

Abstract
Rat superior cervical ganglion (SCG) neurons express low-threshold noninactivating M-type potassium channels (IK(M)), which can be inhibited by activation of M1 muscarinic receptors. This inhibition occurs via pertussis toxin-insensitive G-proteins belonging to the Gαq family (Caulfield et al., 1994). We have used DNA plasmids encoding antisense sequences against the 3′ untranslated regions of Gα subunits (antisense plasmids) to investigate the specific G-protein subunits involved in muscarinic inhibition of IK(M). These antisense plasmids specifically reduced levels of the target G-protein 48 hr after intranuclear injection. In cells depleted of Gαq, muscarinic inhibition of IK(M) was attenuated compared both with uninjected neurons and with neurons injected with an inappropriate GαoAantisense plasmid. In contrast, depletion of Gα11 protein did not alter IK(M) inhibition. To determine whether the α or βγ subunits of the G-protein mediated this inhibition, we have overexpressed the C terminus of β adrenergic receptor kinase 1 (βARK1), which binds free βγ subunits. βARK1 did not reduce muscarinic inhibition of IK(M) at a concentration of plasmid that can reduce βγ-mediated inhibition of calcium current (Delmas et al., 1998a). Also, expression of β1γ2 dimers did not alter the IK(M) density in SCG neurons. In contrast, IK(M) was virtually abolished in cells expressing GTPase-deficient, constitutively active forms of Gαq and Gα11. These data suggest that Gαq is the principal mediator of muscarinic IK(M) inhibition in rat SCG neurons and that this more likely results from an effect of the α subunit than the βγ subunits of the Gqheterotrimer.
Keywords: M-current, G-protein, antisense, muscarinic receptor, superior cervical ganglion neuron, β adrenergic receptor kinase
The M-type potassium current (IK(M)) is a noninactivating, voltage-gated potassium current found in various peripheral and central neurons, including rat superior cervical ganglion (SCG) neurons, and in some cell lines (for review, see Brown, 1988). It is activated in the subthreshold voltage range for action potentials and increases with membrane depolarization. Thus, cells remain clamped around rest, display spike adaptation, and have limited excitability. Inhibition of IK(M) results in depolarization with increased action potential discharge (Brown and Selyanko, 1985) and provides a switch between phasic and tonic firing properties (Wang and McKinnon, 1995). IK(M) in rat SCG neurons can be inhibited after activation of various receptors, including M1 muscarinic receptors [M1 mAChR (Marrion et al., 1989; Bernheim et al., 1992)] and bradykinin B2 receptors (Jones et al., 1995), coupled through Bordetella pertussis toxin-insensitive GTP-binding proteins (G-proteins) (Brown et al., 1989; Caulfield et al., 1994;Jones et al., 1995).
Using antibodies raised against the C-terminal domain of different Gα subunits, we have previously obtained evidence to suggest that the G-protein α subunits involved in M1 mAChR-mediated inhibition of IK(M) in rat SCG neurons include Gαq or Gα11 or both (Caulfield et al., 1994). However, the antibodies that were used could not distinguish between Gq and G11 because they have identical C-terminal sequences (Strathmann and Simon, 1990). Because the C terminus is thought to be a locus of G-protein GDP-bound α subunit/receptor and GTP-bound α subunit/phospholipase C-β1 (PLC-β1) interactions (Conklin and Bourne, 1993; Conklin et al., 1993; Arkinstall et al., 1995), Gαq and Gα11 can couple to the same receptors (Aragay et al., 1992; Wu et al., 1992b; Nakamura et al., 1995; Dippel et al., 1996), and the cloned subunits stimulate the different PLC-β isoforms to a similar degree (Taylor et al., 1991; Hepler et al., 1993; Jhon et al., 1993). However, they are not invariably equivalent, because in rat portal vein myocytes, Gαq and Gα11 elevate intracellular calcium levels after α1-adrenoceptor activation by coupling to very different mechanisms (Macrez-Leprêtre et al., 1997).
In the present experiments, we have therefore tried to find out whether either or both of these two G-proteins (Gq and G11) were involved in muscarinic inhibition of IK(M) in rat SCG neurons by using Gα antisense-generating plasmids to deplete cells of specific subunits. We have also sought evidence to determine whether the α subunit or the βγ dimer of the activated dissociated heterotrimer acted as the primary intermediary (Wickman and Clapham, 1995; Clapham and Neer, 1997) by selectively overexpressing βγ subunits or GTPase-deficient forms of the α subunits and by testing whether a βγ-sequestering agent [C-terminal peptide of β adrenergic receptor kinase 1 (βARK1)] modified the effect of mAChR stimulation.
Our results suggest that Gαq, but not Gα11, couples the M1 mAChR to IK(M)inhibition in SCG neurons and that α, rather than βγ, subunits are the mediators of this response.
MATERIALS AND METHODS
Cell culture. Sympathetic neurons were isolated from SCG of 15- to 19-d-old Sprague Dawley rats and cultured using standard procedures as described previously (Delmas et al., 1998a).
DNA plasmids. The constructs used in this study were made by PCR-cloning using standard molecular techniques (Abogadie et al., 1997). These were designed antisense to sequences in the 3′ untranslated (3′UT) regions of the rat target genes and subcloned into pCR3 or pCR3.1 (Invitrogen, San Diego, CA) unless stated otherwise. The cloned 3′UT sequences share no significant homology with any other rat G-protein α subunits. The nucleotide sequences reported in this paper have been submitted to the GenBank/EMBL Data Bank with accession numbers Y17161, Y17162, Y17163, and Y17164. The clones are as follows, in 5′ to 3′ orientation [nucleotide (nt); coding region (CR); numbers indicate position relative to stop or start codon]: GαoA(clone 207–8) 3′UT nt 2–169: CTCTTGTCCTGTATAGCAACCTATTTGACTGCTTCATGGACTCTTTGCTGTTGATGTTGATCTCCTGGTAGCATGACCTTTGGCCTTTGTAAGACACACAGCCTTTCTGTACCAAGCCCCTGTCTAACCTACGACCCCAGAGTGACTGACGGCTGTGTATTTCTGTA; Gαq/11 common (clone 107–6 in pBK-CMV, Stratagene, La Jolla, CA) CR nt 484–741: ATGACTTGGACCGTGTAGCCGACCCTTCCTATCTGCCTACACAACAAGATGTGCTTAGAGTTCGAGTCCCCACCACAGGGATCATTGAGTACCCCTTCGACTTACAGAGTGTCATCTTCAGAATGGTCGATGTAGGAGGCCAAAGGTCAGAGAGAAGAAAATGGATACACTGCTTTGAAAACGTCACCTCGATCATGTTTCTGGTAGCGCTTAGCGAATACGATCAAGTTCTTGTGGAGTCAGACAATGAGAACCGCA; Gα11 antisense clones: 243–7, 3′UT nt 4–104; C97–4, 3′UT nt 82–123. Gαq antisense clones: C23–24, 3′UT nt 6–289; C6–6, 3′UT nt 6–129; C23-D7, 3′UT nt 193–289; C23–16, 3′UT nt 29–129. Targeted sequences are shown in Figure1.
Fig. 1.

DNA Sequences of Gαq and Gα11 3′ untranslated regions. Sequences of rat Gαq and Gα11 in the 3′ untranslated region immediately after the stop codon. Homology between the two proteins is very low in this region, with only 19% identity, although this rises to 31% when the two sequences are aligned for maximum homology. Theunderlined areas represent the sequences targeted by the Gα11 antisense plasmids; the closed arrowheads correspond to clone 243–7 and the open arrowheads to clone C97–4.
The constitutively active, GTPase-deficient form of hamster Gαq (Q209L) (Wu et al., 1992a) was subcloned into the pCMV5 vector, the GTPase-deficient Gα11 (Q209L, also known as 11QL) (Wu et al., 1992a; from S. Offermanns) was provided in the pCIS vector, and the GTPase-deficient GαoA (Q205L) (Wong et al., 1992; from B. R. Conklin) was provided in the pCDNA1 vector. Bovine β1 and γ2 subunits were subcloned into pCDNA3 (Invitrogen). The C-terminal Gly495 to Leu689 of human βARK1 (also called GRK2) (Koch et al., 1994; from C. Scorer and C. Harris) was supplied in the vector pCIN1 engineered with newNotI and EcoRI sites. All plasmids were propagated in either XL-1 blue or DH5α Escherichia coliand purified using maxiprep columns (Qiagen, Hilden, Germany). All clones were verified by sequencing. RNA synthesis was driven by a strong viral promoter (cytomegalovirus) to ensure sustained high intracellular levels of transcripts after delivery of plasmids.
In situ hybridization. In situ hybridization was performed on 12 μm cryostat sections of 17-d-old rat SCGs using digoxigenin-labeled riboprobes, essentially as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993). Sense and antisense cRNAs were transcribed from the same clones used in the electrophysiological experiments using SP6 and T7 polymerase according to standard protocols.
Microinjection. DNA plasmids were diluted to 400 μg/ml in calcium- and glucose-free Krebs’ solution (290 mOsm/l, pH 7.3) containing 0.5% FITC-dextran and pressure-injected into the nucleus of SCG neurons 2 d in culture, either as described previously (Abogadie et al., 1997) or with a microinjector (Eppendorf, Hamburg, Germany). Cells were maintained in culture for an additional 2 d, and a survival rate of 75–85% was obtained.
Electrophysiology. M-currents were measured from SCG neurons cultured for 4 d, using the amphotericin-B perforated-patch technique (Horn and Marty, 1988; Rae et al., 1991). Patch electrodes (2–4 MΩ) were filled by dipping the tip for 40 sec into a filtered internal solution containing (in mm): potassium acetate 80, KCl 30, HEPES 40, MgCl2 3 (adjusted to pH 7.3–7.4 with KOH and to 280 mOsm/l with potassium acetate). The pipette was then back-filled with the above solution containing 0.1 mg/ml amphotericin-B. High-resistance seals (>2 GΩ) were initially achieved, and after amphotericin-B permeabilization, access resistances were <25 MΩ. SCG neurons were perfused at 5–10 ml/min at 32°C with an external solution consisting of (in mm): NaCl 120, KCl 3, HEPES 5, NaHCO3 23, glucose 11, MgCl21.2, CaCl2 2.5, tetrodotoxin (TTX) 0.0005, pH 7.4. Cells were voltage-clamped at approximately −25 mV using either an Axopatch 200A amplifier (data sampling rate 4–10 kHz, filter 1 kHz) or a switching amplifier (Axoclamp-2A, switching frequencies 3–5 kHz, filter 0.1 kHz), both from Axon instruments (Foster City, CA). IK(M) was measured as a slowly developing inward deactivation relaxation after a 1 sec jump to a command potential of approximately −55 mV (Caulfield et. al., 1994). Inhibition was measured as the fractional reduction in the amplitude of the IK(M) deactivation relaxation in response to cumulative increases in concentrations of oxotremorine methiodide (Oxo-M) (Research Biochemicals International, Natick, MA) (see Fig. 4). Steady-state current–voltage relationships were obtained by applying slow (3.3 mV/sec) voltage ramps from −20 mV to −100 mV. For experiments with Bordetella pertussis toxin (PTX) (Speywood, Maidenhead, Berkshire, UK), SCG neurons were incubated with 1 μg/ml PTX in the culture medium for at least 24 hr before recording.
Fig. 4.

Time course of cumulative Oxo-M application; effect on IK(M) amplitude. A, IK(M) deactivation relaxation elicited by a −30 mV step for 1 sec from a holding potential of approximately −25 mV. Waveforms (average of 3 traces) are from cells injected with Gα11(C97–4) or Gαq (C23–16) antisense plasmids and whose time courses are shown in B. IK(M)relaxations are shown in the absence and presence of 1 μmand 10 μm Oxo-M. Dotted lines represent 0 pA. B, Time course of normalized IK(M)amplitude during application of increasing concentrations of Oxo-M, as indicated, for neurons injected with GαoA, Gαq, and Gα11 antisense plasmids. IK(M) was recorded every 10 sec, and each Oxo-M concentration was applied for 1 min.
Data were collected and analyzed using PClamp6 software (Axon Instruments) and expressed as mean ± SEM. An estimate of the mean log IC50 for each antisense plasmid treatment was obtained by fitting the data from each individual cell with a best-fit dose–response curve and determining the log IC50 for each cell. IK(M) deactivation relaxations were best-fit by a double exponential with fast (τ1) and slow (τ2) components. Statistics used the two-way ANOVA comparing plasmid treatments across four agonist concentrations for all antisense samples (including uninjected groups). If a significant effect of plasmid treatment was found overall, further analysis was performed using the two-way ANOVA to determine which treatments contributed to this significance. The constitutively active GαoA*, Gαq*, Gα11*, and β1γ2 expression data were analyzed with one-way ANOVA, as was the log IC50 data, and if an overall significant effect of plasmid treatment was found, this was followed by Bonferroni’s multiple comparison test. p values < 0.05 were considered significant.
Immunocytochemistry. SCG cells, cultured and injected as described above, were fixed in acetone and stained for GαoA+B, Gαq, Gα11, and C terminus of βARK1 using selective antibodies and the alkaline phosphatase substrate 5-bromo-4-chloro-3-indoxyl phosphate and nitro blue tetrazolium chloride (BCIP/NBT) (Dako, Carpinteria, CA), as described by Abogadie et al., (1997). The polyclonal antibodies anti-GαoA+B(sc-387), anti-Gα11 (sc-394), and anti-βARK1 C terminus (sc-562) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and the anti-Gβ antibody (3B-200) was from Gramsch Laboratories (Schwabhausen, Germany). The specific polyclonal antibody anti-Gαq (IQB2) was raised against a synthetic peptide fragment of Gαq (Milligan et al., 1993). Specificity of the antibodies was determined by competing out the staining by preabsorbing the antibody with the relevant immunogenic peptide. All dishes of SCG neurons recorded in the electrophysiology experiments were subsequently fixed and stained. The BCIP/NBT purple/blue product was too dark to quantitate photometrically, so we assessed whether there was an overall qualitative reduction in staining by comparing each injected cell with its nearest uninjected neighbor and determining (by eye) whether the level of staining was equal to or less than that of the uninjected cell. Using this method we have therefore estimated the proportion of cells with a visible reduction in staining (regardless of the magnitude of this reduction) 48 hr after injection of the antisense plasmid.
RESULTS
Gαq and Gα11 expression in SCG neurons
Both in situ hybridization and RT-PCR clearly showed the presence of Gαq and Gα11 mRNAs in rat SCG tissue where they were expressed mainly in neurons (Fig.2). The specificity of the hybridization probes was confirmed when no signal was seen after competition with unlabeled probes (data not shown) or after use of sense, rather than antisense, probes (Fig. 2). Staining with specific antibodies against Gαq and Gα11 demonstrated the presence of Gαq and Gα11 protein in most cultured SCG neurons (see below). We have constructed plasmids encoding for RNA antisense (antisense plasmids) directed against the 3′ untranslated region of these G-proteins to specifically deplete cells of each of the α-subunits. Gαq and Gα11 share 81% homology in coding region sequence (based on mouse sequence), and this drops to a maximum of 31% (when rat sequences are aligned for maximum homology) in the first 200 bases of the 3′ untranslated region in rat (Fig. 1).
Fig. 2.
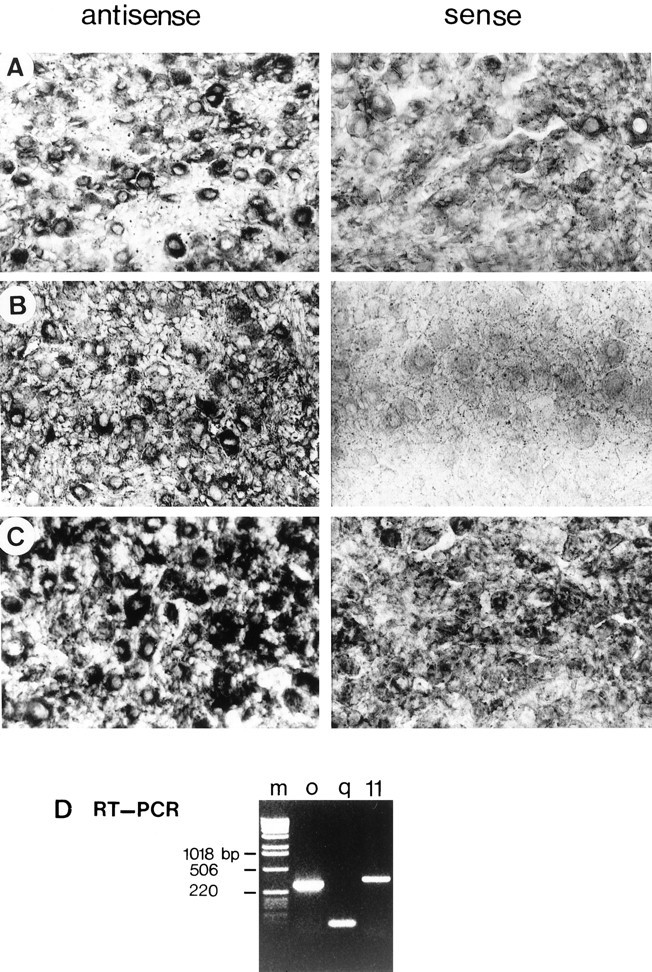
In situ hybridization and RT-PCR demonstrate the presence of Gαq and Gα11mRNA in rat SCG. In situ hybridization (ISH) and RT-PCR demonstrate the presence of Gαq and Gα11 in rat SCG. ISH of Gαq (B) and Gα11 (C) shows neuronal staining. GαoA ISH (A) was used as a positive control. All probes used were against the 3′ untranslated region (3′ UTR) of the gene. The black arrows indicate representative individual SCG neurons expressing the relevant mRNA.D, RT-PCR using rat SCG DNA as a template.m, Marker lane; o, Gαo with primers 266s/849a; q, Gαq with primers Gαq u6s/u111a, where “u” denotes sequence in the 3′ UTR; 11, Gα11 with primers Gα11 488s/u103a; bp, base pair.
Direct intranuclear injection of SCG neurons with various antisense plasmids (Gαq, Gα11, and GαoA) resulted in a marked reduction in the respective Gα subunit staining 48–72 hr later (Fig.3). This protein depletion was specific, with GαoA antisense not altering Gαq or Gα11 staining, Gαq/11 common antisense not touching GαoA+B staining, Gαq not altering GαoA+B or Gα11 staining, and Gα11 antisense leaving GαoA+B and Gαq staining intact. The two Gα11 antisense plasmids, however, were not equally effective. Thus, clone C97–4 reduced visible Gα11 protein staining in 9 of 19 cells (47%; n = 7 dishes of cells) (see Materials and Methods) (Fig. 3C), whereas 243–7 reduced Gα11 staining in only 18 of 63 cells (29%;n = 17 dishes). Similarly, the specific Gαq antisense plasmids were not all effective. C6–6 and C23–24 reduced staining in 23 of 71 cells (32%; n = 8 dishes) and 8 of 32 cells (25%; n = 6 dishes), respectively (Fig. 3B), whereas C23-D7 and C23–16 were more effective, reducing staining in 31 of 65 cells (48%; n= 10 dishes) and 55 of 109 cells (50%; n = 21 dishes), respectively (Fig. 3B). The antisense plasmids were maximally effective in reducing Gα subunit staining 2 d after injection, and their effects on IK(M) modulation were therefore assessed 2 d after injection.
Fig. 3.

Reduction of G-protein α subunit staining in cells expressing antisense. Complementary fluorescence and Gα immunostaining photographs of cells intranuclearly injected with antisense plasmids and a fluorescent marker. A, Cells immunostained with GαoA+B antibody and injected with (i) GαoA antisense plasmid and (ii) Gαq antisense plasmid (clone C23-D7).B, Cells immunostained with Gαq antibody and neurons injected with (i) Gαqantisense plasmid (C23-D7), (ii) GαoAantisense plasmid, (iii) Gαq antisense plasmid (C23–24), and (iv) Gα11 antisense plasmid (C97–4). C, Cells immunostained with Gα11 antibody and neurons injected with (i) Gα11 antisense plasmid (C97–4) and (ii) Gαq antisense plasmid (C23-D7).
Effect of antisense plasmids on IK(M) modulation by a muscarinic agonist
Injection of DNA plasmids encoding antisense to GαoAslightly reduced inhibition of IK(M) by the muscarinic agonist Oxo-M. Thus, the inhibition of IK(M) by 300 nm Oxo-M was 24.6 ± 5.0% (n = 6) in GαoA antisense-treated cells compared with 34.2 ± 2.6% (n = 7) in uninjected cells (p = 0.007 across all Oxo-M concentrations) (see Fig. 5B). To investigate further this effect of GαoA antisense, we pretreated several dishes of SCG neurons with 1 μg/ml PTX, which ADP-ribosylates and inactivates members of the Gαo/i G-protein family. There was no significant difference in Oxo-M inhibition of IK(M) between the treated and untreated cells (see Fig. 5D), confirming previous findings with other mAChR agonists (Brown et al., 1989). A comparable treatment strongly attenuated the Go-mediated inhibition of the Ca2+ current by noradrenaline (Caulfield et al., 1994) and Oxo-M (Delmas et al., 1998b). Furthermore, overexpression of a constitutively active, GTPase-deficient form of GαoA (Wong et al., 1992) did not alter IK(M)density (see Fig. 8 and below). It seems unlikely, therefore, that the GαoA antisense-induced reduction in IK(M)inhibition results directly from the loss of GαoA or that GαoA participates in IK(M) inhibition, a conclusion supported by previous studies using specific antibodies (Caulfield et al., 1994). This reduction is also unlikely to be caused by the plasmid injection per se, because cells injected with antisense constructs that were ineffective in reducing protein had no effect on the Oxo-M dose–response curves (see Fig. 5B,C and below). Hence, we do not yet understand why the GαoA antisense plasmid reduced IK(M) inhibition. Nevertheless, because the most suitable control group for comparison with the Gαqand Gα11 antisense plasmids is the expression of an inappropriate antisense, we have taken the effect of the GαoA antisense as our baseline for assessing the effect of the Gαq and Gα11 antisense plasmids, because at the very least this would mitigate against any “nonspecific” effects of antisense plasmid injection. Thus, allp values quoted are compared against GαoAantisense-expressing neurons unless stated otherwise (in practice, the same outcome of the experiments below would be obtained if the comparison were with uninjected cells).
Fig. 5.

Dose–response curves for Oxo-M inhibition of IK(M) in antisense plasmid-injected SCG neurons.A, Schematic diagram demonstrating length [in base pairs (bp)] and relative positions of the four antisense sequences targeted at the 3′ untranslated region of rat Gαq. Only C23-D7 and C23–16 consistently reduced Gαq protein levels in immunocytochemical staining with a Gαqantibody. B, Dose–response curves (mean ± SEM, plus best-fit curve) for uninjected neurons compared with GαoA antisense, Gα11 antisense, and Gαq/11 antisense plasmid-injected cells. The dose–response curve for GαoA (n = 6) antisense is significantly different from the uninjected dose–response curve (n = 6; p = 0.007) but not the Gα11 antisense plasmid curve (n = 9). The dose–response curve for cells injected with the Gαq/11 common antisense plasmid (n = 6) is significantly different from those of cells injected with GαoA and Gα11 antisense plasmids (p = 0.005 andp < 0.0001, respectively). C, Oxo-M dose–response curves for neurons injected with Gα11 antisense plasmid and four different antisense plasmids against Gαq. The dose–response curves for C6–6 (n = 8) and C23–24 (n = 5) are not significantly different from the dose–response curves for GαoA antisense-expressing (B) or Gα11 antisense-expressing neurons. C23-D7 (n = 6) and C23–16 (n = 9) dose–response curves are both significantly different from GαoA antisense-expressing neurons (p = 0.004 and p= 0.001, respectively) and from Gα11 antisense-expressing neurons (p < 0.0001 andp < 0.0001, respectively). D, Oxo-M inhibition of IK(M) is not altered by pretreatment with 1 μg/ml PTX (n = 6; n = 4 for untreated neurons).
Fig. 8.

Expression of GTPase-deficient forms of Gαq and Gα11 tonically inhibits IK(M), whereas β1γ2dimers have no effect. A, Representative waveforms from cells in C. Neurons expressing constitutively active Gαq* or Gα11* have very little holding current at −20 mV and no IK(M) deactivation relaxation in response to a −30 mV voltage step (bottom trace). IK(M) is normal in cells expressing β1γ2 dimers compared with injected neurons (e.g., GαoA antisense-expressing cells). Waveforms are the average of three traces, and the dotted linerepresents 0 pA. B, Current–voltage curves in response to a voltage ramp from −20 to −100 mV at 3.3 mV/sec (displayed ininsert) in GαoA antisense-expressing cells and neurons expressing Gαq* or Gα11*.I–V plot is in reverse direction from the ramp applied, and the dotted line represents 0 pA. These traces have not been leak-subtracted; leak current in the Gαq* and Gα11* cells is less than in the GαoA antisense-expressing neurons. C, Scatter-plot of IK(M) densities in GαoAantisense-expressing cells and cells expressing GαoA*, Gαq*, Gα11*, and β1γ2dimers. Horizontal lines represent means of each group. Gαq* (n = 13) and Gα11*(n = 8) are significantly different from GαoA antisense-expressing neurons (n= 7) (p < 0.001 and p< 0.001, respectively), GαoA*-expressing neurons (n = 6) (p < 0.001 andp < 0.001, respectively), and β1γ2 dimer-expressing neurons (n = 6) (p < 0.01 andp < 0.01, respectively).
Injection of SCG neurons with the Gαq/11 common antisense plasmid significantly reduced Oxo-M inhibition of IK(M)when compared with GαoA antisense-expressing cells (GαoA antisense: 24.6 ± 5.0% inhibition with 300 nm Oxo-M, n = 6; Gαq/11antisense: 13.9 ± 2.2%, n = 6; p= 0.005 across all Oxo-M concentrations) (see Fig. 5B). This confirms previous observations, using functionally inactivating antibodies, that either Gαq or Gα11 or both mediate muscarinic inhibition of IK(M) (Caulfield et al., 1994). To determine which (or whether both) of these G-protein α subunits is responsible for mediating this response, cells were injected with antisense plasmids that specifically reduced Gαq and Gα11 levels (see above). Four plasmids encoding different Gαq antisense sequences were investigated (see Fig. 5A). Of these, two significantly reduced muscarinic inhibition of IK(M) (C23-D7 and C23–16) (percentage inhibition with 300 nm Oxo-M: C23-D7: 15.5 ± 4.7%, n = 6, p = 0.004 compared with GαoA dose–response curve; C23–16: 15.7 ± 3.7%, n = 9, p = 0.001) (Figs.4,5C). This is in agreement with the immunocytochemical data where the clones C23-D7 and C23–16 selectively reduced immunocytochemical staining of Gαq. The other two clones, C6–6 and C23–24, however, were less effective at reducing either Gαq staining or muscarinic inhibition of IK(M) (percentage inhibition of IK(M) with 300 nm Oxo-M: C6–6: 24.3 ± 5.0%, n= 8; C23–24: 24.3 ± 3.0%, n = 5) (Fig.5C). The attenuation of IK(M) inhibition by the Gαq antisense plasmids C23-D7 and C23–16 was reflected in an increase in the log IC50 for these groups. Although the log IC50 values for neurons injected with C6–6 (−6.14 ± 0.09; n = 8) and C23–24 (−6.07 ± 0.11; n = 5) were close to that for uninjected neurons (−6.31 ± 0.06; n = 6), those for SCG neurons injected with the Gαq antisense plasmids C23-D7 (−5.64 ± 0.23; n = 6) and C23–16 (−5.16 ± 0.37; n = 7; p < 0.05 compared with Gα11 antisense-expressing neurons) were greater (Fig.6). Gαq depletion did not significantly change the maximum response or Hill slope. In contrast with the Gαq antisense plasmids, IK(M)inhibition in cells depleted of Gα11 with C97–4 antisense plasmid (29.7 ± 4.1% inhibition at 300 nmOxo-M; log IC50 = −6.13 ± 0.09; n = 9) was no different from that seen in GαoAantisense-expressing neurons (Figs. 4, 5B, 6). In agreement with the time course of Gα subunit depletion, the reduction of Oxo-M inhibition was maximal at 48 hr for the concentration of the plasmid injected (400 μg/ml), because no further reduction was seen 72 hr after injection [e.g., at 72 hr, 300 nm Oxo-M produced 17.4 ± 2.7% (n = 9) inhibition of IK(M) in Gαq antisense (C23–16)-expressing cells)]. The resting membrane potential was not altered in neurons injected with the Gαq or Gα11 antisenses compared with GαoA antisense constructs (e.g., GαoA antisense: −63.5 ± 2.6 mV, n= 6; Gαq antisense, C23-D7: −63.8 ± 1.1 mV,n = 5; Gα11 antisense, C97–4: −62.0 ± 4.0 mV, n = 4).
Fig. 6.

Gαq antisense plasmids increase the log IC50 for Oxo-M inhibition of IK(M). Scatter-plot is shown of the log IC50for each neuron included in the mean dose–response curves in Figure 5. Log IC50 was calculated from the best-fit curve for Oxo-M inhibition of IK(M) for every neuron recorded with the injected antisense sequences indicated. Horizontal linesrepresent the mean of each group. The Gαq antisense plasmid C23–16 significantly increased the log IC50compared with uninjected neurons (p < 0.01), Gα11 antisense plasmid injected cells (p < 0.05), and cells injected with the Gαq antisense plasmid C6–6 (p< 0.05).
Expression of the C-terminal βARK1 peptide in SCG neurons
The above results suggest that Gαq is primarily responsible for M1 mAChR-induced inhibition of IK(M), but they do not indicate which subunit(s) of the heterotrimer mediates the inhibition. To determine the role of endogenous Gαq-linked βγ dimers, we overexpressed the C-terminal domain of βARK1, which has been shown to sequester free βγ subunits (Koch et al., 1994). Expression of the peptide was routinely detected 24–48 hr after injection as a strong increase in βARK1 peptide immunoreactivity. Injection of the βARK1 construct at 200 μg/ml, a concentration that has been found to be effective at attenuating noradrenergic inhibition of the calcium current (Delmas et al., 1998a), a presumed βγ-mediated pathway (Herlitze et al., 1996;Ikeda, 1996), did not alter inhibition of IK(M) (300 nm Oxo-M produced 21.8 ± 3.2% inhibition in βARK1-expressing cells; n = 8) (Fig.7). Increasing the plasmid concentration to 400 μg/ml, however, resulted in a reduction of M1 mAChR inhibition of IK(M) (300 nm Oxo-M resulted in 17.7 ± 2.4% inhibition, n = 7, p = 0.0002, compared with GαoA antisense plasmid cells, across all concentrations of Oxo-M) (Fig. 7). This effect was not a result of a use-dependent sequestration of βγ subunits by βARK1 peptide, because repetitive application of 1 μm Oxo-M did not result in an accumulated loss of inhibition in neurons injected with 400 μg/ml βARK1-encoding plasmid (first application, 40.5 ± 4.0% inhibition; fourth application, 37.7 ± 4.1%;n = 3). Furthermore, neither IK(M) current density (GαoA antisense: 2.8 ± 0.4 pA/pF,n = 7; 400 μg/ml βARK1: 4.7 ± 1.1 pA/pF,n = 7) nor IK(M) deactivation relaxation (GαoA antisense: τ1 40.3 ± 2.3 msec, τ2 263 ± 21 msec, n = 9; 400 μg/ml βARK1: τ1 37.2 ± 2.2 msec, τ2 248 ± 30 msec, n = 10) was significantly altered by the βARK1-encoding plasmid.
Fig. 7.

βARK1-injected cells show some attenuation of IK(M) inhibition by Oxo-M. Dose–response curves for Oxo-M inhibition of IK(M) in cells injected with either GαoA antisense plasmid (n = 6) or C-terminal βARK1 plasmid at 200 μg/ml (n = 8) or 400 μg/ml (n = 7) are shown. Only the βARK1 400 μg/ml dose–response curve is significantly different from the GαoA antisense dose–response curve (p = 0.0002).
Expression of GTPase-deficient forms of Gαq and Gα11 subunits, but not β1γ2dimers, inhibits IK(M)
The above experiments with βARK1 peptide expression suggest that α, rather than βγ, subunits mediate M1 mAChR inhibition of IK(M). To test this further, we overexpressed GTPase-deficient, constitutively active forms of GαoA(GαoA*, Q205L), Gαq(Gαq*, Q209L), Gα11(Gα11*, Q209L), and β1γ2 dimers. Overexpression of Gαq* and Gα11* resulted in a dramatic decrease in IK(M) current density 24–48 hr after injection, compared with cells injected with GαoAantisense plasmid or GαoA* (Fig.8A,C) (GαoA antisense: 2.8 ± 0.4 pA/pF, n = 7; GαoA*: 4.0 ± 0.6 pA/pF, n = 6; Gαq*: 0.2 ± 0.02 pA/pF, n = 13,p < 0.001, compared with either GαoAantisense or GαoA*; Gα11*: 0.1 ± 0.02 pA/pF, n = 8, p < 0.001, compared with either GαoA antisense or GαoA*). This loss of IK(M) was also clear in the steady-state current–voltage relationships by the absence of outward rectification positive to −60 mV in Gαq*- and Gα11*-expressing cells (Fig. 8B). Consistent with the suppression of IK(M), the resting membrane potential of these cells was more depolarized than in cells injected with GαoA antisense and GαoA* plasmids (GαoA antisense: −63.5 ± 2.6 mV, n = 6; GαoA*: −62.7 ± 0.7 mV, n = 6; Gαq*: −48.4 ± 1.7 mV, n = 13, p < 0.001, compared with either GαoA antisense or GαoA*; Gα11*: −50.3 ± 2.7 mV, n = 8,p < 0.01, compared with GαoA antisense or GαoA*). In contrast, overexpression of free βγ subunits, by coexpressing β1 and γ2subunits, had no significant effect on either IK(M) current density (2.03 ± 0.4 pA/pF, n = 6;p > 0.05 compared with GαoA antisense) (Fig. 8C) or resting membrane potential (−59.2 ± 1.8,n = 6; p > 0.05, compared with GαoA antisense).
DISCUSSION
Our data clearly demonstrate that direct intranuclear injection of antisense-generating plasmids is an effective method for reducing levels of G-protein subunits in neurons (Fig. 3). These antisense sequences, designed against the 3′ UTR for increased specificity, are thought to bind to their target regions and destabilize the whole mRNA (Phillips and Gyurko, 1997), resulting in a reduced level of expression of the target protein. It is interesting to note that not all Gα 3′ UTR antisense sequences were effective in reducing protein expression. Of four Gαq antisense sequences tested, only two were effective (C23-D7 and C23–16). Similarly, of two Gα11antisense sequences tested, only one (C97–4) consistently reduced Gα11 protein levels. This difference in effectiveness of the antisense sequences could perhaps arise from some unknown secondary structure in the target mRNA transcript (Phillips and Gyurko, 1997). Such structures may determine how accessible the target region is for binding by antisense transcripts. The two less effective Gαq antisenses include a short region between nt 6 and 28 in the 3′ UTR not covered by the other antisense sequences (Fig. 5). It is therefore tempting to speculate that this particular region or sequence might not be amenable to antisense action.
Using these specific antisense plasmids, we have shown that depletion of Gαq, but not Gα11, subunits significantly reduced muscarinic inhibition of IK(M) (Figs. 4, 5). Indeed, although both in situ hybridization and immunocytochemical experiments clearly demonstrated the presence of Gα11 in SCG neurons (Figs.2, 3), muscarinic inhibition of IK(M) was not altered in cells specifically depleted of Gα11 by the antisense plasmid (Figs. 3-6). This suggests that Gαq rather than Gα11 preferentially mediates M1 mAChR inhibition of IK(M). This idea is supported by the finding that the common Gαq/11 antisense plasmid produced no greater reduction in IK(M) inhibition than the specific Gαq antisense plasmids. Although Gαqantisense plasmids clearly shifted the dose–response curve for Oxo-M inhibition of IK(M), they did not completely prevent inhibition by this mAChR agonist. This partly results from the variable response between neurons expressing the Gαq antisense: some cells display very little inhibition when Oxo-M is applied, whereas others resemble uninjected neurons and robust inhibitions are observed (Fig. 6). This finding mirrors the results obtained when antibodies directed against Gαq/11 were directly injected into SCG neurons. In these neurons there was an overall reduction in mean inhibition but a wide range of responses, from no reduction to total suppression in individual cells (Caulfield et al., 1994; Brown et al., 1995). The variability seen with the Gαq antisense plasmids may most reasonably be attributed to a variable reduction in endogenous Gαq protein. Although the participation of additional G-proteins cannot be totally excluded, this seems less likely because one would then have to postulate that the component of inhibition mediated by Gq varied greatly from one cell to another in an apparently arbitrary manner. Certainly, if another G-protein is involved, it must be insensitive to PTX (Fig.5D), and it cannot be G11 because (1) Gα11 antisense plasmids were unable to alter IK(M) inhibition and (2) the Gαq/11 commonantisense had no more effect than the specific Gαqantisense plasmids (Figs. 5B,C, 6).
The inequality between Gαq and Gα11function in these cells is somewhat surprising, because the bulk of evidence implies that Gαq and Gα11 are indistinguishable in both receptor-coupling and effector-coupling preference. These studies have been based mainly on purified protein in cell-free systems (Taylor et al., 1991) or cloned wild-type and constitutively active proteins expressed in cell lines (Aragay et al., 1992; Wu et al., 1992a,b; Hepler et al., 1993). Studies using antisense oligonucleotides, however, have implicated (1) both Gαqand Gα11 in M1 mAChR activation of PLC-β in RBL-2H3 cells (Dippel et al., 1996) but( 2) neuromedin B receptor activation of PLC occurring via Gαq, and not Gα11, in Xenopus oocytes (Shapira et al., 1994). Furthermore, in both rat myocytes and Xenopusoocytes, endogenous Gαq and Gα11 appear to have distinctly different functions after thyrotropin-releasing hormone or α1-adrenergic receptor activation (Lipinsky et al., 1992; Macrez-Leprêtre et al., 1997). These studies, and our results, suggest that the coupling preferences suggested by in vitro and overexpression studies may not reflect those found in native cells.
The lack of involvement of Gα11 in mediating IK(M) inhibition does not seem to arise from an inability of the subunit to couple to appropriate effector systems, because both Gα11* and Gαq* virtually abolished IK(M). It is more likely, therefore, that the M1 muscarinic receptor preferentially links to Gαq, rather than Gα11, in these neurons. Because studies with recombinant Gαq and Gα11 have demonstrated that both of these subunits are capable of coupling to M1 receptors (Nakamura et al., 1995), differential receptor coupling might arise from a greater abundance of Gαq relative to Gα11. Lower levels of Gα11, relative to Gαq, have been observed in most cell lines (Milligan et al., 1993) and all regions of brain examined (Milligan, 1993), and Western blots from whole ganglia indicate that these proteins may not be equally expressed in SCG (Caulfield et al., 1994). Alternatively, differences in membrane compartmentalization of Gαq and Gα11 could result in differential access to the receptor, as has been suggested for Gαi and Gαo (Neubig, 1994; Gudermann et al., 1996). Membrane association of Gαq and Gα11 is primarily determined by their N-terminal regions where the palmitoylation sites and the regions essential for βγ subunit interaction are situated (Conklin and Bourne, 1993; Milligan et al., 1995; Hepler et al., 1996). Because this region also contains the greatest amino acid diversity between Gαq and Gα11 (Strathmann and Simon, 1990), it is possible that they may differ in their membrane association or distribution. Recent work by Umemori et al. (1997)indicates that Gαq and Gα11 can undergo phosphorylation by tyrosine kinase after M1 mAChR activation and that this is required for activation of the subunits and leads to disassociation of the receptor–G-protein complex. A final possibility, therefore, could be that the phosphorylation states of Gαq and Gα11 in SCGs may differ, thereby altering their ability to interact with the receptor. Nevertheless, the effectiveness with which the GTPase-resistant Gα11inhibits IK(M) leaves open the possibility that Gα11 may mediate PTX-insensitive inhibition of IK(M) via other receptors such as angiotensin II (Shapiro et al., 1994) or bradykinin (the effect of which is also inhibited by the Gαq/11 antibody) (Jones et al., 1995).
The strong suppression of IK(M) after overexpression of GTPase-resistant αq (and α11), but not αoA, suggests that inhibition might well be mediated by dissociated GTP-bound αq subunits but does not, of itself, exclude the possibility that βγ subunits released from the endogenous αβγ heterotrimer might be the physiological mediator of inhibition. However, this possibility seems unlikely for two reasons. First, co-overexpression of β1 with γ2 subunits did not significantly reduce IK(M) (Fig. 8). In parallel experiments, this procedure effectively inhibited the N-type voltage-gated Ca2+current in these neurons (Delmas et al., 1998a), an inhibitory process considered from previous work to be driven by free βγ subunits (Ikeda, 1996; Herlitze et al., 1996). Second, muscarinic inhibition of IK(M) was unaffected in neurons injected with 200 μg/ml of a construct expressing the C-terminal sequence of βARK1 (also known as GRK2), which binds and sequesters free βγ subunits (Koch et al., 1994) (Fig. 7). Again, in parallel experiments, this concentration of the βARK1 construct reduced ICainhibition by noradrenaline and Oxo-M (Delmas et al., 1998a,b). Although increasing the plasmid concentration (to 400 μg/ml) did attenuate IK(M), this might be a nonspecific effect. We cannot exclude the possibility that the βγ dimer associated with Gαq might have a low affinity for the βARK1 βγ-binding domain [for instance, β3 subunits cannot interact with βARK1 (Daaka et al., 1997)]. However, Gαq and Gα11 can form heterotrimers with β1γ2 dimers (Nakamura et al., 1995), and βARK1 abolishes muscarinic-activated calcium release inXenopus oocytes, a response involving both Gαqand Gα11 (Stehno-Bittel et al., 1995).
Hence, and in conclusion, our results suggest that the pathway involved in muscarinic inhibition of IK(M) in rat SCG neurons requires Gαq but not Gα11 and that it is the α subunit of the heterotrimer Gq, rather than the βγ dimer, that acts as the primary transducer. To this extent, they also provide further evidence that coupling between receptors and G-proteins in neurons is highly specific and more subtle than experiments with reconstituted subunits might indicate.
Footnotes
We thank Drs. C. Scorer and C. Harris, Receptor Systems, Glaxo Wellcome, for the gift of βARK1 plasmid; Dr. S. Offermanns, Institut für Pharmakologie, Freie Universität Berlin, for the gift of constitutively active Gα11 plasmid; Dr. B. R. Conklin, The Gladstone Institute of Cardiovascular Disease, Departments of Medicine and Pharmacology, University of California San Francisco, for the gift of constitutively active GαoA cDNA; and Professor C. Hopkins for allowing us to use the Eppendorf microinjector in the Medical Research Council Laboratory for Molecular and Cellular Biology, University College London. This work was supported by the Wellcome Trust and the U.K. Medical Research Council.
Correspondence should be addressed to Jane Haley, Laboratory for Molecular Pharmacology, Department of Pharmacology, University College London, Gower Street, London WC1E 6BT, United Kingdom.
Ms. Vallis’s present address: Laboratory for Molecular Biology, Medical Research Council Centre, Hills Road, Cambridge CB2 2QH, United Kingdom.
REFERENCES
- 1.Abogadie FC, Vallis Y, Buckley NJ, Caulfield MP. Use of antisense-generating plasmids to probe the function of signal transduction proteins in primary neurons. In: Challiss RAJ, editor. Methods in molecular biology, Vol 83: Receptor signal transduction protocols. Humana Press; Totowa, NJ: 1997. pp. 217–225. [DOI] [PubMed] [Google Scholar]
- 2.Aragay AM, Katz A, Simon MI. The Gαq and Gα11 proteins couple the thyrotropin-releasing hormone receptor to phospholipase C in GH3 rat pituitary cells. J Biol Chem. 1992;267:24983–24988. [PubMed] [Google Scholar]
- 3.Arkinstall S, Chabert C, Maundrell K, Peitsch M. Mapping regions of Gαq interacting with PLCβ1 using multiple overlapping synthetic peptides. FEBS Lett. 1995;364:45–50. doi: 10.1016/0014-5793(95)00351-9. [DOI] [PubMed] [Google Scholar]
- 4.Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown DA. M-current. In: Narahashi T, editor. Ion channels. Plenum; New York: 1988. pp. 55–99. [DOI] [PubMed] [Google Scholar]
- 6.Brown DA, Selyanko AA. Membrane currents underlying the cholinergic slow excitatory post-synaptic potential in the rat sympathetic ganglion. J Physiol (Lond) 1985;365:365–387. doi: 10.1113/jphysiol.1985.sp015777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown DA, Marrion NV, Smart TG. On the transduction mechanism for muscarine-induced inhibition of M-current in cultured rat sympathetic neurons. J Physiol (Lond) 1989;413:469–488. doi: 10.1113/jphysiol.1989.sp017664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown DA, Buckley NJ, Caulfield MP, Duffy SM, Jones S, Lamas JA, Marsh SJ, Robbins J, Selyanko AA. Coupling of muscarinic acetylcholine receptors to neural ion channels: Closure of K+ channels. In: Wess J, editor. Molecular mechanisms of muscarinic acetylcholine receptor function. RG Landes; Austin, TX: 1995. pp. 165–182. [Google Scholar]
- 9.Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim G-D, Milligan G, Brown DA. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. J Physiol (Lond) 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clapham DE, Neer EJ. G protein βγ subunits. Annu Rev Pharmacol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 11.Conklin BR, Bourne HR. Structural elements of Gα subunits that interact with Gβγ, receptors, and effectors. Cell. 1993;73:631–641. doi: 10.1016/0092-8674(93)90245-l. [DOI] [PubMed] [Google Scholar]
- 12.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gqα to that of Giα. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 13.Daaka Y, Pitcher JA, Richardson M, Stoffel RH, Robishaw JD, Lefkowitz RJ. Receptor and Gβγ isoform-specific interactions with G protein-coupled receptor kinases. Proc Natl Acad Sci USA. 1997;94:2180–2185. doi: 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J Physiol (Lond) 1998a;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. Eur J Neurosci. 1998b;10:1654–1666. doi: 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- 16.Dippel E, Kalkbrenner F, Wittig B, Schultz G. A heterotrimeric G protein complex couples the muscarinic m1 receptor to phospholipase C-β. Proc Natl Acad Sci USA. 1996;93:1391–1396. doi: 10.1073/pnas.93.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudermann T, Kalkbrenner F, Schultz G. Diversity and selectivity of receptor-G protein interaction. Annu Rev Pharmacol Toxicol. 1996;36:429–459. doi: 10.1146/annurev.pa.36.040196.002241. [DOI] [PubMed] [Google Scholar]
- 18.Hepler JR, Kozasa T, Smrcka AV, Simon MI, Rhee SG, Sternweis PC, Gilman AG. Purification from Sf9 cells and characterization of recombinant Gqα and G11α. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- 19.Hepler JR, Biddlecome GH, Kleuss C, Camp LA, Hofmann SL, Ross EM, Gilman AG. Functional importance of the amino terminus of Gqα. J Biol Chem. 1996;271:496–504. doi: 10.1074/jbc.271.1.496. [DOI] [PubMed] [Google Scholar]
- 20.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 21.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 23.Jhon D-Y, Lee H-H, Park D, Lee C-W, Lee K-H, Yoo OJ, Rhee SG. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-β3. J Biol Chem. 1993;268:6654–6661. [PubMed] [Google Scholar]
- 24.Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gαq/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- 25.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 26.Lipinsky D, Gershengorn MC, Oron Y. Gα11 and Gαq guanine nucleotide regulatory proteins differentially modulate the response to thyrotropin-releasing hormone in Xenopus oocytes. FEBS Lett. 1992;307:237–240. doi: 10.1016/0014-5793(92)80775-c. [DOI] [PubMed] [Google Scholar]
- 27.Macrez-Leprêtre N, Kalkbrenner F, Schultz G, Mironneau J. Distinct functions of Gq and G11 proteins in coupling α1-adrenoreceptors to Ca2+ release and Ca2+ entry in rat portal vein myocytes. J Biol Chem. 1997;272:5261–5268. doi: 10.1074/jbc.272.8.5261. [DOI] [PubMed] [Google Scholar]
- 28.Marrion NV, Smart TG, Marsh SJ, Brown DA. Muscarinic suppression of the M-current in the rat sympathetic ganglion is mediated by receptors of the M1-subtype. Br J Pharmacol. 1989;98:557–573. doi: 10.1111/j.1476-5381.1989.tb12630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milligan G. Regional distribution and quantitative measurement of the phosphoinositidase C-linked guanine nucleotide binding proteins G11α and Gqα in rat brain. J Neurochem. 1993;61:845–851. doi: 10.1111/j.1471-4159.1993.tb03595.x. [DOI] [PubMed] [Google Scholar]
- 30.Milligan G, Mullaney I, McCallum JF. Distribution and relative levels of expression of the phosphoinositidase-C-linked G-proteins Gqα and G11α: absence of G11α in human platelets and haemopoietically derived cell lines. Biochim Biophys Acta. 1993;1179:208–212. doi: 10.1016/0167-4889(93)90143-d. [DOI] [PubMed] [Google Scholar]
- 31.Milligan G, Parenti M, Magee AI. The dynamic role of palmitoylation in signal transduction. Trends Biol Sci. 1995;20:181–186. doi: 10.1016/s0968-0004(00)89004-0. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura F, Kato M, Kameyama K, Nukada T, Haga T, Kato H, Takenawa T, Kikkawa U. Characterization of Gq family G proteins GL1α (G14α), GL2α (G11α), and Gqα expressed in the baculovirus-insect cell system. J Biol Chem. 1995;270:6246–6253. doi: 10.1074/jbc.270.11.6246. [DOI] [PubMed] [Google Scholar]
- 33.Neubig RR. Membrane organization in G-protein mechanisms. FASEB J. 1994;8:939–946. doi: 10.1096/fasebj.8.12.8088459. [DOI] [PubMed] [Google Scholar]
- 34.Phillips MI, Gyurko R. Antisense oligonucleotides: new tools for physiology. News Physiol Sci. 1997;12:99–105. [Google Scholar]
- 35.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 36.Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- 37.Shapira H, Way J, Lipinsky D, Oron Y, Battey JF. Neuromedin B receptor, expressed in Xenopus laevis oocytes, selectively couples to Gαq and not Gα11. FEBS Lett. 1994;348:89–92. doi: 10.1016/0014-5793(94)00570-2. [DOI] [PubMed] [Google Scholar]
- 38.Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12:1319–1329. doi: 10.1016/0896-6273(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 39.Stehno-Bittel L, Krapivinsky G, Krapivinsky L, Perez-terzic C, Clapham DE. The G protein βγ subunit transduces the muscarinic receptor signal for Ca2+ release in Xenopus oocytes. J Biol Chem. 1995;270:30068–30074. doi: 10.1074/jbc.270.50.30068. [DOI] [PubMed] [Google Scholar]
- 40.Strathmann M, Simon MI. G protein diversity: a distinct class of α subunits is present in vertebrates and invertebrates. Proc Natl Acad Sci USA. 1990;87:9113–9117. doi: 10.1073/pnas.87.23.9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor SJ, Chae HZ, Rhee SG, Exton JH. Activation of the β1 isozyme of phospholipase C by α subunits of the Gq class of G proteins. Nature. 1991;350:516–518. doi: 10.1038/350516a0. [DOI] [PubMed] [Google Scholar]
- 42.Umemori H, Inoue T, Kume S, Sekiyama N, Nagao M, Itoh H, Nakanishi S, Mikoshiba K, Yamamoto T. Activation of the G protein Gq/11 through tyrosine phosphorylation of the α subunit. Science. 1997;276:1878–1881. doi: 10.1126/science.276.5320.1878. [DOI] [PubMed] [Google Scholar]
- 43.Wang H-S, McKinnon D. Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J Physiol (Lond) 1995;485:319–335. doi: 10.1113/jphysiol.1995.sp020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wickman K, Clapham DE. Ion channel regulation by G proteins. Physiol Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 45.Wong YH, Conklin BR, Bourne HR. GZ-mediated hormonal inhibition of cyclic AMP accumulation. Science. 1992;255:339–342. doi: 10.1126/science.1347957. [DOI] [PubMed] [Google Scholar]
- 46.Wu D, Lee C-H, Rhee SG, Simon MI. Activation of phospholipase C by the α subunits of the Gq and G11 proteins in transfected Cos-7 cells. J Biol Chem. 1992a;267:1811–1817. [PubMed] [Google Scholar]
- 47.Wu D, Katz A, Lee C-H, Simon MI. Activation of phospholipase C by α1-adrenergic receptors is mediated by the α subunits of Gq family. J Biol Chem. 1992b;267:25798–25802. [PubMed] [Google Scholar]