

Abstract
We report the characterization of a new sodium channel blocker, μ-conotoxin PIIIA (μ-PIIIA). The peptide has been synthesized chemically and its disulfide bridging pattern determined. The structure of the new peptide is:

where Z = pyroglutamate andO = 4-trans-hydroxyproline.
We demonstrate that Arginine-14 (Arg14) is a key residue; substitution by alanine significantly decreases affinity and results in a toxin unable to block channel conductance completely. Thus, like all toxins that block at Site I, μ-PIIIA has a critical guanidinium group.
This peptide is of exceptional interest because, unlike the previously characterized μ-conotoxin GIIIA (μ-GIIIA), it irreversibly blocks amphibian muscle Na channels, providing a useful tool for synaptic electrophysiology. Furthermore, the discovery of μ-PIIIA permits the resolution of tetrodotoxin-sensitive sodium channels into three categories: (1) sensitive to μ-PIIIA and μ-conotoxin GIIIA, (2) sensitive to μ-PIIIA but not to μ-GIIIA, and (3) resistant to μ-PIIIA and μ-GIIIA (examples in each category are skeletal muscle, rat brain Type II, and many mammalian CNS subtypes, respectively). Thus, μ-conotoxin PIIIA provides a key for further discriminating pharmacologically among different sodium channel subtypes.
Keywords: Na channels, μ-conotoxin, tetrodotoxin, neuromuscular transmission, ion channel subtype, peptide
Several potent toxins target voltage-gated sodium channels; the different sites of binding and modes of activity of these toxins have been described (Catterall, 1992). These ligands have been indispensable for investigating the structure and function of these ion channels, which play a key role in excitable tissues. The demonstration (Narahashi et al., 1964) that tetrodotoxin specifically inhibited voltage-gated sodium currents without effect on potassium currents provided crucial experimental support for the Hodgkin–Huxley formulation of the action potential. Sodium channel toxins continue to be important pharmacological tools for neuroscientists.
Channel blockers, notably the guanidinium toxins saxitoxin and tetrodotoxin, target a site generally postulated to be at the extracellular end of the channel pore (Site I). Only one family of polypeptide toxins, the μ-conotoxins, has been shown to act at this site and functionally affect voltage-gated sodium currents. These were isolated originally from the venom of the marine snail Conus geographus (Stone and Gray, 1982; Sato et al., 1983; Cruz et al., 1985; Olivera et al., 1985).
Other families of Conus peptides (notably the ω-conotoxins, which target calcium channels, and the α-conotoxins, which target nicotinic acetylcholine receptors) have been found in the venoms of many Conus species that have been examined. Members within a given family of peptides from differentConus species have homologous structures but show extreme sequence hypervariability, and comparison of their activities has provided insightful structure–function information. In particular, the wide diversity among natural toxins within each family has been instrumental in identifying new subclasses of receptors (Olivera et al., 1990, 1994). By contrast, because the μ-conotoxins so far have been described only from the venom of C. geographus, most structure–function information for this peptide family has come from experiments with synthetic analogs.
In this report we describe the first new member of the μ-conotoxin peptide family to be characterized in over a decade, μ-conotoxin PIIIA from Conus purpurascens, an Eastern Pacific fish-hunting species. As expected, the new μ-conotoxin shows considerable sequence divergence from the μ-conotoxins of Conus geographus. In addition to a comprehensive biochemical characterization of the peptide, we provide electrophysiological and binding data, which demonstrate that μ-conotoxin PIIIA has considerable potential, both as a novel pharmacological tool for electrophysiology of the neuromuscular junction and for distinguishing among different tetrodotoxin-sensitive Na channel subtypes in the CNS.
MATERIALS AND METHODS
Molecular cloning. Conus purpurascensvenom ducts were collected, and mRNA was prepared by methods previously described (Woodward et al., 1990; Colledge et al., 1992; Hopkins et al., 1995). The analysis of cDNA clones from Conus venom ducts was performed as detailed previously (Colledge et al., 1992;Hillyard et al., 1992; McIntosh et al., 1995).
Solid-phase peptide synthesis. The peptide was built without the N-terminal pyroglutamate on Rink amide resin, using standard Fmoc chemistry. All amino acids were purchased from Bachem (Torrance, CA), and side chains were protected as follows: Arg(pmc), His(trt), Hyp(t-bu), Lys(Boc), Ser(t-bu), Gln(trt), and Cys(trt). Peptide bond coupling was performed with equimolar amounts of amino acid, dicyclohexylcarbodimide (DCC), and hydroxybenzotriazole (HOBT) on an ABI model 430A synthesizer. The terminal Fmoc group was removed by treatment with 1:4 piperidine/N-methylpyrrolidone (NMP) (v/v). To complete the predicted sequence of the peptide, we manually coupled pyroglutamate to portions of the resin before peptide cleavage. Resin (50 mg) was deprotonated by treatment with 1 ml of piperidine (20% in NMP) for 1 min and washed three times each with alternating methanol and NMP. Pyroglutamate (0.5 mmol) was activated in 1 ml of 1m diisopropylcarbodimide (DIC)/1 mhydroxybenzotriazole (HOBT) in NMP for 30 min; the solution was added to the deprotonated resin, and the reaction mixture was stirred for 2.5 hr. Then the resin was centrifuged and washed with NMP five times, followed by three washes with methanol. Pyroglutamate was not protected with Fmoc, and so further deprotection was not necessary. The final resin was subjected to peptide cleavage as described previously (Shon et al., 1995). The cleavage mixture was filtered into tert-butyl methyl ether at −10°C. The peptide immediately precipitated, and the solution was centrifuged to separate the pellet, which was washed once with the ether. The pellet dissolved in 60% acetonitrile (ACN)/0.1% trifluoroacetic acid (TFA) in H20 and was purified by HPLC on a Vydac C18 preparative column (2.5 cm; flow = 20 ml/min). The linear peptide was oxidized with glutathione as described previously (Dudley et al., 1995) and yielded a mixture of isomers. The major isomer, accounting for 20–30% of the total absorbance, proved to be biologically active. After preparative purification, this isomer was purified to homogeneity, using a Vydac C18semi-preparative column (7.0 mm × 240 cm; flow = 3 ml/min). All HPLC was done by using a 12–30% linear gradient of ACN in 0.1% TFA and water. Analysis of the purified, biologically active peptide by electrospray mass spectrometry gave a monoisotopic MH+ = 2604.05 (calculated MH+ = 2604.12).
The same methods were used to obtain the R14A analog of μ-PIIIA. To obtain the unblocked analog, μ-PIIIA[2–22], we performed the same cleavage and oxidation procedures with resin to which pyroglutamate was not coupled manually.
Disulfide bridge analysis. The disulfide connectivity of μ-PIIIA was analyzed by the partial reduction strategy of Gray (1993), using an analog without the N-terminal pyroglutamate and the mono-iodohistidine derivative of the same peptide (see Results). As detailed in Shon et al. (1995), peptides were partially reduced, using tris (2-carboxyethyl) phosphine (TCEP), and then alkylated with iodoacetamide. Yields of partially alkylated peptide were low. To avoid further loss of peptide, we performed amino acid sequencing with the remaining disulfide bonds intact, using automated Edman degradation on an ABI Model 477A instrument. All purification was done with a Vydac C18 analytical HPLC column (218TP54; 4.6 × 250 mm), and peptides were eluted under the conditions previously described above in Solid-Phase Peptide Synthesis.
Iodination of μ-conotoxin PIIIA[2–22]. Peptide solution (5–10 nmol in ∼700 μl of HPLC eluant) was combined with an equal volume of 0.1 m Tris, pH 8. Iodine in methanol (2 mm) was added to make a final iodine concentration of 20 μm. After a 10 min incubation at room temperature, the reaction was quenched by the addition of 0.75m ascorbic acid (1:100 by volume). The mixture was applied to an analytical column and eluted by using the HPLC conditions described above. The mono- and di-iodo derivatives of μ-PIIIA[2–22] eluted as discrete peaks ∼1.5 and 3 min after the noniodinated form, using a gradient of 0.6% ACN increase per minute. The expected masses of the iodinated derivatives were confirmed by electrospray mass spectrometry.
Electrophysiology. The cutaneous pectoris muscle dissected from ∼7 cm of Rana pipiens frogs was trimmed longitudinally so that only the lateral one-quarter of muscle remained (Yoshikami et al., 1989). The trimmed muscle was pinned flat on the bottom of a shallow trough fabricated from SYLGARD (a silicone elastomer, Dow Chemical, Midland, MI). To examine the response of the muscle to direct electrical stimulation, we partitioned the trough as illustrated in Figure 3A. Current was injected into the muscle across partition 1; the stimulating electrodes were connected to a stimulus isolation unit, and supramaximal 1-msec-long rectangular pulses were used to elicit action potentials in the muscle directly. Stimuli were applied at a frequency of 1/min or less. The recording electrodes monitored the potential across partition 3 (partition 2 served to isolate the recording electrically from the stimulating electrodes). When the action potential propagated into chamber C, a positive response was recorded by the preamplifier, and the further propagation of the action potential into chamber D was recorded as a negative response. Thus, the extracellularly recorded action potential from the population of fibers in the muscle was recorded as a biphasic response, with the phases separated from each other by only a few milliseconds (see Fig. 3B). To examine the effect of the toxin, we replaced the normal frog Ringer’s solution in chamber D by one containing toxin. If the toxin blocked sodium channels, attenuation of only the late negative phase should be observed. The early positive phase should remain mainly unaltered, reflecting the fact that portions of the muscle not exposed to toxin remained normal. Thus, there are two advantages of exposing only the solution in chamber D to toxin: one, this allows the response in chamber C to serve as an internal control for the overall vitality of the muscle preparation as well as to insure that the stimulus remains supramaximal; two, the volume of toxin solution necessary is reduced, and in these experiments 25 μl sufficed.
Fig. 3.

A, Sketch of electrophysiological recording chamber for testing toxin on frog’s cutaneous pectoris muscle response to direct electrical stimulation. The rectangular SYLGARD trough (∼4 × 16 × 1 mm) was partitioned into four compartments (A–D) by three Mylar sheets (1–3). The sheets were inserted into slots in the wall of the trough after the muscle had been pinned to the floor of the trough. Two Mylar strips (∼1 × 55 × 0.1 mm) were placed on either side of the muscle to serve as stops to prevent the partitions from cutting into the muscle. The cutaneous end of the muscle was located in A, and the xiphisternum (cartilage) was in D. Stimulating electrodes were inA and B, and a ground electrode was inB. The recording electrode in C was connected to the negative input, and that in D was connected to the positive input of a differential AC preamplifier. Compartment D served as the test chamber; only the muscle segment in this compartment was exposed to toxin. All compartments contained Ringer’s solution, and all electrodes were bare platinum wires. B, PIIIA (1 μm) blocks directly evoked action potentials in frog muscle. Shown are superimposed traces of responses before, during, and after exposure to toxin. Stimulus was applied at t = 0. Thin solid curve, Control response; bold solid curve, response after exposure to toxin for 23 min and just before toxin was washed out; bold dashed curve, response 20 min after toxin washout; thin dashed curve, response after >4.5 hr of washing. Toxin was placed only in compartment D(see panel A), which contained the portion of the muscle that produced the negative phase of the response in the control trace.C, Time course of the block of the directly evoked action potentials. Maximum amplitudes of the positive phase (open circles) and negative phase (filled circles) of the response are plotted as a function of time. The solution in compartment D was replaced with 1 μm PIIIA at time 0 (downward arrow), and the toxin was washed out 23 min later (upward arrow). D, μ-Conotoxin GIIIA (5 μm) reversibly blocks directly evoked action potentials in frog muscle. Shown are superimposed traces of responses before, during, and after exposure to toxin. Thin solid curve, Control response; bold solid curve, response after 20 min of exposure to toxin; dashed curve, response 45 min after toxin is washed out. Experimental conditions were essentially the same as those for Figure 4A, except that GIIIA instead of PIIIA was used and that the experiment was conducted with a different cutaneous pectoris muscle preparation. E, Time course of the block of the directly evoked action potentials by μ-conotoxin GIIIA. Maximum amplitudes of the positive phase (open circles) and of the negative phase (filled circles) of responses are plotted as a function of time. The solution in compartment D was replaced by 5 μm toxin at t = 0 (downward arrow), and the toxin was washed out 20 min later (upward arrow). Responses behaved essentially the same as those obtained with exposure to μ-conotoxin PIIIA (see Fig.4B), except that here the effect of the toxin GIIIA was reversible.
To examine synaptically evoked responses, we used an arrangement similar to that previously described (Yoshikami et al., 1989) (see Fig.4A). The trough was similar to that used for direct stimulation but had no partitions, and the motor nerve was draped into a two-compartment well adjacent to the trough. Both compartments of the well were filled with Ringer’s solution. The portions of the nerve exposed to air were covered with Vaseline. Each compartment of the well had an electrode to allow for electrical stimulation (0.1 msec rectangular pulses) of the nerve. The recording and ground electrodes were essentially the same as those for direct stimulation, except that the negative recording electrode was centered near the middle of the muscle where the endplates were located. This placement of the electrodes allowed the extracellular endplate currents from the population of fibers to be recorded readily (Yoshikami et al., 1989) (see also Fig. 4B). When the action potentials of the muscle were blocked irreversibly with PIIIA, no muscle movement occurred when the nerve was stimulated, so an extracellular recording microelectrode could be placed close to the endplate of a selected fiber to record more focal extracellular synaptic currents (see Fig.4C).
Fig. 4.
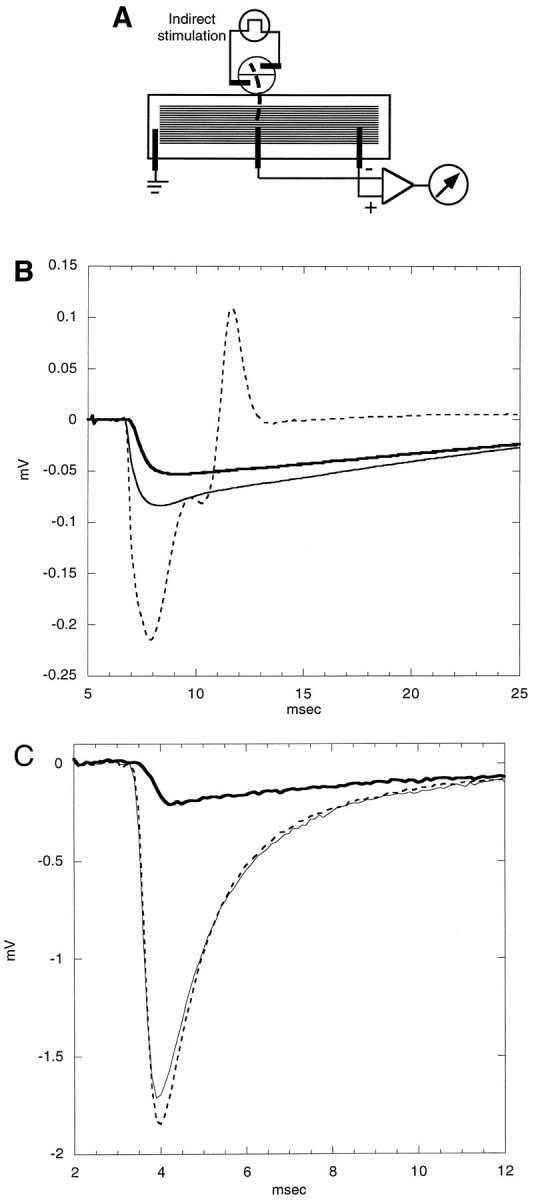
A, Sketch of recording chamber for indirect stimulation. For indirect stimulation the frog motor nerve (bold dashed line) was draped into a circular, two-compartment well adjoining the trough in which the muscle resided. The compartments of the well contained stimulating electrodes, and the wire recording electrodes that are illustrated were used to acquire the extracellular responses shown in B. The trough contained no partitions so that the entire muscle could be exposed to toxin. B, Extracellularly recorded responses before, during, and after exposure to 5 μm PIIIA. The motor nerve was stimulated at t = 5 msec while the response was recorded with a wire electrode from the endplate region of a population of muscle fibers. The “before” trace (dashed) has an early negative and late positive phase, the former having contributions from both action and synaptic potentials and the latter consisting of the propagated action potential. The “during” (bold) and “after” (thin solid) responses have only a negative phase, indicating that only the endplate current remains after treatment with toxin. Note that the latency of the response is reversibly increased by the peptide, suggesting that PIIIA reversibly reduces the conduction velocity of the action potential of the motor nerve. PIIIA also reversibly attenuates the postsynaptic response, an action reminiscent of the effect of focal tetrodotoxin application (Katz and Miledi, 1968). C, Extracellular synaptic currents recorded before, during, and after exposure to ∼20 μm PIIIA. The motor nerve was stimulated (att = 2 msec) every minute, and the responses were recorded with a focal extracellular electrode, the tip of which was placed near the endplate of a fiber of a muscle that previously had been exposed to ∼2 μm PIIIA for 15 min, to preblock the sodium channels of the muscle irreversibly; then it was washed. Each trace represents the average of five evoked responses acquired before toxin exposure (dashed curve), after the preparation had been exposed to 20 μm PIIIA for 15 min (bold curve), or 50 min after the washout of toxin was initiated (solid curve). The response was mainly from a single endplate; hence, its time course was briefer than the synaptic responses in B. As in B, the toxin reversibly attenuated the amplitude of the response, albeit more strongly, and reversibly increased its latency.
Recording from cloned channels in oocytes. Oocytes fromXenopus laevis were prepared as described previously (Stühmer, 1992). cRNA encoding rat Type II sodium channel α-subunit (Noda et al., 1986) or rat μ1 skeletal muscle Na channel (Trimmer et al., 1989) was injected into stage VI oocytes (30–50 ng/oocyte). The vitelline membranes of the oocytes were removed mechanically with fine forceps, and currents were recorded in frog Ringer’s solution 2–6 d after injection under two-electrode voltage-clamp control with a Turbo-Tec amplifier (NPI Elektronik, Tamm, Germany) driven by the Pulse+PulseFit software package (HEKA Elektronik, Lambrecht, Germany). Intracellular electrodes were filled with 2 m KCl and had a resistance between 0.6 and 0.8 MΩ. Current records were low-pass-filtered at 3 kHz and were sampled at 10 kHz. Leak and capacitive currents were corrected on-line by using a P/N method. To estimate the IC50 for the block of PIIIA, we measured whole-cell currents of oocytes expressing rat Type II or μ1 Na channels, and we successively increased the toxin concentrations in the bath. The peak inward current was measured and plotted against the toxin concentration. Dose–response curves were fit by using the equation: y = (1 + (T/IC50)n)−1, where T is the toxin concentration and n is the Hill coefficient.
Binding experiments. [3H]Saxitoxin binding to rat brain membranes was performed by the protocol of Doyle et al. (1993) except that the assays were scaled down to a volume of 0.25 ml, and 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 μm leupeptin, and 1 μm pepstatin were present. Electric eel membranes were prepared as described by Becker et al. (1989) except that the homogenizing buffer used was (in mm) 10 HEPES-Tris, 10 EDTA, 10 EGTA, and 1 PMSF plus 1 μm leupeptin and pepstatin, pH 7.0.
RESULTS
Identification of a cDNA clone from Conus purpurascensencoding a putative μ-conotoxin: chemical synthesis
The feasibility of discovering new Conus peptides from the predicted amino acid sequences encoded by cDNA clones was demonstrated previously with ω-conotoxins (Hillyard et al., 1992). As part of a comprehensive program to characterize the toxins in the venom of Conus purpurascens systematically, a large number of cDNA clones derived from the mRNA of the venom duct of C. purpurascens have been sequenced. Several cDNA clones contain the nucleotide sequence shown in Figure 1; the predicted amino acid sequence from this nucleotide sequence strongly suggests that the clone might encode a μ-conotoxin. There are a number of important features similar to those of the previously characterized μ-conotoxins (μ-GIIIA, μ-GIIIB, and μ-GIIIC fromConus geographus), despite the significant sequence divergence: the pattern of Cys residues, the high net positive charge, and the apparent conservation of the critical Arg residue (residue 14 of the predicted mature peptide) believed to be essential for μ-conotoxin function (Sato et al., 1991; Becker et al., 1992).
Fig. 1.

A, The nucleic acid sequence derived by analyzing cDNA clones from a Conus purpurascens venom duct library. The sequence encoding the inferred C-terminal end of an open reading frame is shown. The pattern of Cys residues suggests that the encoded C-terminal peptide might be a μ-conotoxin. The arrow indicates the predicted site of proteolytic cleavage to generate the mature toxin: a -Lys-Arg- sequence is the most common motif for proteolytic cleavage of conotoxin precursors. B, The predicted sequence of the post-translationally processed mature peptide. The amino acid sequence shown in A would be predicted to be post-translationally processed at the four indicated sites as follows: the encoded glutamine residue would be converted to pyroglutamate after proteolysis (site1), proline would be hydroxylated to 4-trans-hydroxyproline (sites 2 and3), and the C-terminal -Cys-Gly-Arg- sequence would be processed by an exopeptidase and amidation enzymes to a -Cys-NH2 moiety (site 4). These post-translational processing events would yield the indicatedbold sequence, where Z = pyroglutamate and O = 4-trans-hydroxyproline.
We therefore synthesized the predicted 22-residue peptide, incorporating post-translational modifications modeled on other related peptides: (1) Gln1 to pyroglutamate (all glutamines at the termini of Conus peptides so far have been found as pyroglutamates), (2) prolines to hydroxyproline (all prolines in known μ-conotoxins are hydroxylated), and (3) C-terminal -Cys.Cys.Gly.Arg- to -Cys.Cys.NH2 (the presence of -Gly.Arg- is a signal for C-terminal amidation). This predicted peptide is referred to as μ-conotoxin PIIIA (μ-PIIIA), based on the physiological evidence detailed below. A detailed description of the chemical synthesis and oxidation of μ-PIIIA is given under Materials and Methods. The pure synthetic peptide caused flaccid paralysis in both mice and fish, as expected for a μ-conotoxin (Cruz et al., 1985).
Disulfide bridge analysis
The putative mature peptide has a pyroglutamate residue at the N terminus. Because disulfide bridge analysis requires that the partially reduced intermediates be sequenced by Edman degradation, each blocked intermediate would need to be unblocked enzymatically. Instead, we analyzed the analog lacking the N-terminal pyroglutamate. This was readily available from cleavage of the resin before the final addition of pyroglutamate. The major μ-conotoxin PIIIA[2–22] peptide obtained after the Cys residues were oxidized was bioassayed and proved to cause paralysis in fish. A comparison of the efficacy of the two peptides on frog skeletal muscle (see Electrophysiology Using Amphibian Muscle, below) also shows that both peptides blocked muscle action potentials, with no recovery after toxin washout for at least 3 hr in both cases. This strongly indicated that the major oxidation product of both the original peptide and the analog shares the same disulfide pattern.
Partial reduction of μ-PIIIA[2–22] produced only one species that could be separated from the fully oxidized peptide (indicated by thearrow in Fig.2A). Alkylation and sequence analysis revealed that this peptide had a single intact disulfide bridge (shown as sequence 2 in Fig. 2). A second reduction intermediate was required to determine the remaining disulfide linkages. Repeated attempts to obtain another intermediate under the reaction conditions in Figure 2A were unsuccessful; however, partial reduction of the mono-iodohistidine derivative of μ-PIIIA[2–22] produced three species, as shown in Figure2B, that could be separated from the oxidized peptide in quantities sufficient for analysis. Each peak in Figure2B is numbered to show the corresponding peptide structure in Figure 2C, as revealed by alkylation and sequence analysis. These data reveal that μ-conotoxin PIIIA has the same disulfide pattern as μ-conotoxin GIIIA as well as the following structure:

Fig. 2.

Disulfide bridge analysis. Reverse-phase HPLC chromatograms (12–30% acetonitrile gradient in 30 min; flow rate = 1 ml/min) of native (N), fully reduced (R), and partially reduced μ-PIIIA[2–22] after incubation with 20 mm TCEP, pH 3.A, Before iodination, the partially reduced species is indicated with an arrow (1 min incubation at 65°C).B, After mono-iodination of His residue, intermediates are labeled 1–3 (5 min incubation at room temperature).C, Schematic diagrams of disulfide connectivity in fully oxidized and partially reduced peptides, as labeled inB.
where Z = pyroglutamate and O = 4-trans-hydroxyproline.
Electrophysiology using amphibian muscle
The effects of the peptide on the response of the frog muscle to direct electrical stimulation were investigated with the recording chamber illustrated in Figure3A. A control response before toxin addition is shown in Figure 3B. The progression of the action potential between muscle segments in compartments C and D is readily apparent; the biphasic waveform that was generated represents the propagation of the action potential from C to D. When μ-PIIIA was added to segment D, the action potential clearly propagated into segment C, causing the voltage change characteristic of the first half of the biphasic waveform in Figure 3B; however, the negative phase was abolished completely, indicating that propagation in segment D of the muscle was abolished. These results are consistent with inhibition of voltage-gated sodium channels in the muscle plasma membrane.
On exposure to toxin, the positive phase initially becomes larger as the counteracting negative phase is attenuated. The initial rising phase of the positive phase also is delayed slightly after exposure to toxin; this is thought to be attributable to leakage of the toxin into compartment C with an attendant decrease in the propagation velocity of the action potential in that compartment. The leak of toxin into compartment C is also thought to be responsible for the decrement in the amplitude of the positive phase as well as delayed time to peak observed in the response taken >4.5 hr later.
The peaks of the responses as a function of time before, during, and after toxin addition are shown in Figure 3C. The negative phase is completely and irreversibly obliterated by exposure to μ-conotoxin PIIIA, whereas the positive phase remains mainly intact, indicating that no untoward systemic changes occurred. Even with washing for many hours in the absence of toxin, no recovery was observed in segment D, although action potential propagation to segment C was essentially normal (a slight rundown was observed with time). Similar results also were observed with the μ-PIIIA[2–22] analog of the toxin (results not shown).
The results are consistent with the activity of a μ-conotoxin; the homologous peptides from Conus geographus previously have been shown to be highly specific for the skeletal muscle Na channel subtype in peripheral systems. Although μ-GIIIA and μ-PIIIA selectively inhibit skeletal muscle action potentials, a notable difference is that the latter peptide appears to act much more irreversibly in the frog neuromuscular preparation (see Fig.3D,E for the results with μ-GIIIA).
Indirect stimulation experiments
Synaptically evoked responses also were examined (Fig.4). When the frog motor nerve was stimulated electrically, a muscle twitch was observed and muscle action potential was recorded. After exposure to μ-conotoxin PIIIA, muscle twitches and action potentials were abolished completely; in contrast, endplate currents still were observed. Thus, the propagation of action potentials in the motor axon is not blocked, although action potentials in the muscle were abolished.
However, in the presence of high concentrations of PIIIA, an effect on the excitatory postsynaptic response was observed. The latency of the response was increased, and its amplitude was attenuated. These effects on the synaptic response, unlike the block of the action potential of the muscle, were reversible. The attenuation of the synaptic response was greater during exposure to 20 than to 5 μm PIIIA (Fig. 4B,C). At lower concentrations of toxin (∼1 μm) the muscle action potential still could be abolished irreversibly, but the delay in response latency was barely detectable (data not shown). These changes in the synaptic response are presumed to be a presynaptic effect of PIIIA. They clearly require a relatively high concentration of toxin and are reversible, as compared with the irreversible high-affinity effects of the toxin on the muscle action potential.
The new peptide, μ-PIIIA, should serve as a most convenient pharmacological agent for irreversibly preventing muscle twitching when the synaptic electrophysiology of amphibian neuromuscular junctions is investigated. Application of the toxin, followed by washout, yields a frog motor nerve/skeletal muscle preparation with muscle action potentials selectively blocked, an ideal preparation for examining synaptic events.
Effect of μ-PIIIA on two different mammalian Na channel subtypes
We investigated whether μ-conotoxin PIIIA could affect voltage-gated sodium channels in the mammalian CNS. The effect of the toxin was tested on a major subtype of voltage-gated sodium channels found in central neurons expressed in Xenopus oocytes, the tetrodotoxin (TTX)-sensitive Type II voltage-gated sodium channels (Fig. 5A–C). μ-PIIIA blocked Type II Na channels from rat; the presence of μ-PIIIA (2 μm) in the bath solution abolished nearly all Na current, but in a reversible manner (Fig. 5B,C). Under the same conditions μ-GIIIA did not affect Na currents (Fig. 5D). A comparison of the dose–response for the two toxins is shown in Figure6; the inhibition by μ-GIIIA is incomplete even at the highest concentration tested. The data indicate that μ-PIIIA has a ∼50-fold greater affinity for the channel than does μ-GIIIA.
Fig. 5.

μ-PIIIA blocks rat Type II Na channel expressed in Xenopus oocytes. A, Whole-cell current recorded from an oocyte expressing rat Type II Na channels. Voltage steps ranging from −80 to + 60 mV, in 10 mV increments, were generated from a holding potential of −100 mV. B, The addition of 2 μm μ-PIIIA to the bath solution resulted in a profound block of the currents. C, Wash with frog Ringer’s solution. D, The addition of 2 μm μ-GIIIA did not block the sodium currents.
Fig. 6.

Inhibition of sodium currents by μ-conotoxin PIIIA and the R14A analog. Shown are dose–response curves for the block of μ-conotoxin GIIIA (top panel) and PIIIA (middle panel) on skeletal muscle sodium channels (open circles) and on rat brain IIA sodium currents (filled circles). Bottom panel, Block of rat skeletal muscle sodium currents by the analog μ-PIIIA[R14A]. For the R14A analog tested on the rat brain IIA sodium channel only, the data point for 50 μm is given, which blocked the currents by only ∼30%.
The effects of μ-conotoxins PIIIA and GIIIA on a cloned mammalian skeletal muscle sodium channel subtype expressed in Xenopusoocytes also were evaluated; a comparison between the muscle and Type II subtypes is shown for both μ-GIIIA and μ-PIIIA in Figure 6(top and middle panels); both toxins block the conductance of this cloned channel. The affinity of μ-PIIIA for the mammalian muscle sodium channel is higher than for the CNS Type II subtype (IC50 ∼44 vs 640 nm); however, the affinity of μ-PIIIA for the mammalian muscle channel does not seem to be as high as for fish and amphibian channels of the same subtype. Although both toxins are high-affinity antagonists when tested on the skeletal muscle Na channel subtypes from a variety of vertebrate systems, μ-PIIIA binds more irreversibly in the amphibian system, whereas μ-GIIIA is the more potent toxin for the mammalian skeletal muscle subtype.
Structure–activity studies: effects of mutations in a critical arginine residue
Structure–activity studies done on μ-conotoxins fromConus geographus suggested that the guanidinium group of a critical Arg residue is responsible for occluding the ion channel pore. Although μ-conotoxin PIIIA is highly divergent in sequence from μ-conotoxins from Conus geographus, the alignment of conserved cysteine residues suggests that Arginine-14 (Arg14) is homologous to the critical Arg defined in the Conus geographus μ-conotoxins. We therefore evaluated the effects of an alanine substituted for the Arg residue in this position. The R14A-μ-conotoxin PIIIA homolog was tested first on the Type II rat brain sodium channel; no effects on channel conductance were seen at concentrations >10-fold greater than theKD of the wild-type toxin for the Type II sodium channel. Although we did not have sufficient toxin to carry out a complete curve, an extrapolation suggests that the μ-PIIIA[R14A] homolog has a IC50 >150 μm for antagonizing the Type II rat brain sodium channel (Fig. 6, bottom panel).
The μ-PIIIA[R14A] homolog also was evaluated, using rat skeletal muscle sodium channel subtype expressed in oocytes (Fig. 6,bottom panel). Clearly, the homolog has a lower affinity for this cloned channel than does the wild-type toxin. However, the data in Figure 6 do not fit a simple monotonic inhibition curve; it appears that, even at 100 μm μ-PIIIA[R14A], significant conductance is still detected (>20% of the control). Thus, although an apparent IC50 of ∼1.5 μmis obtained in this experiment, the data suggest that a sodium channel bound by the μ-PIIIA[R14A] homolog has significantly reduced but measurable conductance. These data are qualitatively similar to results obtained with the R13Q homolog of μ-conotoxin GIIIA (French et al., 1996). Thus, the R14A substitution caused a decrease in the affinity of the toxin for the μ1 rat muscle sodium channel and resulted in a channel with some residual conductance even when toxin was bound.
Binding experiments
Binding displacement experiments were performed with [3H]saxitoxin as the radiolabeled ligand and withElectrophorus electricus electric organ membranes as the source of the receptors (Fig. 7). The electric organ has a high density of Na channels more closely resembling a skeletal muscle subtype than the neuronal Na channel subtypes. As expected, μ-conotoxin PIIIA completely displaced [3H]saxitoxin ([3H]STX) binding to electric organ membranes. Clearly, μ-conotoxin PIIIA has a high affinity (IC50 ∼3 × 10−9M) for the STX binding site of Electrophorus electric organ.
Fig. 7.

Binding competition experiments with [3H]saxitoxin. The displacement of specific [3H]STX binding by μ-conotoxin in rat brain and in eel electroplax was determined as described in Materials and Methods. Specific binding was determined by subtracting the nonspecific binding of [3H]saxitoxin from the total binding; the nonspecific binding was measured by using 12 μm TTX to displace [3H]saxitoxin binding. Open circles, μ-PIIIA displacement for eel electroplax sites;squares, μ-PIIIA displacement for rat brain sites;triangles, μ-GIIIA displacement for rat brain sites. Error bars indicate SEM.
In contrast, it was found that μ-PIIIA displaced only a fraction (>50%) of specific [3H]STX binding to crude membranes from rat brain. A comparison of μ-PIIIA and μ-GIIIA displacement of specific [3H]STX binding to rat brain sites is shown in Figure 7. These data indicate that μ-PIIIA displaces more than one-half of the [3H]STX high-affinity sites in rat brain; in contrast, μ-GIIIA displaces ∼20% of specific [3H]STX binding at the same concentrations. These results suggest the presence of a significant number of μ-PIIIA-sensitive, μ-GIIIA-resistant Na channels in the mammalian CNS.
DISCUSSION
The studies above establish that Conus purpurascensvenom ducts express a μ-conotoxin. Although this peptide, μ-conotoxin PIIIA, has clear structural homology with the three previously characterized μ-conotoxins from Conus geographus venom, it exhibits significant sequence divergence. There are many parallels between this work and previous work on the Ca channel blocker ω-conotoxin MVIIC ((Hillyard et al., 1992; Olivera et al., 1994). Both peptides were synthesized directly from predicted sequences of cDNA clones and were not purified from venom. Both peptides, although being relatively specific, exhibited somewhat broader target specificity than their previously characterized homologs (ω-conotoxin GVIA in the case of ω-MVIIC and μ-conotoxin GIIIA for μ-PIIIA).
The peptide from Conus purpurascens, μ-conotoxin PIIIA, like the μ-conotoxins from Conus geographus (Table1) is highly positively charged and has the same disulfide framework. Of the 16 noncysteine amino acids in μ-conotoxin PIIIA, only five are identical in all four peptides (Arg2, Hyp8, Arg14, Lys17, and Hyp18). Some of the most divergent substitutions involve the replacement of the two aspartic acid residues by Leu3 and Ser13. Sato et al. (1991) reported that individual replacements of these aspartate residues by Ala increased the potency of μ-GIIIA analogs in rat diaphragm muscle two- to threefold. Of great significance is the conservation of Arg14, the amino acid residue previously reported to be critical for activity (Sato et al., 1991;Becker et al., 1992). We have demonstrated that the R14A analog of μ-PIIIA has much lower affinity for muscle Na channels expressed in oocytes and, even at saturating concentrations, is unable to block channel conductance completely. A detailed model of μ-GIIIA with its homologous Arg residue placed within the vestibule of the sodium channel has been published (Dudley et al., 1995); our results with the R14A homolog are consistent with this postulated role for Arg14.
Table 1.
Comparison of sequence of μ-PIII to previously known μ-conotoxins

Z = pyroglutamate; 0 = 4-trans-hydroxyproline. Arrow, Arg residue postulated to be critical for biological activity.
Asterisk indicates amidated C terminus; the amidation for GIIIC was not directly determined but is inferred by homology.
Potentially, some of the most useful results of the present study arise from the differences in affinity and Na channel subtype specificity between μ-conotoxin PIIIA and μ-GIIIA. In amphibians, the inhibition of muscle action potentials by μ-GIIIA is reversible, whereas that by μ-PIIIA is essentially irreversible (see Fig. 3). At high concentrations PIIIA appears to have presynaptic effects analogous to the reduction and delay of transmitter release produced by the focal application of TTX to the presynaptic nerve terminal (Katz and Miledi, 1968). Thus, our working assumption is that high concentrations of PIIIA can attenuate the action potential in the nerve terminal. If the reduction in the endplate current is used as an index of the potency of PIIIA in this regard, it can be estimated from the data in Figure 4 that the IC50 of the toxin is ∼10 μm. These effects of PIIIA, unlike its block of the muscle action potential, are reversible, however. This makes μ-PIIIA a most convenient pharmacological tool for studying synaptic events at the amphibian neuromuscular junction—it is the only known agent to inhibit muscle Na channels irreversibly (and therefore muscle action potential and attendant muscle twitching) in a selective manner.
Compared with μ-GIIIA, μ-conotoxin PIIIA appears to target a wider spectrum of mammalian voltage-gated sodium channel subtypes in the mammalian CNS. μ-PIIIA reversibly blocked the TTX-sensitive rat brain Type II Na channel with an IC50 of 0.64 μm; in contrast, this channel was relatively μ-GIIIA-resistant (IC50 ∼29.4 μm). In addition, μ-conotoxin PIIIA was able to displace a larger fraction of specific [3H]STX binding to high-affinity rat brain sites than could μ-GIIIA. However, not all [3H]STX binding sites could be displaced by μ-PIIIA even at high peptide concentrations, suggesting that μ-PIIIA discriminates among different classes of [3H]STX binding sites in the mammalian CNS.
At the present time, voltage-gated sodium channels are distinguished primarily in situ by their tetrodotoxin sensitivity. The discovery and characterization of μ-conotoxin PIIIA described above provide the basis for subdividing the tetrodotoxin-sensitive sodium channels into three categories, distinguishable by their differential sensitivity to two μ-conotoxins:
(1) Voltage-gated sodium channels that are sensitive to both μ-PIIIA and μ-GIIIA. An example of this subtype is the skeletal muscle subtype in both frog and mammalian systems. The binding data in Figure7 are suggestive that there are CNS sodium channels that also may fit into this category, but this would represent only a minor fraction of the total STX/TTX-sensitive voltage-gated sodium channels present in adult rat brain.
(2) Voltage-gated sodium channels that are sensitive to both TTX and μ-PIIIA but that are significantly more resistant to μ-GIIIA. Rat brain Type II sodium channels apparently belong to this category.
(3) Finally, both the binding and electrophysiological data strongly suggest that a significant fraction of tetrodotoxin-sensitive sodium channels will be resistant to both μ-PIIIA and μ-GIIIA at micromolar concentrations of these toxins. The binding data indicate that a major fraction of the total CNS sodium channels falls into this category.
The discovery of μ-conotoxin PIIIA suggests that the μ-conotoxin peptide family may be broadly distributed in Conus species. Different μ-conotoxin sequence variants that remain to be discovered in the ∼500 species of Conus may be expected to exhibit different affinities for the various subtypes of voltage-gated sodium channels. The μ-conotoxins should prove to be a useful class of ligands for dissecting the role of Na channel subtypes in neurons or circuits when multiple molecular forms of voltage-gated Na channels are present.
Footnotes
This work was supported by Grant PO1 GM 48677 from the United States Public Health Service (USPHS), grants from the SFB Synaptische Interaktionen in neuronalen Zellverbänden (A.B. and H.T.), DOST-ESEP, Philippines (C.Z.F.), and USPHS Grant GM 54710 (K.S.). Rat Type II Na channel mRNA was prepared by Dr. M. Stocker. Some of the binding experiments were performed by J. S. Imperial.
Correspondence should be addressed to Dr. Baldomero M. Olivera, Department of Biology, University of Utah, Salt Lake City, Utah 84112.
REFERENCES
- 1.Becker S, Atherton E, Gordon RD. Synthesis and characterization of μ-conotoxin IIIA. Eur J Biochem. 1989;185:79–84. doi: 10.1111/j.1432-1033.1989.tb15084.x. [DOI] [PubMed] [Google Scholar]
- 2.Becker S, Prusak-Sochaczewski E, Zamponi G, Beck-Sickinger AG, Gordon RD, French RJ. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry. 1992;31:8229–8238. doi: 10.1021/bi00150a016. [DOI] [PubMed] [Google Scholar]
- 3.Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- 4.Colledge CJ, Hunsperger JP, Imperial JS, Hillyard DR. Precursor structure of ω-conotoxin GVIA determined from a cDNA clone. Toxicon. 1992;30:1111–1116. doi: 10.1016/0041-0101(92)90056-b. [DOI] [PubMed] [Google Scholar]
- 5.Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, Moczydlowski E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J Biol Chem. 1985;260:9280–9288. [PubMed] [Google Scholar]
- 6.Doyle DD, Guo Y, Lustig SL, Satin J, Rogart RB, Fozzard HA. Divalent cation competition with [3H]saxitoxin binding to tetrodotoxin-resistant and -sensitive sodium channels. J Gen Physiol. 1993;101:153–182. doi: 10.1085/jgp.101.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley SC, Todt H, Lipkind G, Fozzard HA. A μ-conotoxin-insensitive Na channel mutant: possible localization of a binding site at the outer vestibule. Biophys J. 1995;69:1657–1665. doi: 10.1016/S0006-3495(95)80045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.French RJ, Prusak-Sochaczewski E, Zamponi GW, Becker S, Kularatna AS, Horn R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 1996;16:407–413. doi: 10.1016/s0896-6273(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 9.Gray WR. Disulfide structures of highly bridged peptides: a new strategy for analysis. Protein Sci. 1993;2:1732–1748. doi: 10.1002/pro.5560021017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, Miljanich G, Azimi-Zoonooz A, McIntosh JM, Cruz LJ, Imperial JS, Olivera BM. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins C, Grilley M, Miller C, Shon K-J, Cruz LJ, Gray WR, Dykert J, Rivier J, Yoshikami D, Olivera BM. A new family of Conus peptides targeted to the nicotinic acetylcholine receptor. J Biol Chem. 1995;270:22361–22367. doi: 10.1074/jbc.270.38.22361. [DOI] [PubMed] [Google Scholar]
- 12.Katz B, Miledi R. The effect of local blockage of motor nerve terminals. J Physiol (Lond) 1968;199:729–741. doi: 10.1113/jphysiol.1968.sp008675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McIntosh JM, Hasson A, Spira ME, Li W, Marsh M, Hillyard DR, Olivera BM. A new family of conotoxins which block sodium channels. J Biol Chem. 1995;270:16796–16802. doi: 10.1074/jbc.270.28.16796. [DOI] [PubMed] [Google Scholar]
- 14.Narahashi T, Moore JW, Scott WR. Tetrodotoxin blockage of sodium conductance increase in lobster giant neurons. J Gen Physiol. 1964;47:965–974. doi: 10.1085/jgp.47.5.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noda M, Ikeda T, Kayano T, Suzuki H, Takeshima H, Karusaki M, Takahashi H, Numa S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature. 1986;320:188–191. doi: 10.1038/320188a0. [DOI] [PubMed] [Google Scholar]
- 16.Olivera BM, Gray WR, Zeikus R, McIntosh JM, Varga J, Rivier J, de Santos V, Cruz LJ. Peptide neurotoxins from fish-hunting cone snails. Science. 1985;230:1338–1343. doi: 10.1126/science.4071055. [DOI] [PubMed] [Google Scholar]
- 17.Olivera BM, Hillyard DR, Rivier J, Woodward S, Gray WR, Corpuz G, Cruz LJ. Conotoxins: targeted peptide ligands from snail venoms. In: Hall S, Strichartz G, editors. Marine toxins: origin, structure and molecular pharmacology. American Chemical Society; Washington, DC: 1990. pp. 256–278. [Google Scholar]
- 18.Olivera BM, Miljanich G, Ramachandran J, Adams ME. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu Rev Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- 19.Sato K, Ishida Y, Wakamatsu K, Kato R, Honda H, Ohizumi Y, Nakamura H, Ohya M, Lancelin J-M, Kohda D, Inagaki F. Active site μ-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J Biol Chem. 1991;266:16989–16991. [PubMed] [Google Scholar]
- 20.Sato S, Nakamura H, Ohizumi Y, Kobayashi J, Hirata Y. The amino acid sequences of homologous hydroxyproline containing myotoxins from the marine snail Conus geographus venom. FEBS Lett. 1983;155:277–280. doi: 10.1016/0014-5793(82)80620-0. [DOI] [PubMed] [Google Scholar]
- 21.Shon K, Grilley MM, Marsh M, Yoshikami D, Hall AR, Kurz B, Gray WR, Imperial JS, Hillyard DR, Olivera BM. Purification, characterization, and cloning of the lockjaw peptide from Conus purpurascens venom. Biochemistry. 1995;34:4913–4918. doi: 10.1021/bi00015a002. [DOI] [PubMed] [Google Scholar]
- 22.Stone BL, Gray WR. Occurrence of hydroxyproline in a toxin from the marine snail Conus geographus. Arch Biochem Biophys. 1982;216:756–767. doi: 10.1016/0003-9861(82)90268-5. [DOI] [PubMed] [Google Scholar]
- 23.Stühmer W. Electrophysiological recordings from Xenopus oocytes. Methods Enzymol. 1992;207:319–339. doi: 10.1016/0076-6879(92)07021-f. [DOI] [PubMed] [Google Scholar]
- 24.Trimmer JS, Cooperman SS, Tomiko SA, Zhou J, Crean SM, Boyle MB, Kallen RG, Sheng Z, Barchi L, Sigworth FJ, Goodman RH, Agnew WS, Mandel G. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- 25.Woodward SR, Cruz LJ, Olivera BM, Hillyard DR. Constant and hypervariable regions in conotoxin propeptides. EMBO J. 1990;1:1015–1020. doi: 10.1002/j.1460-2075.1990.tb08204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshikami D, Bagabaldo Z, Olivera BM. The inhibitory effects of omega-conotoxins on calcium channels and synapses. Ann NY Acad Sci. 1989;560:230–248. doi: 10.1111/j.1749-6632.1989.tb24100.x. [DOI] [PubMed] [Google Scholar]