls (Figs. 1C, 2A). In contrast, apoE-KO mice did not respond to manipulations of E2, as measured by SYN immunoreactivity in the inner molecular layer (Figs.1D–F,2A).
Optical density for SYN in the outer molecular layer could not be quantified because of a ceiling effect (i.e., staining too intense and not linear). The width of the outer molecular layer, however, was significantly increased in E2-replaced WT mice (Figs.1C, 2B). ApoE-KO mice did not show any difference in outer molecular layer width between groups (Figs.1D–F, 2B).
Apolipoprotein J mRNA responses
ApoJ mRNA at the lesion site was increased threefold to fourfold in all mice. However, OVX- and E2-treated WT mice showed a twofold greater increase over sham-OVX and OVX-WT mice, whereas apoE-KO mice did not show E2 influences (Fig. 3A). In the molecular layer of the dentate gyrus of WT mice, apoJ mRNA levels were unaffected by OVX and E2 replacement, whereas in apoE-KO mice (Fig. 3B) OVX increased levels irrespective of E2 replacement. In ovary-bearing apoE-KO mice, apoJ mRNA levels in the deafferented dentate gyrus were not induced by ECL above levels in the contralateral dentate gyrus. Ovariectomized apoE-KO mice, with or without E2 replacement, had apoJ mRNA levels in the molecular layer such as those of WT controls. Although the present analysis at 2 weeks after ECL did not allow examination of the peak in the apoE mRNA response (6 d after ECL; Poirier et al., 1991), apoE mRNA levels still showed E2-dependent trends at both the lesion site and in the deafferented dentate gyrus of WT mice (data not shown).
Fig. 3.
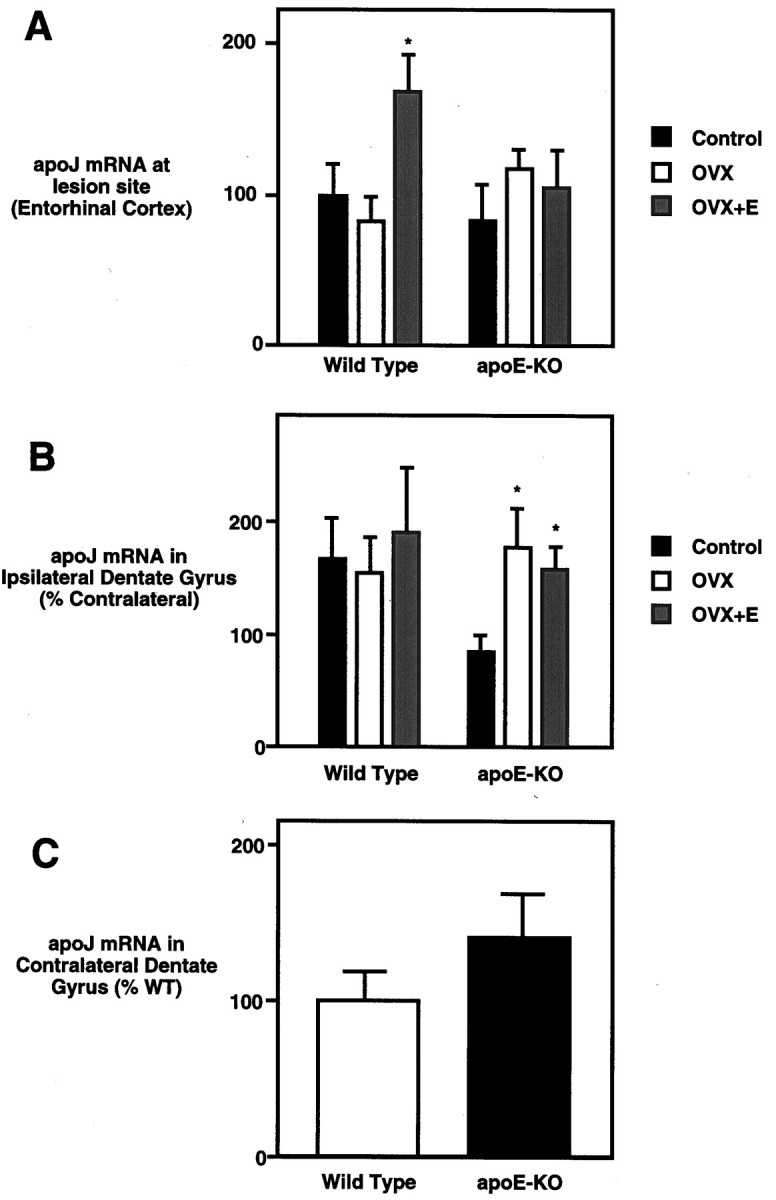
ApoJ mRNA levels show E2-dependent effects in wild-type but not ApoE-KO mice. A, At the lesion site (entorhinal cortex), apoJ mRNA level shows a nonsignificant decrease in response to OVX and a 1.7-fold increase with E2replacement. No significant changes were observed in apoE-KO mice. They-axis gives in situ grain density as a percent of WT, ovary-bearing (control) mice. B, In the deafferented dentate gyrus (ipsilateral to lesion), apoJ mRNA levels did not change in WT mice, whereas apoE-KO mice showed increased apoJ mRNA levels in response to ovariectomy. E2 replacement did not have an effect. Levels are given as a percent of contralateral values. The y-axis gives in situ grain density expressed as a percent of that in the contralateral (unlesioned) dentate gyrus of the same brain. C, In the unlesioned hippocampus (contralateral to lesion) of sham-ovariectomized mice, apoE-KO mice showed a nonsignificant increase in apoJ mRNA, suggesting a possible compensatory increase. This trend did not extend to the apoJ responses to E2 or lesion. They-axis gives the in situ grain density in the unlesioned dentate gyrus, expressed as a percent of that in WT mice. *Significantly different from sham-OVX mice;p < 0.05.
The diminished responsiveness of apoE-KO mice to E2 during sprouting responses to hippocampal deafferentation does not extend to reproductive tract functions. ApoE-KO mice are indistinguishable from WT mice in fecundity, e.g., number of corpus lutea or fetuses (Jablonka-Shariff et al., 1996) and estrogen-dependent uterine weights (Table 1). Thus the congenital deficiency of apoE does not impair some fundamental actions of E2outside of the brain.
Table 1.
Uterine weights of wild-type and ApoE-KO mice
| Treatment | Wild-type C57B1/6J | ApoE-KO | ||
|---|---|---|---|---|
| Weight (mg) | SE | Weight (mg) | SE | |
| Control | 72 | 9 | 65 | 6 |
| OVX | 20* | 2 | 18* | 2 |
| OVX + E2 | 183* | 16 | 165** | 15 |
OVX caused a threefold decrease, and E2 replacement caused a 2.5-fold increase in uterine weight in both WT and apoE-KO mice. There were no significant strain or strain × treatment interaction effects.
*p < 0.0001.
**p < 0.01.
These experiments thus demonstrate two trends: apoE-dependent effects of E2 in WT mice and E2-independent effects in the outer molecular layer of the dentate gyrus of apoE-KO mice revealed by OVX. In the unlesioned hippocampus of sham-OVX mice, apoJ mRNA levels showed a nonsignificant 1.4-fold increase in apoE-KO mice over WT (Fig. 3C). Because this effect did not extend to apoJ mRNA levels at the lesion site or in the deafferented hippocampus, loss of the apoE gene may not cause a compensatory apoJ increase in activated astrocytes.
DISCUSSION
Hormonal effects on synaptic sprouting
These studies show that SYN, a presynaptic protein that mediates neurotransmitter release, is regulated by E2. The enhanced outgrowth of afferent fibers by E2 (Morse et al., 1986,1992) is thus extended to presynaptic terminals. These findings also are consistent with the steroidal control of other mRNAs encoding presynaptic proteins (synaptosomal-associated protein 25) and growth cones (GAP-43) during development (Lustig et al., 1993).
These data demonstrate that E2 influences synaptic sprouting in response to injury via an apoE-dependent mechanism. In all brain parameters measured, apoE-KO mice did not respond to E2. The present findings, in conjunction with the induction of apoE by E2 (Srivastava et al., 1996; Stone et al., 1997), suggest that E2 increases compensatory synaptic sprouting by upregulating local transporters of cholesterol and other hydrophobic membrane components. Thus E2 could increase synaptic sprouting via increased production of apoE protein or increased uptake of apoE-containing lipoproteins.
Sprouting to the outer molecular layer of the dentate gyrus has been attributed to the septohippocampal pathway, because these fibers are acetylcholinesterase (AChE)-positive (Scheff, 1989). However, various markers associated with cholinergic neurons do not increase in accordance with AChE in the outer molecular layer of the dentate gyrus after ECL; thus the neurochemistry of the compensatory synapses in question is not fully characterized (Aubert et al., 1994). Because AChE-positive, noncholinergic interneurons are present in the hippocampus, it is possible that much of the reafferentation to the outer molecular layer is from a local source (Shute and Lewis, 1966). Unlike C/A sprouting, sprouting in the outer molecular layer was not decreased in WT mice by OVX (although E2 replacement caused an increase). The increased sprouting in the E2-replaced mice may be caused by the actions of estrogen when not interacting with progesterone or may be a result of exposure to high-physiological (proestrus) levels of estrogen on a daily basis, rather than once every 4 d as in normally cycling mice.
Both the E2-induced increase in apoJ mRNA at the lesion site (Fig. 3A) and the lesion-induced apoJ mRNA increase in the deafferented dentate gyrus (Fig. 3B, sham-OVX mice) appear to be apoE-dependent. A possible explanation is that the production of the two proteins is linked. In the optic tract cholesterol transport involves lipoprotein particles with both apoE and apoJ components (Shanmugaratnam et al., 1997). This finding predicts that production of apoE and apoJ in the CNS will be closely coregulated. The removal of apoE could thus alter production of these particles, resulting in changes in apoJ expression. Examples of this alteration would be the loss of both the estrogen effect at the lesion site and the lesion-induced increase in the deafferented dentate gyrus observed in the present study. The small, possibly compensatory increase in apoJ mRNA in apoE-KO mice over WT is only observed in the unlesioned dentate gyrus and does not apply to glia responding to steroids or damage.
Estrogen, apoE, ECL, and Alzheimer’s disease
ECL is used as a model of the deafferenting aspects of AD (see introductory remarks). In ECL, the outer and middle molecular layers of the dentate gyrus are deafferented by transection or ablation of the perforant path. The outer two-thirds of the molecular layer of the dentate gyrus receive the majority of their input from the stellate neurons from layer 2 of the entorhinal cortex, which make up the majority of the perforant path (Amaral and Witter, 1995). Likewise, one aspect of AD is the death of entorhinal cortex neurons and the loss of input to the dentate gyrus. As in ECL, the AD brain responds to the deafferentation with an increase in the C/A pathway and AChE-positive fibers in the outer molecular layer (Geddes et al., 1985).
These results thus pertain to the etiology and treatment of AD. The apoE ε4 allele is a risk factor for AD (Corder et al., 1993; Poirier et al., 1993b), and AD patients with the ε4 allele have less neuronal remodeling than those without the ε4 allele (Arendt et al., 1997). In cultured neurons, addition of the human apoE ε4 isoform also inhibits neurite outgrowth relative to ε3 (Nathan et al., 1994). Thus the ε4 allele may increase AD risk by impairing neuronal sprouting (Poirier et al., 1993a). Evidence that ε4 carriers with AD have less hippocampal apoE than noncarriers (Bertrand et al., 1995) supports this theory.
Estrogen increases activity of choline acetyltransferase (ChAT) and AChE (Luine and McEwen, 1983; Liune et al., 1986), high-affinity choline uptake (Singh et al., 1994), and ChAT mRNA levels (Gibbs et al., 1994; McMillan et al., 1996). Because cholinergic deficits are a hallmark of AD (Whitehouse et al., 1982), estrogen may slow the progress of AD by cholinergic upregulation. In AD patients, an allele dose of apoE ε4 is inversely correlated with ChAT activity, clinical responses to tacrine (a cholinomimetic drug), and the density of cholinergic forebrain neurons (Poirier et al., 1995). It has been hypothesized that the decreased apoE protein levels associated with the ε4 genotype (Bertrand et al., 1995) inhibit transport of phospholipids necessary for the production of choline (Poirier et al., 1995). Growing evidence suggests that estrogen replacement therapy slows the progression and delays the onset of AD (Simpkins et al., 1994; Paganini-Hill and Henderson, 1996; Tang et al., 1996). Increased apoE production or uptake in response to estrogen could ameliorate the effects of AD through two pathways: increased compensatory synaptic sprouting and increased ChAT activity.
The present data suggest a mechanism for this neuroprotective effect and a link between two important risk factors in AD. Moreover, the apparently greater risk of AD in female carriers of ε4 than in males (Poirier et al., 1993b; Payami et al., 1996; Rao et al., 1996) could be attributed to the combined effects of an estrogen-deficient state and the ε4 genotype on the ability for neuronal reorganization and ChAT activity. These data further suggest that estrogen replacement therapy might be less effective against AD in rare humans who have hereditary apoE deficiencies (Schaefer et al., 1986) and those with the apoE ε4 genotype.
Footnotes
This work was supported by National Institutes of Health Grants AG05766-0151 (D.J.S.) and AG-13499 (C.E.F.) and Alzheimer’s Association/Estate of Ann Clark Hobson Grant FSA 95033 (T.E.M.).
Correspondence should be addressed to David J. Stone, Andrus Gerontology Center and the Department of Biological Sciences, University of Southern California, 3715 McClintock Avenue, Los Angeles, CA 90089-0191.
REFERENCES
- 1.Amaral DG, Witter MP. Hippocampal formation. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1995. pp. 443–493. [Google Scholar]
- 2.Arendt T, Schindler C, Bruckner MK, Eschrich K, Bigl V, Zedlick D, Marcova L. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein e4 allele. J Neurosci. 1997;17:516–529. doi: 10.1523/JNEUROSCI.17-02-00516.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aubert I, Poirier J, Gauthier S, Quirion R. Multiple cholinergic markers are unexpectedly not altered in the rat dentate gyrus after entorhinal cortex lesions. J Neurosci. 1994;14:2476–2484. doi: 10.1523/JNEUROSCI.14-05-02476.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM. Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer’s disease. Mol Brain Res. 1995;33:174–178. doi: 10.1016/0169-328x(95)00097-c. [DOI] [PubMed] [Google Scholar]
- 5.Boyles JK, Notterpeck LM, Anderson LJ. Accumulation of apolipoproteins in the regenerating and remyelinating mammalian peripheral nerve. Identification of apolipoprotein D, apolipoprotein A-IV, apolipoprotein E, and apolipoprotein A-I. J Biol Chem. 1990;265:17805–17815. [PubMed] [Google Scholar]
- 6.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 7.Day JR, Laping NJ, McNeil TH, Schreiber SS, Pasinetti G, Finch CE. Castration enhances expression of glial fibrillary acidic protein and sulfated glycoprotein-2 in the intact and lesion-altered hippocampus of the adult male rat. Mol Endocrinol. 1990;4:1995–2002. doi: 10.1210/mend-4-12-1995. [DOI] [PubMed] [Google Scholar]
- 8.Fagan AM, Bu G, Sun Y, Daughtery A, Holtzman DM. Apolipoprotein E containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J Biol Chem. 1996;271:30121–30125. doi: 10.1074/jbc.271.47.30121. [DOI] [PubMed] [Google Scholar]
- 9.Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC. Plasticity of hippocampal circuitry in Alzheimer’s disease. Science. 1985;230:1179–1181. doi: 10.1126/science.4071042. [DOI] [PubMed] [Google Scholar]
- 10.Gibbs RB, Wu D, Hersh LB, Pfaff DW. Effects of estrogen replacement on the relative levels of choline acetyltransferase, trkA, and nerve growth factor messenger RNAs in the basal forebrain and hippocampal formation of adult rats. Exp Neurol. 1994;129:70–80. doi: 10.1006/exnr.1994.1148. [DOI] [PubMed] [Google Scholar]
- 11.Gordon MN, Osterburg HH, May PC, Finch CE. Effective oral administration of 17β-estradiol to female C57BL/6J mice through drinking water. Biol Reprod. 1986;35:1088–1095. doi: 10.1095/biolreprod35.5.1088. [DOI] [PubMed] [Google Scholar]
- 12. Jablonka-Shariff A, Schreiber JR, Wong TW. Reproductive capacity of male apolipoprotein E knock-out mice. Soc Study Repro 54[Suppl] 1996. 69 (abstract). [Google Scholar]
- 13.Jordan-Stark TC, Lund SD, Witte DP, Aronow BJ, Ley CA, Stuart WD, Swertfeger DK, Clayton LR, Sells SF, Paigen B, Harmony JAK. Mouse apolipoprotein J: characterization of a gene implicated in atherosclerosis. J Lipid Res. 1994;35:194–210. [PubMed] [Google Scholar]
- 14.Koo C, Innerarity TL, Mahley RW. Obligatory role of cholesterol and apolipoprotein E in the formation of large cholesterol enriched and receptor active high density lipoproteins. J Biol Chem. 1985;260:11934–11943. [PubMed] [Google Scholar]
- 15.Luine VN, McEwen BS. Sex differences in cholinergic enzymes of diagonal band nuclei in the rat preoptic area. Neuroendocrinology. 1983;36:475–482. doi: 10.1159/000123501. [DOI] [PubMed] [Google Scholar]
- 16.Luine VN, Renner KJ, Heady S, Jones KJ. Age and sex-dependent decreases in ChAT in basal forebrain nuclei. Neurobiol Aging. 1986;7:193–198. doi: 10.1016/0197-4580(86)90042-4. [DOI] [PubMed] [Google Scholar]
- 17.Lustig RH, Hua P, Wilson MC, Federoff HJ. Ontogeny, sexual dimorphism, and neonatal sex hormone determination of synapse-associated messenger RNAs in rat brain. Mol Brain Res. 1993;20:101–110. doi: 10.1016/0169-328x(93)90114-5. [DOI] [PubMed] [Google Scholar]
- 18.Masliah E, Fagan AM, Terry RD, DeTeresa R, Mallory M, Gage FH. Reactive synaptogenesis assessed by synaptophysin immunoreactivity is associated with GAP-43 in the dentate gyrus of the adult rat. Exp Neurol. 1991;113:131–142. doi: 10.1016/0014-4886(91)90169-d. [DOI] [PubMed] [Google Scholar]
- 19.Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the CNS of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- 20.May PC, Lampert-Etchells M, Johnson SA, Poirier J, Masters JM, Finch CE. Dynamics of gene expression for a hippocampal glycoprotein elevated in Alzheimer’s disease and in response to experimental lesions in rat. Neuron. 1990;5:831–839. doi: 10.1016/0896-6273(90)90342-d. [DOI] [PubMed] [Google Scholar]
- 21.McMillan PJ, Singer CA, Dorsa DM. The effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mRNA expression in the basal forebrain of the adult female Sprague Dawley rat. J Neurosci. 1996;16:1860–1865. doi: 10.1523/JNEUROSCI.16-05-01860.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morse JK, Scheff SW, DeKosky ST. Gonadal steroids influence axon sprouting in the hippocampal dentate gyrus: a sexually dimorphic response. Exp Neurol. 1986;94:649–658. doi: 10.1016/0014-4886(86)90244-x. [DOI] [PubMed] [Google Scholar]
- 23.Morse JK, DeKosky ST, Scheff SW. Neurotrophic effects of steroids on lesion-induced growth in the hippocampus. II. Hormone replacement. Exp Neurol. 1992;118:47–52. doi: 10.1016/0014-4886(92)90021-h. [DOI] [PubMed] [Google Scholar]
- 24.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 25.Paganini-Hill A, Henderson V. Estrogen replacement therapy and the risk of Alzheimer’s disease. Arch Intern Med. 1996;156:2213–2217. [PubMed] [Google Scholar]
- 26.Payami H, Zareparsi S, Montee KR, Sexton GJ, Kaye JA, Bird TD, Yu CE, Wijsman EM, Heston LL. Gender differences in apolipoprotein E-associated risk for familial Alzheimer-disease: a possible clue to the higher incidence of Alzheimer disease in women. Am J Hum Genet. 1996;58:803–811. [PMC free article] [PubMed] [Google Scholar]
- 27.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poirier J. Apolipoprotein E in animal models of CNS injury and in Alzheimer’s disease. Trends Neurosci. 1994;17:525–530. doi: 10.1016/0166-2236(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 29.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 30.Poirier J, Baccichet A, Dea D, Gauthier S. Cholesterol synthesis and reuptake during synaptic remodeling in hippocampus in adult rats. Neuroscience. 1993a;55:81–90. doi: 10.1016/0306-4522(93)90456-p. [DOI] [PubMed] [Google Scholar]
- 31.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993b;342:697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 32.Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, Hui S, Bertrand P, Nalbantoglu J, Gilfix BM, Gauthier S. Apolipoprotein E4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Nat Acad Sci USA. 1995;92:12260–12264. doi: 10.1073/pnas.92.26.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao VS, Cupples LA, Vandujin CM, Kurz A, Green RC, Chui H, Duara R, Auerbach SA, Volicer L. Evidence for major gene inheritance of Alzheimer disease in families of patients with and without apolipoprotein-E epsilon-4. Am J Hum Genet. 1996;59:664–675. [PMC free article] [PubMed] [Google Scholar]
- 34.Schaefer EJ, Gregg RE, Ghiselli G, Forte TM, Ordovas JM, Zech LA, Brewer HB. Familial apolipoprotein E deficiency. J Clin Invest. 1986;78:1206–1219. doi: 10.1172/JCI112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheff SW. Synaptic reorganization after injury: the hippocampus as a model system. In: Seil FJ, editor. Neural regeneration and transplantation. Liss; New York: 1989. pp. 137–156. [Google Scholar]
- 36.Shanmugaratnam J, Berg E, Kimmer L, Johnson RJ, Amaratunga A, Schreiber BM, Fine RE. Retional Muller glia secrete apolipoproteins E and J which are efficiently assembled into lipoprotein particles. Mol Brain Res. 1997;50:113–120. doi: 10.1016/s0169-328x(97)00176-9. [DOI] [PubMed] [Google Scholar]
- 37.Shute CCD, Lewis PR. Electron microscopy of cholinergic terminals and acetylcholine esterase containing neurons in the hippocampus of rats. Z Zellforsch Mikrosk Anat. 1966;69:334–343. doi: 10.1007/BF00406286. [DOI] [PubMed] [Google Scholar]
- 38.Simic G, Kostovic I, Winblad B, Bogdanovic N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J Comp Neurol. 1997;379:482–494. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 39.Simpkins J, Singh M, Bishop J. The potential role for estrogen therapy in treatment of cognitive decline and neurodegeneration associated with Alzheimer’s disease. Neurobiol Aging. 1994;15:195–197. doi: 10.1016/0197-4580(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 40.Singh M, Meyer EM, Millard WJ, Simpkins JW. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague Dawley rats. Brain Res. 1994;644:305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava RA, Bhasin N, Srivastava N. Apolipoprotein E gene expression in various tissues of mouse and regulation by estrogen. Biochem Mol Biol Int. 1996;38:91–101. [PubMed] [Google Scholar]
- 42.Steward O, Loesche J. Quantitative autoradiographic analysis of the time course of proliferation of contralateral entorhinal efferents in the dentate gyrus denervated by ipsilateral entorhinal lesions. Brain Res. 1977;125:11–21. doi: 10.1016/0006-8993(77)90356-0. [DOI] [PubMed] [Google Scholar]
- 43.Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Hajian H, Finch CE. Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol. 1997;143:313–318. doi: 10.1006/exnr.1996.6360. [DOI] [PubMed] [Google Scholar]
- 44.Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeaux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. Lancet. 1996;388:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 45.Whitehouse PJ, Price DL, Struble RG, Clark AW. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- 46.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. JComp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]