

Abstract
Amyotrophic lateral sclerosis (ALS) involves motor neuron degeneration, skeletal muscle atrophy, paralysis, and death. Mutations in Cu,Zn superoxide dismutase (SOD1) are one cause of the disease. Mice transgenic for mutated SOD1 develop symptoms and pathology similar to those in human ALS. To understand the disease mechanism, we developed a simple behavioral assay for disease progression in mice. Using this assay, we defined four stages of the disease in mice expressing G93A mutant SOD1. By studying mice with defined disease stages, we tied several pathological features into a coherent sequence of events leading to motor neuron death. We show that onset of the disease involves a sharp decline of muscle strength and a transient explosive increase in vacuoles derived from degenerating mitochondria, but little motor neuron death. Most motor neurons do not die until the terminal stage, ∼9 weeks after disease onset. These results indicate that mutant SOD1 toxicity is mediated by damage to mitochondria in motor neurons, and this damage triggers the functional decline of motor neurons and the clinical onset of ALS. The absence of massive motor neuron death at the early stages of the disease indicates that the majority of motor neurons could be rescued after clinical diagnosis.
Keywords: ALS, mitochondria, SOD1, paralysis, motor neuron, degeneration, spinal cord
Amyotrophic lateral sclerosis (ALS) involves motor neuron loss leading to progressive skeletal muscle atrophy and death (Mulder et al., 1986; Munsat, 1989). Despite a long history of clinical and pathological studies, the pathological progression for ALS has not been clearly defined. This is largely attributable to difficulties in correlating clinical symptoms and motor neuron loss in human patients. At least two possible models could explain the progressive nature of clinical symptoms. The first is that at the onset of muscle weakness the patient has already gone through an extended period of gradual motor neuron death. The emergence of symptoms reflects an exhausted functional compensation by the remaining motor neurons. Therefore, a large fraction of motor neurons have already been lost, and the clinical course reflects the loss of the remaining motor neurons. The second possibility is that at the onset of muscle weakness (or clinical disease) there is little or no motor neuron death, but there is a decline of motor neuron function. As the degenerative process proceeds, motor neurons gradually die, eventually leading to paralysis and death of the patient. The distinction between these two models has important implications for prognosis and therapy. New animal models developed recently for ALS have made it possible to distinguish between these two models.
ALS occurs in sporadic (SALS) and familial (FALS) forms (Mulder et al., 1986; Munsat, 1989). Mutations in the SOD1 gene are one genetic cause for FALS (Rosen et al., 1993; Brown, 1995). Several mouse models that express the mutated genes develop motor neuron degeneration similar to that in humans (Gurney et al., 1994; Ripps et al., 1995; Wong et al., 1995; Bruijn et al., 1997). The initial characterization of these mouse lines has proven that a dominant gain of an “adverse property” by the mutated enzymes causes motor neuron degeneration (for review, seeBruijn and Cleveland, 1996; Tu et al., 1997). In addition, these analyses confirmed numerous pathological features that have been observed in humans (Hirano, 1991; Chou, 1992), including axonal spheroids (Tu et al., 1996), increase of ubiquitin (Wong et al., 1995;Bruijn et al., 1997), Lewy body-like inclusions (Wong et al., 1995;Bruijn et al., 1997), fragmentation of Golgi apparatus (Mourelatos et al., 1996), and selective loss of motor neurons (Gurney et al., 1994;Ripps et al., 1995; Wong et al., 1995; Bruijn et al., 1997). The most prominent new feature is the large number of membrane-bound vacuoles in G93A and G37R lines (Dal Canto and Gurney, 1995; Wong et al., 1995). These vacuoles emerge before the end stage of the disease and appear to be derived from dilated mitochondria (Dal Canto and Gurney, 1995; Wong et al., 1995) and endoplasmic reticulum (Dal Canto and Gurney, 1995).
In these early studies, pathological changes were correlated with the age of animals but not with disease stage. Because of the highly heterogeneous onset and duration of the disease in individual animals (see below), the subjects of study at any age were a mixture of individuals at different disease stages. Consequently, pathological changes seen in animals of the same age were not necessarily the ones that develop at the same disease stage. Similarly, pathological features that appear in younger animals do not necessarily precede those seen in older animals in disease progression. Transient changes, which correspond to specific disease stages, are particularly difficult to recognize. In short, pathological studies without definition of the disease stage cannot delineate the sequence of events leading to motor neuron death and thus do not use the full potential of these transgenic animal models.
To effectively study these models, we have conducted the first experiments to analyze pathological changes in the context of clinical progression. We first developed a simple, objective assay to measure muscle strength in mice and used this assay to follow the clinical progression in a transgenic line that expresses the G93A SOD1 mutation (Gurney et al., 1994). We then quantitatively measured several pathological features in the mice with defined disease stages. We show that the disease progression goes through four stages, each with specific and time-dependent features. Of particular interest are the early stages in which mitochondrial abnormalities are the most prominent feature and neuronal function declines sharply, whereas neuronal death is minimal. Thus, mitochondrial degeneration is an important early event in triggering the decline of motor neuron function and, consequently, clinical disease. The minor motor neuron loss for a substantial period after the onset of clinical disease offers a period of time after diagnosis to rescue the majority of motor neurons.
MATERIALS AND METHODS
Human SOD1 transgenic mice. Mice transgenic for the mutated human SOD1 G93A (TgN[SOD1-G93A]1Gur) and wild-type human SOD1 (TgN[SOD1]2Gur) were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in the University of Massachusetts Medical School animal facility. Both lines were originally made and characterized byGurney et al. (1994). The survival of the purchased lines was prolonged in comparison with the original line because of a reduced number of transgene copies (lena.jax.org/resources/documents/imr/SODletter.html). The transgenic mice were identified using the PCR method described byGurney et al. (1994).
Muscle strength test. The test was conducted using a device designed in this laboratory. Mice were allowed to grab onto a vertical wire (2 mm in diameter) with a small loop at the lower end. A vertical wire allows mice to use both fore- and hindlimbs to grab onto the wire. Although in the first few tests some mice used forelimbs predominantly, they usually learned to use all four limbs after a few trials. This results in a significant improvement during the first three trials, after which the performances stabilize. Thus, both the fore- and hindlimbs contribute to the measured muscle strength in this assay.
The wire was maintained in a vertically oriented circular motion (the circle radius was 10 cm) at 24 rpm. Early tests indicated that maintaining the wire in motion gave much more consistent measurements than a stationary wire. The time that the mouse was able to hang onto the wire was recorded with a timer. Because most mice fell within 5 min, we cut off testing at 300 sec to test more animals in a limited time period. Mice were usually tested once a week starting when they were 90 d old, and testing continued until they could no longer hang onto the wire.
Morphological analysis. Animal fixation, tissue dissection, and microscopic analysis were performed as described previously (Xu et al., 1993). In brief, mice were anesthetized and perfused with a solution of 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1m phosphate buffer, pH 7.6. Tissues were kept in the same solution for further fixation. The L4 and L5 spinal nerve roots and lumbar spinal cords (a 2 mm segment centered at the L5 root entry level) were dissected out and post-fixed with 2% osmium tetroxide in 100 mm cacodylate buffer, pH 7.6. After dehydration in graded alcohol, the tissue blocks were embedded in Epon. Sections (1 μm) were stained with toluidine blue and examined with light microscopy.
For quantification of ventral roots, complete microscopic pictures of L4 and L5 ventral roots were taken from 1 μm transverse sections and digitized using a Nikon LS-1000 scanner. The area and equivalent radius of all axons in a ventral root larger than 1.5 μm in diameter were measured using MetaMorph software (Universal Imaging Corporation, West Chester, PA). Degenerating axons were counted directly under light microscope.
To quantify motor neurons in spinal cord, serial transverse sections 8 μm thick were cut from Epon-embedded L5 lumbar spinal cord. Every fifth section (five sections total from each animal) was collected and stained with toluidine blue. All neurons with nuclei and located in the lateral motor column (as defined in Fig.1) were drawn using a Nikon drawing tube. The drawings were scanned into computer using an HP flat-bed scanner. Several parameters, including the number and size of motor neurons and their nuclear size, were measured from these drawings using MetaMorph software.
Fig. 1.

The definition of L5 lateral ventral horn where neuronal numbers were quantified was derived as follows. Perpendicular to the midline M, line Npassing the central canal was drawn. Parallel to line Nand across point B was line L. PointB was the first point on the contour of the ventral gray matter (tracing from the midline) at which the contour made a >180° turn. Connecting point C (the cross-point of lineN and the contour of gray matter) and pointD (at one-third the distance from point Bto A), line P was drawn. All neurons within the area encompassed by line P and the contour of ventral horn (hatched area) were drawn and measured.
To measure vacuoles, two ventral horns were photographed from a 1 μm transverse section of the spinal cord. The picture frame contained an area 280 × 433 μm in size and covered about three-quarters of the ventral horn. These images were digitized using a Nikon LS-1000 scanner. Measurements were performed using MetaMorph image analysis software. Vacuoles were quantified using the threshold function. Nonvacuolar structures such as nuclei and large blood vessels were manually eliminated from the measurement. Capillary vessels were usually indistinguishable from the vacuoles under the light microscope and therefore were included in the measurement. However, this background was estimated by making the same measurements in wild-type animals.
For electron microscopy, thin sections of ventral horn were cut from the Epon tissue blocks, stained with uranyl acetate and lead citrate, and visualized using a Philips EM-400 transmission electron microscope.
RESULTS
Mouse ALS undergoes four functional declining stages
To follow disease progression in G93A mice, we developed a simple and objective assay to measure muscle strength. In this assay, the time that a mouse was able to hang onto a wire was measured as an indication of muscle strength (see Materials and Methods). We first tested mice in different age groups, which revealed an age-dependent decline of hanging time for G93A mice, but not for wild-type mice (WT) and wild type human SOD1 (WS) transgenic mice (Fig.2A).
Fig. 2.

Four stages of muscle strength change in G93A mice. The time that mice were capable of hanging onto a wire was measured as an indication of muscle strength. A, Age-dependent decline of muscle strength in G93A mice. Groups of mice at different ages were tested. The number of mice in each age group ranged from 3 to 20. G93A, Mutant SOD1 mice;WT, nontransgenic (wild-type) mice; WS, wild-type SOD1 transgenic mice. Error bars indicate SEM.B, Highly variable onset of muscle weakness among different G93A mice. Each trace represents measurements from one animal. For clarity, only measurements around the RD stage in 20 animals are shown. C, Synchronized plot of muscle strength decline in individual G93A mice. Zero week represents the time point just before the decline begins. For clarity, only 10 traces are shown. D, Average time course of muscle strength decline in G93A mice (n = 12). PMW, Pre-muscle weakness stage; RD, rapid declining stage;SD, slow declining stage; Para., paralysis stage. Error bars represent SEM.
To determine how muscle weakness develops in individual animals, we tested G93A and wild-type animals once a week beginning at 90 d of age and continued the tests until the G93A mice could no longer hold onto the wire. The results revealed several interesting aspects of muscle strength change. First, both the onset and the duration of muscle strength decline were highly variable among different individuals, ranging from age 113 to 188 d for the former and 35 to 94 d for the latter (Fig. 2B, Table1). Second, the time course of muscle strength change apparently went through four different stages (Fig.2C,D): a pre-muscle weakness stage (PMW), during which muscle strength was maintained at the normal level; a rapid declining stage (RD), during which the hanging time declined sharply (usually by >50% within a period of 2 weeks); a slow declining stage (SD), which followed the RD stage and lasted for 4–11 weeks; and a paralysis stage (Para), during which paralysis of limbs began and the mouse could no longer hold onto the wire (Fig. 2C,D). The sharp decline in muscle strength during the RD stage suggests that there may be a synchronized pathological change at the onset of the disease.
Table 1.
ALS onset and duration in the G93A mice
| Range | Average ± SD | n | |
|---|---|---|---|
| RD onset1-a | 113–188 | 151 ± 17 | 72 |
| Para onset1-b | 183–253 | 211 ± 18 | 23 |
| Duration1-c | 35–94 | 65 ± 19 | 23 |
The age (days) at which a sharp decline in hanging time was first detected.
The age (days) at which the mouse could no longer hold onto the wire.
The period between the last day that the mouse displayed a normal hanging time and the first day that the mouse could no longer hold onto the wire.
Pathological progression in motor axons at different stages
To elucidate the sequence of events leading to motor neuron death, and in particular to see whether there is a synchronized loss of motor neurons at the onset of the disease, we examined the motor axons in ventral roots (Fig. 3). We found few pathological changes in the PMW stage (Fig. 3C) compared with the WT (Fig. 3A) or WS (Fig. 3B) mice. Surprisingly, the sharp muscle strength decline during the RD stage was not correlated with a loss of a large number of motor axons (Fig.3D). Instead, for a long period during the SD stage (up to 50 d after the onset of RD), a significant fraction of motor axons remained (Fig. 3E–G). It was not until the paralysis stage that the vast majority of motor axons became degenerated (Fig.3H).
Fig. 3.
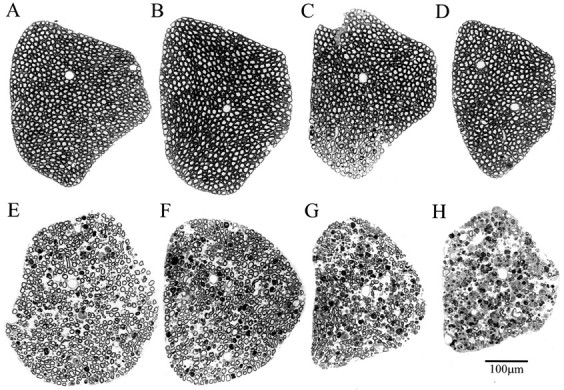
Axonal degeneration in L5 ventral roots at different stages of ALS in G93A mice. Plastic sections (1 μm) were stained with toluidine blue. A, WT; B, WS; (C–H), G93A at the following stages: (C) PMW, (D) RD, (E–G) 20, 33, and 50 d after the onset of RD, respectively; H, paralysis.
To quantify the changes in motor axons, we measured the axon size and number in the L4 and L5 ventral roots. As the disease developed, there was a progressive loss of large motor axons after the onset of muscle weakness (Fig. 4). Two features of this loss have emerged from this analysis. First, two processes, atrophy and degeneration, contributed to the loss of large axons during the early stages. This was particularly evident when the axons were divided into two groups, one larger and the other smaller than 4.5 μm in diameter. Beginning from the RD stage, there was a significant loss of large motor axons (Fig. 5). Meanwhile, the number of small axons was increased up to 20 d after the onset of muscle weakness, before it reverted back to the original number (Fig.5). These results indicate that large motor axons undergo atrophy before degeneration.
Fig. 4.

Axon size distribution in L4 and L5 ventral roots. The distribution of axons in the two roots was similar, and therefore the measurements were pooled together in one plot. Three animals were measured for each plot. The three PMW animals (C) were between 120 and 160 d old and therefore were near the RD stage. A–H, Same as in Figure 2.
Fig. 5.
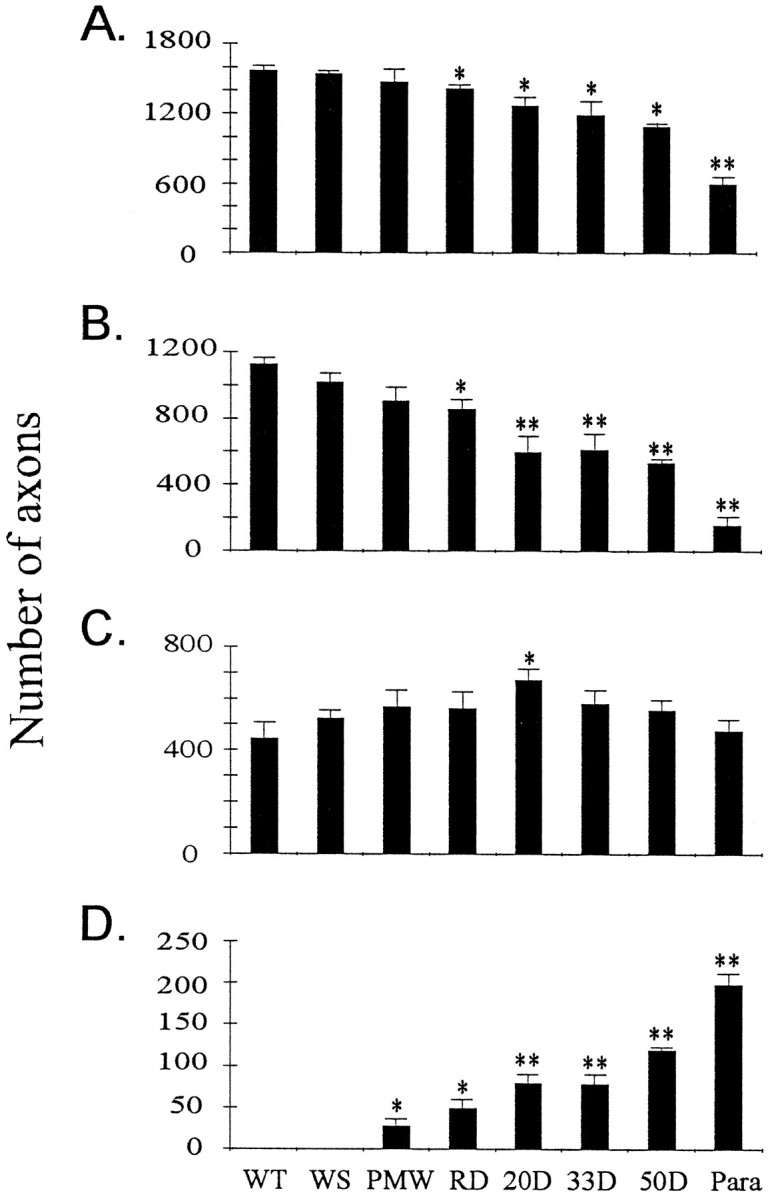
Motor axons undergo atrophy before degeneration.A, Total number of axons in L4 and L5 ventral roots at different disease stages. B, Number of axons larger than 4.5 μm in diameter at different disease stages. C, Number of axons smaller than 4.5 μm in diameter at different disease stages. D, Number of degenerating axons in L4 and L5 ventral roots at different disease stages. Asterisksindicate significance level in comparison with the WT using two-tailed Student’s t test. One asteriskrepresents p < 0.05; two asterisksrepresent p < 0.01. n = 3 for all stages.
Second, taking into account the substantial fraction of atrophic large axons, the actual fraction of degenerated large axons during the SD phase was relatively low (Table 2). As long as paralysis had not begun, even at 50 d after the onset of muscle weakness, the fraction of degenerated large axons was <50% (Figs. 3-5, Table 2), which implies that more than half of the large motor neurons were still alive. The largest single loss of motor axons occurred at the paralysis stage (Fig. 5, Table 2), when the majority of large motor neurons were lost permanently. These data indicate that the vast majority of motor neurons are still alive when muscle weakness develops, and the initial muscle weakness reflects motor neuron dysfunction rather than loss (see below). Thus, there is a period of time to rescue motor neurons after muscle weakness begins.
Table 2.
Axon number changes (%) in L4 and L5 ventral roots
| Stage | PMW | RD | 20 d | 33 d | 50 d | Para |
|---|---|---|---|---|---|---|
| Total axons | −6.2 | −9.8 | −19.3 | −24.1 | −30.6 | −61.9 |
| Large axons | −19.5 | −23.9 | −47.1 | −45.7 | −52.5 | −86.0 |
| Large → small | 10.9 | 10.3 | 20.2 | 12.1 | 9.9 | 2.8 |
| Degenerated large axons | −8.6 | −13.6 | −26.9 | −33.6 | −42.6 | −83.3 |
The percent reduction in total axon number and large axon number is calculated in reference to wild-type mice. The definition of large and small axons is described in the text (also see Fig. 5). The fraction of large axons that become small (large → small) is estimated by subtracting the small axon number in the wild-type mice from that of the G93A mice at different stages. The difference between the total lost large axons and the fraction of the large axons that become small (large → small) is the fraction of large axons that are degenerated.
Pathological progression in ventral horn motor neurons at different stages
To investigate the sequence of events in the vicinity of motor neuron cell bodies, we analyzed changes in the ventral horn spinal cord at different disease stages. Consistent with changes in motor axons, there were considerable numbers of neurons remaining in the ventral horn after muscle weakness began (Fig.6D–G). Quantification of neuronal number and size in the lateral ventral horn showed a selective loss of large neurons (Fig.7A). However, this was not evident until 33 d after the onset of muscle weakness (Fig.7A). At the paralysis stage, most of large motor neurons were lost, whereas the number of small neurons was unaffected (Figs.6H, 7A).
Fig. 6.
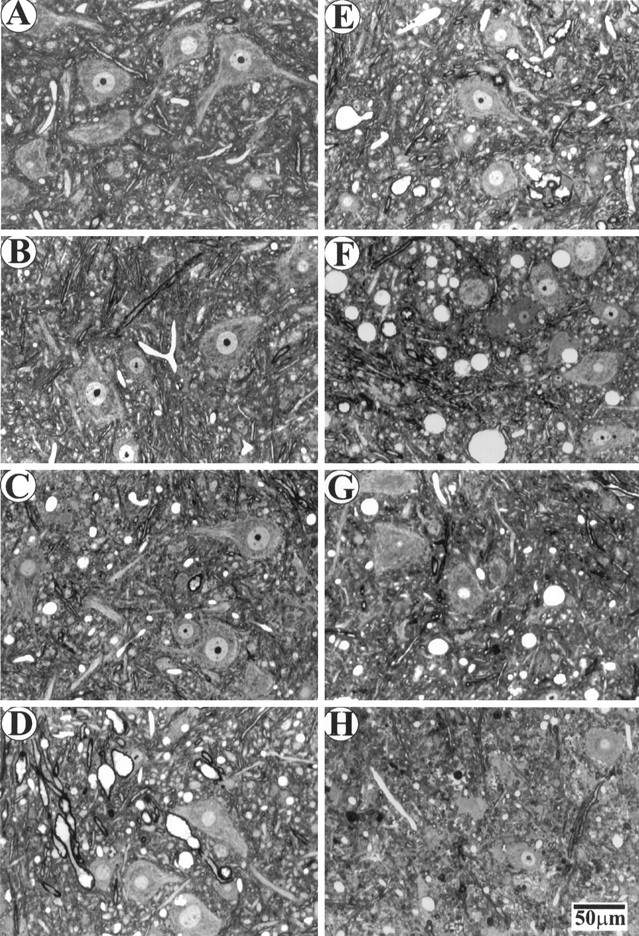
Changes in ventral horn at different disease stages. Plastic sections (1 μm) were stained with toluidine blue.A–H, Same as in Figure 2.
Fig. 7.

Changes in spinal cord at different disease stages. A, Changes in the number of neurons in the ventral horn. All numbers were averages from two animals and were normalized to the average number of neurons in each wild-type mouse (145 neurons). Filled bars, Total number of neurons;open bars, number of neurons with diameters <25 μm;shaded bars, number of neurons with diameters >25 μm. Note that the division of large and small neuronal groups at 25 μm diameter is arbitrary because there was not a clear division of large and small neuron peaks in the size distribution (data not shown). Changes in vacuole number (B) and vacuole size (C) at different disease stages. Two ventral horns were measured in each animal. The small number of vacuoles in the WT mice are capillary blood vessels that could not be distinguished from the real vacuoles under the light microscope.Asterisks indicate significance level in comparison with the WT, using two-tailed Student’s t test. One asterisk represents p < 0.05; two asterisks represent p < 0.01.n = 3 for all stages.
The most prominent change in the early stages of the disease was vacuolation. At the PMW stage, there was a slight increase of vacuoles (Figs. 6C, 7B) compared with WT (Figs.6A, 7B) and WS (Figs.6B, 7B) mice. However, vacuoles were greatly increased at the onset of the disease (RD stage) (Figs.6D, 7B). Although conspicuous throughout the SD stage, the number of vacuoles gradually declined toward the end stage of the disease (Figs. 6E–H, 7B). But the size of vacuoles increased slightly from the RD stage until 33 d after the onset of muscle weakness, and then declined as the disease progressed toward the paralysis stage (Fig. 7C). These results indicate that the massive vacuolation in motor neurons is transient in the disease process and suggest that small vacuoles may fuse or cluster before being lost with the motor neurons.
Mitochondrial damage precedes the onset of the disease
The light microscopic data described above showed only a minor increase in vacuoles during the PMW stage. To determine what change(s) triggers the onset of RD stage, we used electron microscopy (EM) to examine the spinal cords. The most prominent feature at the PMW stage was abnormal mitochondria, which were present in abundance in dendrites and axons but scarce in the cell bodies. Shown in Figure8 are several examples of mitochondrial changes, including those with dilated and disorganized cristae (Fig.8A,B), leakage of the outer membrane (Fig.8A, arrow), broken outer membrane (Fig.8C, arrows), and early vacuoles that still carry remnants of mitochondria (Fig. 8D,E, arrows). The abnormal mitochondria often appeared swollen compared with the normal ones in adjacent synaptic terminals (Fig. 8C,arrowheads). Densely accumulated neurofilaments can be seen in axons in close proximity with vacuoles (Fig. 8D) and also at many other proximal axon sites without vacuoles (data not shown), suggesting that slow axonal transport begins to fail before the onset of the RD stage. At the RD stage, abundant vacuoles in dendrites as well as in axons were seen, some of which were no longer carrying the remnants of mitochondria (Fig. 8F). Only rarely were vacuoles present in cell bodies.
Fig. 8.

Mitochondrial abnormalities at PMW (A–E) and RD (F) stages.A, A swollen dendritic mitochondrion with dilated cristae (asterisk) and leaking outer membrane (arrow). B, Swollen dendritic mitochondria with dilated and disorganized cristae. A synaptic terminal on the dendrite contains normal mitochondria (arrowhead). C, A proximal dendrite containing mitochondria with broken outer membranes (arrows). Adjacent synaptic terminals contain normal mitochondria (arrowheads). D, Early vacuoles in a proximal axon. Arrows point to mitochondrial remnants. E, Early vacuoles in a dendrite.Arrows point to mitochondrial remnants.F, Massive dendritic vacuolation at the RD stage.
DISCUSSION
Pathological observations without correlation with disease stages are largely a description of scattered phenomena and provide only limited information regarding the mechanism of disease progression. Because of difficulties involved in studying human cases, particularly because of the near impossibility of obtaining pathological specimens at the early stages of the disease, descriptions of end-stage disease have dominated the study of ALS since the original identification of the disease more than a century ago (Chou, 1992; Hirano, 1991). Transgenic mice expressing mutated SOD1 have provided an unprecedented opportunity to study the early stages of the disease. Indeed, several new pathological features including mitochondrial vacuolation and early astrogliosis were revealed (Dal Canto and Gurney, 1995; Wong et al., 1995; Morrison et al., 1996; Bruijn et al., 1997).
In all studies preceding the current work, pathological features were correlated with age of animals but not with stage of the disease. As shown in Figure 1, the onset and duration of disease in mice mirrors human disease (Cudkowicz et al., 1997; Juneja et al., 1997) and are highly heterogeneous. As a result, it was difficult to determine the sequence of events in the disease progression and the significance of the pathological features that were observed. To solve this problem, we used a novel approach by developing an assay to determine the disease stages and analyzing pathological changes in mice with defined disease stages. By this approach, a coherent picture of the functional and pathological progression of ALS in the G93A mice begins to emerge (Fig.9).
Fig. 9.

Sequence of pathological events leading to motor neuron death in G93A mice. All the data points were derived from the same set of animals. Each data point represents the average of three animals. All the values are normalized against the highest measure (100%) in the sequence. For the hanging test, only the last hanging times before the animals were killed are shown. Error bars have been shown in Figures 3 and 6 and are omitted here for clarity.
The progression of ALS in G93A mice can be divided into four stages based on changes in muscle strength, and each of these stages is correlated with specific pathological features. The PMW stage (just before the RD stage) shows little difference from the wild type under the light microscope. However, abundant mitochondrial abnormalities can be found by EM (Fig. 8). The onset of muscle weakness (RD stage) correlates with a massive mitochondrial vacuolation, signifying the beginning of a degenerative process in motor neurons that compromises their function (Fig. 9). The SD stage correlates with axonal atrophy and a gradual loss of motor neurons (Fig. 9). However, the majority of motor neurons are alive until the very end stage of the disease (Fig. 9, Table 2).
Several implications can be drawn from these results. First, abundant mitochondrial abnormalities are the most prominent pathological feature before the onset of muscle strength decline. In addition, the onset of a sharp muscle strength decline is correlated with a massive mitochondrial vacuolation. This is the first demonstration that mitochondrial degeneration is associated with the onset of the disease and suggests that the “gained toxicity” of the mutant SOD1 damages mitochondria, triggering the decline of motor neuron function and the onset of clinical disease. Second, the vacuolation is a transient process. It correlates with the onset of muscle weakness but decreases toward the end stage of the disease. It has been pointed out that vacuoles were not a noted feature in human ALS (Bruijn et al., 1997). Our results raise the possibility that vacuoles are also an integral part of human ALS, but they exist only at the very early stages of the disease. Therefore, the lack of vacuoles in human ALS could be attributed to the fact that the pathological examination is performed on patients at the terminal stage of the disease.
Third, the initial muscle weakness signifies a stage in which the degenerative process in large motor neurons begins to compromise their function. However, it does not represent a massive loss (or death) of these neurons. The loss of the majority of motor neurons does not occur until the paralysis stage, the end stage of the disease. The presence of the majority of motor axons during the RD and SD stages indicates that even after the muscle weakness begins, most motor neurons have not died and thus could be rescued by effective therapeutic intervention. Fourth, axonal atrophy has been noted in human ALS patients (Chou, 1992), but it was difficult to determine whether this decrease was caused by the death of large axons or a shift of large axons to small ones. Our results show that large axons become atrophic before degeneration (Fig. 4, Table 2). This suggests that there is a failure in axonal transport at the early stage of ALS, which is consistent with the proximal accumulation of neurofilaments observed during the PMW and RD stages in this study (Fig. 8), as well as in another study at unspecified disease stages (Tu et al., 1996).
Previous studies have noted the presence of vacuoles. However, the origin of the vacuoles has been controversial (Dal Canto and Gurney, 1995; Wong et al., 1995). One study proposes that vacuoles are derived from dilation of endoplasmic reticulum and mitochondria (Dal Canto and Gurney, 1995), whereas the other study contends that vacuoles are derived from degenerating mitochondria (Wong et al., 1995). By determining the precise time of the disease onset, the current study shows that abundant abnormal mitochondria, including those with dilated cristae and the early vacuoles with mitochondrial remnants, are present before the massive vacuolation at the disease onset. This observation, in conjunction with the absence of dilated endoplasmic reticulum before the massive vacuolation, strongly supports the contention that mitochondria are the source of vacuoles.
Thus, the current study suggests that mitochondrial damage is at the center of the degeneration mechanism. Whether such damage is caused by a direct effect of toxicity from the mutated SOD1 or is mediated through other intermediates remains to be determined. However, the involvement of mitochondrial degeneration in the early stage of the disease is consistent with a direct effect of mutant SOD1 toxicity and perhaps is mediated by properties gained by the mutant enzyme in catalyzing redox reactions (Beckman et al., 1993; Wiedau-Pazos et al., 1996; Yim et al., 1996). Mitochondria are a large source of oxidative free radicals, and the production of free radicals is directly correlated with metabolic activity (Beal, 1996). It is possible that the high metabolic activity in motor neurons, combined with the gained, toxic oxidative properties of the mutant SOD1, causes mitochondria in motor neurons to degenerate earlier than other neurons, triggering the onset of ALS. Consistent with this possibility are the results of a recent report showing that mitochondria in neuroblastoma cells transfected with G93A mutation displayed a significant loss of membrane potential and an increase in cytosolic calcium concentration (Carri et al., 1997).
The assay for disease progression developed in the current work sets the stage for further investigations of the disease mechanism in animal models. This assay enables the study of animals with defined disease stages, thus allowing meaningful comparisons of pathological evolution among different SOD1 mutations. By grouping mice according to their disease stages, inconsistencies caused by heterogeneous disease stages can be avoided, therefore allowing a full construction of the sequence of events (including both biochemical and morphological events) leading to motor neuron death. The assay also provides a useful means to evaluate the effectiveness of potential therapeutic agents.
Footnotes
This work is supported by grants from National Institutes of Health (RO1NS35750-01), the ALS Association, and the Markey Charitable Trust. We thank Vivian Tung for technical assistance and Alonzo Ross and Diane Casey for critically reading this manuscript.
Correspondence should be addressed to Zuoshang Xu, Worcester Foundation, 222 Maple Avenue, Shrewsbury, MA 01545.
REFERENCES
- 1.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 2.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxinitrate. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 3.Brown RH., Jr Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell. 1995;80:687–692. doi: 10.1016/0092-8674(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 4.Bruijn LI, Cleveland DW. Mechanisms of selective motor neuron death in ALS: insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol. 1996;22:373–387. doi: 10.1111/j.1365-2990.1996.tb00907.x. [DOI] [PubMed] [Google Scholar]
- 5.Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 6.Carri MT, Ferri A, Battistoni A, Famhy L, Cabbianelli R, Poccia F, Rotilio G. Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997;414:365–368. doi: 10.1016/s0014-5793(97)01051-x. [DOI] [PubMed] [Google Scholar]
- 7.Chou SM. Pathology-light microscopy of amyotrophic lateral sclerosis. In: Smith RA, editor. Handbook of amyotrophic lateral sclerosis. Marcel Dekker; New York: 1992. pp. 133–181. [Google Scholar]
- 8.Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 9.Dal Canto MC, Gurney ME. Neuropathological changes in two lines of mice carrying a transgene for mutant human Cu, Zn SOD, and in mice overexpressing wild type human SOD: a model of familial amyotrophic lateral sclerosis (FALS). Brain Res. 1995;676:25–40. doi: 10.1016/0006-8993(95)00063-v. [DOI] [PubMed] [Google Scholar]
- 10.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 11.Hirano A. Cytopathology of amyotrophic lateral sclerosis. Adv Neurol. 1991;56:91–101. [PubMed] [Google Scholar]
- 12.Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with glu100gly and ala4val mutations in Cu,Zn superoxide dismutase. Neurology. 1997;48:55–57. doi: 10.1212/wnl.48.1.55. [DOI] [PubMed] [Google Scholar]
- 13.Morrison BM, Gordan JW, Ripps ME, Morrison JH. Quantitative immunocytochemical analysis of the spinal cord in G86R superoxide dismutase transgenic mice: neurochemical correlates of selective vulnerability. J Comp Neurol. 1996;373:619–631. doi: 10.1002/(SICI)1096-9861(19960930)373:4<619::AID-CNE9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 14.Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mulder DW, Kurland LT, Offord KP, Beard CM. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986;36:511–517. doi: 10.1212/wnl.36.4.511. [DOI] [PubMed] [Google Scholar]
- 16.Munsat TL. Adult motor neuron disease. In: Rowland LP, editor. Merritt’s textbook of neurology. Lea & Febiger; Philadelphia: 1989. pp. 682–687. [Google Scholar]
- 17.Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu,Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 19.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VMY. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tu PH, Gurney ME, Julien J-P, Lee VMY, Trojanowski JQ. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab Invest. 1997;76:441–456. [PubMed] [Google Scholar]
- 21.Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, Lee MK, Valentine JS, Bredesen DE. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 22.Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z-S, Cork LC, Griffin JW, Cleveland DW. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73:23–33. doi: 10.1016/0092-8674(93)90157-l. [DOI] [PubMed] [Google Scholar]
- 24.Yim MB, Kang J-H, Yim H-S, Kwak H-S, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]