

Abstract
Many cases of autosomal dominant early onset Alzheimer’s disease (AD) result from mutations in the gene encoding presenilin-1 (PS-1). PS-1 is an integral membrane protein expressed ubiquitously in neurons throughout the brain in which it is located primarily in endoplasmic reticulum (ER). Although the pathogenic mechanism of PS-1 mutations is unknown, recent findings suggest that PS mutations render neurons vulnerable to apoptosis. Because increasing evidence indicates that mitochondrial alterations contribute to neuronal death in AD, we tested the hypothesis that PS-1 mutations sensitize neurons to mitochondrial failure. PC12 cell lines expressing a PS-1 mutation (L286V) exhibited increased sensitivity to apoptosis induced by 3-nitropropionic acid (3-NP) and malonate, inhibitors of succinate dehydrogenase, compared with control cell lines and lines overexpressing wild-type PS-1. The apoptosis-enhancing action of mutant PS-1 was prevented by antioxidants (propyl gallate and glutathione), zVAD-fmk, and cyclosporin A, indicating requirements of reactive oxygen species (ROS), caspases, and mitochondrial permeability transition in the cell death process. 3-NP induced a rapid elevation of [Ca2+]i, which was followed by caspase activation, accumulation of ROS, and decreases in mitochondrial reducing potential and transmembrane potential in cells expressing mutant PS-1. The calcium chelator BAPTA AM and agents that block calcium release from ER and influx through voltage-dependent channels prevented mitochondrial ROS accumulation and membrane depolarization and apoptosis. Our data suggest that by perturbing subcellular calcium homeostasis presenilin mutations sensitize neurons to mitochondria-based forms of apoptosis that involve oxidative stress.
Keywords: Alzheimer’s disease, amyloid, caspase, dantrolene, glutathione, malonate, membrane permeability transition, nifedipine, 3-nitropropionic acid, peroxynitrite
Alzheimer’s disease (AD) is characterized by amyloid deposition and associated loss of neurons in brain regions involved in learning and memory processes (for review, see Yankner, 1996; Mattson, 1997a). Mitochondrial dysfunction (Blass, 1993; Kaneko et al., 1995; Benzi and Moretti, 1997; Mattson, 1997b) and oxidative damage to neuronal membranes, protein, and DNA (Markesbery, 1997) may play prominent roles in the neurodegenerative process in AD. Many cases of early onset autosomal dominant inherited forms of AD are caused by mutations in the genes encoding presenilin-1 (PS-1) on chromosome 14 and presenilin-2 (PS-2) on chromosome 1 (for review, seeHardy, 1997). Presenilins are integral membrane proteins with six to eight membrane-spanning domains (Doan et al., 1996; Li and Greenwald, 1996; Lehman et al., 1997); they are expressed in neurons throughout the brain (Cook et al., 1996; Cribbs et al., 1996; Giannakopoulos et al., 1997), and they are localized mainly in the endoplasmic reticulum (ER) (Kovacs et al., 1996; Walter et al., 1996; De Strooper et al., 1997). Recent findings suggest two possible mechanisms whereby presenilin mutations promote neuronal degeneration in AD. One mechanism involves altered processing of β-amyloid precursor protein; cultured cells and transgenic mice expressing PS mutations and fibroblasts from human carriers of presenilin mutations exhibit increased production of an amyloidogenic form of amyloid β-peptide (Aβ1–42) (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996). A second mechanism involves increased sensitivity of neurons to apoptosis; cultured PC12 cells expressing mutant presenilins exhibit increased vulnerability to apoptosis induced by trophic factor withdrawal and exposure to Aβ (Guo et al., 1996, 1997; Wolozin et al., 1996).
Apoptosis is a complex process involving activation of proteases of the caspase family, alterations in plasma membrane phospholipids, and nuclear DNA condensation and fragmentation (Bredesen, 1995; Thompson, 1995). Among the subcellular events that occur during apoptosis, mitochondrial changes may play a pivotal role in the cell death decision; a decrease in mitochondrial energy charge and redox state, loss of transmembrane potential (depolarization), mitochondrial membrane permeability transition (MPT), and release of substances such as calcium and cytochrome C that induce apoptosis occur in several apoptosis paradigms (Kroemer et al., 1997). One approach to studying the role of mitochondrial impairment in neurodegenerative disorders involves the use of mitochondrial toxins (Beal, 1996). 3-Nitropropionic acid (3-NP) is an irreversible inhibitor of complex II (succinate dehydrogenase) that causes degeneration of striatal and hippocampal neurons in humans and rats (Ludolph et al., 1992; Beal et al., 1993;Ming, 1995; Geddes et al., 1996). Cell culture and in vivostudies indicate that 3-NP can induce neuronal apoptosis (Behrens et al., 1995; Pang and Geddes, 1997; Sato et al., 1997) by a mechanism involving oxidative stress (Schulz et al., 1996). We now report that PC12 cells expressing mutant PS-1 are exquisitely sensitive to apoptosis induced by 3-NP, which is associated with elevation of [Ca2+]i and increased oxyradical production. Agents that suppress elevations of cytoplasmic calcium levels and antioxidants prevented the adverse effects of mutant PS-1, suggesting that the apoptosis-enhancing effect of presenilin mutations involves aberrant calcium regulation and increased oxidative stress. These findings suggest mechanistic links between presenilin mutations, perturbed calcium homeostasis, mitochondrial alterations, and neuronal apoptosis in AD.
MATERIALS AND METHODS
Cell lines, experimental treatments, and quantification of apoptosis. PC12 cell lines (Greene and Tischler, 1976) stably expressing human wild-type PS-1 and mutant PS-1 (L286V) were established using methods described in our previous study (Guo et al., 1996). The untransfected parent cell line, a line transfected with empty vector, and a line overexpressing wild-type PS-1 were used as controls. Three lines expressing PS-1L286V were used; Western blot analysis showed that the line overexpressing wild-type PS-1 and each line expressing PS-1L286V expressed PS-1 protein at a level fivefold to eightfold over background levels (Guo et al., 1997). Cells were maintained at 37°C (5% CO2 atmosphere) in RPMI medium supplemented 10% with heat-inactivated horse serum and 5% with heat-inactivated fetal bovine serum. Three days before experiments the cells were incubated in RPMI medium lacking serum and containing 50 ng/ml NGF. 3-NP, glutathione ethyl ester (GSH), propyl gallate, cyclosporin A, Nω-nitro-l-arginine, and 7-nitroindazole were purchased from Sigma (St. Louis, MO) and were prepared as 100–500× stocks in saline solution (0.15 mNaCl). Nifedipine, dantrolene (Sigma), and BAPTA AM (Molecular Probes, Eugene, OR) were prepared as 500× stocks in dimethylsulfoxide. z-VAD-fmk was purchased from Molecular Probes and prepared as a 200× stock in saline solution.
Methods for quantification of apoptosis are detailed in our previous studies (Guo et al., 1997; Kruman et al., 1997). Briefly, cells were stained with the fluorescent DNA-binding dye Hoechst 33342, and the cellular fluorescence associated with cell nuclei was visualized under epifluorescence illumination. Cells with condensed and fragmented nuclei were counted in 40× fields, and the percentage of apoptotic cells per culture was calculated. Counts were made without knowledge of cell line or treatment history.
Measurements of ATP levels and succinate dehydrogenase activity. Cellular ATP levels were quantified using a luciferin and luciferase-based assay as described previously (Mark et al., 1995). Briefly, after exposure to experimental treatments, cells were rinsed with PBS and lysed with 0.2 ml of ATP-releasing buffer (Sigma); 10 μl of the lysate was taken for protein determination. ATP concentrations in lysates were quantified using a CH II ATP bioluminescence assay kit (Boehringer Mannheim, Indianapolis, IN) and a luminometer (Optocomp I, MGM Instruments) according to the manufacturer’s protocols. A standard curve was generated using solutions of known ATP concentrations; samples were diluted so that readings fell within the linear range. ATP levels were calculated as nanomoles of ATP per microgram of protein and normalized to levels in untreated control cultures. Succinate dehydrogenase activity was measured using the method of Lippold (1982). Briefly, cells were exposed for 30 min to vehicle or 3-NP in the presence of 0.1 mm nitro blue tetrazolium. Cells were then rinsed with medium and scraped in dimethylsulfoxide, and relative absorbance was measured using a plate reader. Values were expressed as a percentage of absorbance in parallel vehicle-treated control cultures.
Assessments of mitochondrial function. Mitochondrial transmembrane potential was assessed using the dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1), the uptake of which is directly related to mitochondrial transmembrane potential, using a protocol similar to that reported previously (Moudy et al., 1995; White and Reynolds, 1996). Briefly, cells were incubated for 30 min in the presence of 5 μmdye, washed twice with Locke’s solution (in mm: 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 10 glucose, and 5 HEPES buffer, pH 7.2) and then imaged 20–50 min later. Images of JC-1 fluorescence were acquired using a confocal laser-scanning microscope with excitation at 488 nm and a 510 nm barrier filter. Levels of JC-1 fluorescence (average pixel intensity per cell) were quantified using ImageSpace Software (Molecular Dynamics, Sunnyvale, CA). The conversion of the dye 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT) to formazan crystals was used as a measure of mitochondrial reducing potential (energy charge and redox state; Musser and Oseroff, 1994;Shearman et al., 1994). Formazan production was measured 40–60 min after adding MTT (1 mg/ml) to the incubation medium and after solubilization of the crystals in dimethylsulfoxide; absorbance (592 nm) was quantified using a plate reader.
Measurements of reactive oxygen species. Levels of cellular oxidative stress were measured using the fluorescent probes 2,7-dichlorofluorescin diacetate (DCF) and dihydrorhodamine 123 (DHR123) as described previously (Goodman and Mattson, 1994; Mattson et al., 1997; Kruman et al., 1998). Briefly, cells were incubated for 30 min in the presence of 50 μm DCF or for 30 min in the presence of 10 μm DHR123, followed by washing in HBSS containing 10 mm HEPES buffer and 10 mmglucose. Cells were loaded with the dyes during the last 30 min of treatment with 3-NP. Cells were imaged using a confocal laser-scanning microscope (Molecular Dynamics) coupled to an inverted microscope (Nikon). Cells were located under bright-field optics and then scanned once with the laser (488 nm excitation and 510 nm emission); cells were not scanned more than once to avoid artifacts resulting from light-induced oxidation of the dyes. The laser beam intensity and photodetector sensitivity were held constant across cultures to allow quantitative comparisons of relative fluorescence intensity of cells between treatment groups. Values of cellular fluorescence (average pixel intensity per cell) were obtained using the software supplied by the manufacturer (Molecular Dynamics). DCF is trapped mainly in the cytoplasm and is oxidized by several ROS, most notably hydrogen peroxide (Page et al., 1993). The dye dihydrorhodamine 123 (DHR) enters mitochondria and fluoresces when oxidized by ROS, particularly peroxynitrite, to the positively charged rhodamine 123 derivative (Kooy et al., 1994; Mattson et al., 1997).
Measurement of intracellular free calcium levels. The [Ca2+]i in individual PC12 cells was quantified by fluorescence imaging of the Ca2+indicator dye fura-2 as described previously (Mattson et al., 1995a;Guo et al., 1996, 1997). Cultures were incubated for 30–40 min in the presence of 5 μm fura-2 AM (Molecular Probes), followed by two washes (2 ml/wash) with fresh medium and a 40–60 min incubation before imaging. Immediately before imaging, the normal culture medium was replaced with HBSS (Life Technologies, Gaithersburg, MD) containing 10 mm HEPES buffer and 10 mm glucose. Cells were imaged on a Zeiss Axiovert inverted microscope (40× oil immersion objective) coupled to an Attofluor imaging system. The ratio of the fluorescence emission using two different excitation wavelengths (340 and 380 nm) was used to determine [Ca2+]i according to the formula: [Ca2+]i =Kd[(R − Rmin)/(Rmax − R)](F0/Fs). The average [Ca2+]i in 10–20 cells per microscope field was quantified in at least six separate cultures per treatment condition.
Measurement of caspase activity. Caspase activity was assessed using the caspase-3 substrate peptide DEVD-biotin using a method modified from that described previously (Armstrong et al., 1997;Yakovlev et al., 1997) and adapted for in situ analysis. Briefly, at designated time points after exposure of cultures to 3-NP, cells were exposed for 10 min to Locke’s solution containing 0.01% digitonin. Cells were then incubated for 20 min in the presence of 10 μg/ml DEVD-biotin (Calbiochem, La Jolla, CA), washed three times with PBS (2 ml/wash), and fixed for 30 min in a cold solution of 4% paraformaldehyde in PBS. Cells were then incubated for 30 min in PBS containing 5 μg/ml Oregon green streptavidin (Molecular Probes) and were washed twice with PBS. Images of cellular fluorescence, corresponding to conjugates of activated caspase-3 with DEVD-biotin, were acquired using a confocal laser scanning microscope and levels of fluorescence (average pixel intensity per cell) were quantified.
RESULTS
PC12 Cells expressing mutant PS-1 are hypersensitive to 3-NP-induced apoptosis and exhibit increased oxyradical production
Basal levels of apoptosis under the culture conditions used were low (2–3%) in each of the PC12 cell lines used in the present study (Fig. 1A). Exposure of untransfected and vector-transfected control PC12 cells lines and a line overexpressing wild-type PS-1 to concentrations of 3-NP from 0.2–10 mm had no significant effect on levels of apoptosis during a 24 hr exposure period (Fig. 1A). In contrast to the control cell lines, exposure of PC12 cells expressing the L286V mutation to increasing concentrations of 3-NP resulted in a concentration-dependent induction of apoptosis with a significant increase occurring with 0.2 mm 3-NP (10% apoptosis) and a maximum of ∼25–30% apoptosis in cultures exposed to 20 mm 3-NP; the dose achieving half-maximal effect was ∼1 mm (Fig. 1A). Two other clonal lines expressing PS-1L286V also exhibited a similar increased sensitivity to 3-NP-induced apoptosis (data not shown). A time course analysis of apoptosis after exposure of each cell line to 2 mm 3-NP revealed only a small (5–10%) increase in levels of apoptosis in untransfected cells, vector-transfected cells and cells overexpressing wild-type PS-1 during a 48 hr exposure period (Fig.1B). In contrast, levels of apoptosis increased to ∼40% during a 48 hr exposure to 3-NP in cells expressing mutant PS-1 (Fig. 1B). To determine whether apoptosis induced by 3-NP in cells expressing mutant PS-1 was specifically related to inhibition of succinate dehydrogenase, we used malonate, another inhibitor of succinate dehydrogenase with a structure different from 3-NP. Malonate induced a significant increase in apoptosis in cells expressing mutant PS-1, but not in control cell lines and lines overexpressing wild-type PS-1 (Fig. 1C).
Fig. 1.
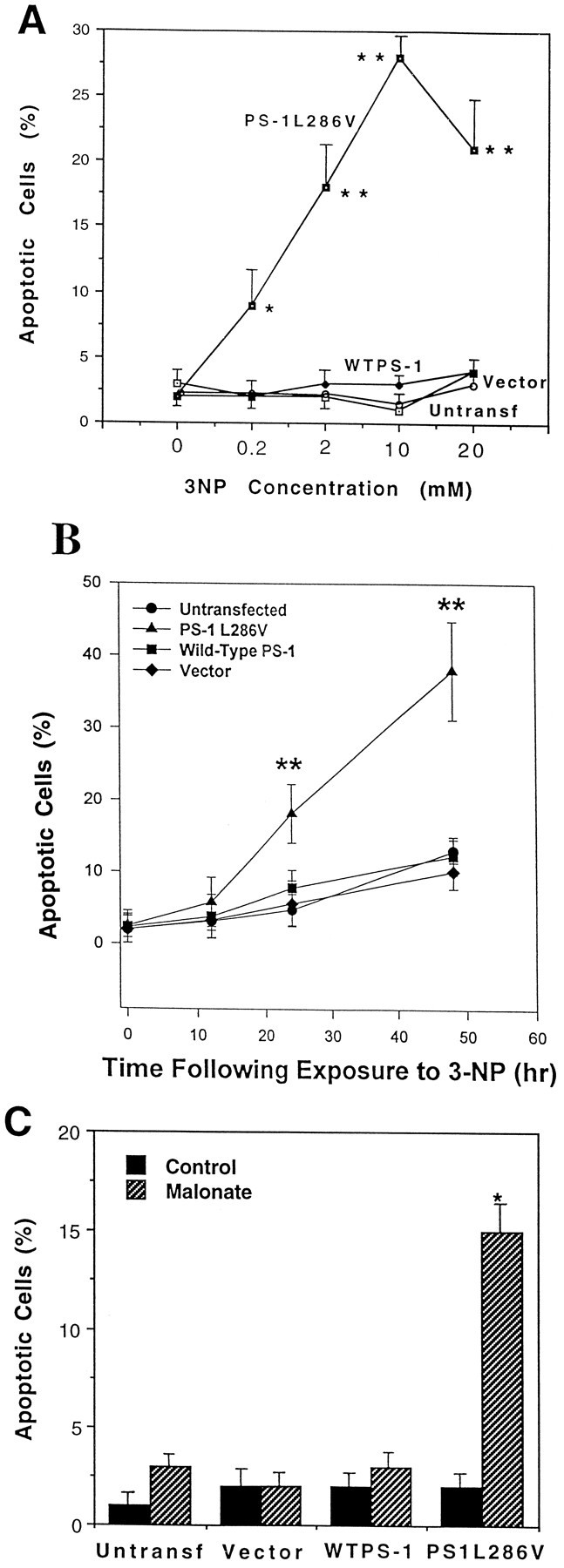
PC12 cells expressing mutant PS-1 exhibit increased sensitivity to apoptosis induced by mitochondrial toxins.A, Untransfected and vector-transfected control PC12 cells lines and lines overexpressing wild-type PS-1 or mutant (L286V) PS-1 were exposed for 24 hr to the indicated concentrations of 3-NP and the percentages of cells with apoptotic nuclei were quantified. Values are the mean and SEM of determinations made in six to eight cultures. **p< 0.01 compared with each of the other values at that concentration of 3-NP (ANOVA with Scheffe’s post hoc test).B, Untransfected and vector-transfected control PC12 cells lines and lines overexpressing wild-type PS-1 or mutant (L286V) PS-1 were exposed to 2 mm 3-NP for the indicated time periods, and the percentages of cells with apoptotic nuclei were quantified. Values are the mean and SEM of determinations made in at least six cultures. **p < 0.01 compared with each of the other values at that time point (ANOVA with Scheffe’spost hoc test). C, Untransfected and vector-transfected control PC12 cells lines and lines overexpressing wild-type PS-1 or mutant (L286V) PS-1 were exposed for 24 hr to saline (Control) or 100 mm malonate and the percentages of cells with apoptotic nuclei were quantified. Values are the mean and SEM of determinations made in six cultures. *p < 0.01 compared with each of the other values (ANOVA with Scheffe’s post hoc test).
Previous studies provided evidence that the neurotoxicity of 3-NP (Schulz et al., 1996) and the apoptosis-enhancing action of mutant PS-1 (Guo et al., 1997) are associated with increased generation of oxyradicals. We therefore used the fluorescent probes DCF and DHR 123 to quantify relative levels of ROS in the different cell lines after exposure to 3-NP. Exposure of untransfected and vector-transfected control PC12 cells lines, and a line overexpressing wild-type PS-1, to 2 mm 3-NP resulted in no increase in the levels of DCF fluorescence (Fig. 2A) or DHR 123 fluorescence (Fig. 2B) during a 2 hr exposure period. In contrast, levels of DCF and DHR 123 fluorescence increased threefold to fourfold during 2 hr exposures to 3-NP in cells expressing mutant PS-1 (Fig. 2). The increases in levels of DCF and DHR 123 fluorescence in cells expressing mutant PS-1 occurred within 30–60 min of exposure to 3-NP. Similar results were obtained when cells were loaded with DCF or DHR123 before exposure to 3-NP for 30 and 60 min (data not shown). These data indicate that one consequence of mutant PS-1 expression is enhanced generation of ROS in response to a mitochondrial toxin.
Fig. 2.
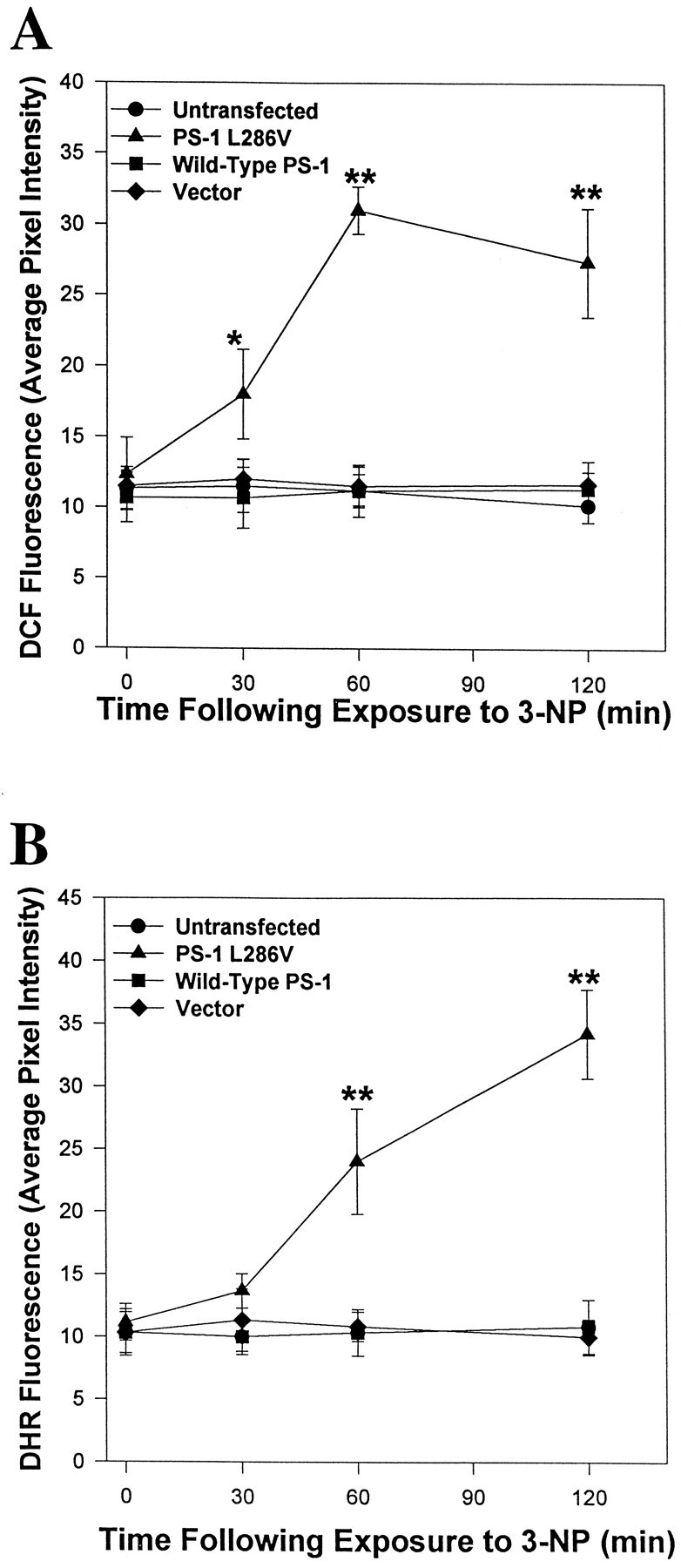
Accumulation of ROS occurs in PC12 cells expressing mutant PS-1, but not in control cell lines, after exposure to 3-NP. Untransfected and vector-transfected control PC12 cells lines and lines overexpressing wild-type PS-1 or mutant (L286V) PS-1 were exposed to 2 mm3-NP for the indicated time periods and levels of DCF fluorescence (A) or DHR 123 fluorescence (B) were quantified. Values are the mean and SEM of determinations made in six to eight cultures (20–30 randomly chosen cells analyzed per culture). *p < 0.05 or **p < 0.01 compared with each of the other values at that time point (ANOVA with Scheffe’s post hoctests).
Effects of 3-NP on succinate dehydrogenase activity and cellular ATP levels
One possible explanation for the increased vulnerability of PC12 cells expressing mutant PS-1 to 3NP toxicity is that 3-NP inhibits succinate dehydrogenase to a greater extent in those cells. We therefore assessed succinate dehydrogenase activity 30 min after exposure to 2 mm 3-NP in untransfected cells, in cells overexpressing wild-type PS-1, and in cells overexpressing mutant PS-1. 3-NP caused significant decreases in levels of succinate dehydrogenase activity in all three cell lines; the magnitude of the decrease ranged from 25–40% and was not significantly different among the cell lines (Fig. 3A). We next measured ATP levels at 30 and 120 min after exposure of each cell line to 2 mm 3-NP. Levels of ATP were maintained at baseline levels during the first 30 min of exposure in all three cell lines (Fig. 3B). However, at 120 min after exposure to 3-NP, ATP levels were significantly decreased to 60% of baseline levels in cells expressing mutant PS-1 but were not decreased in untransfected cells or cells overexpressing wild-type PS-1 (Fig. 3B).
Fig. 3.
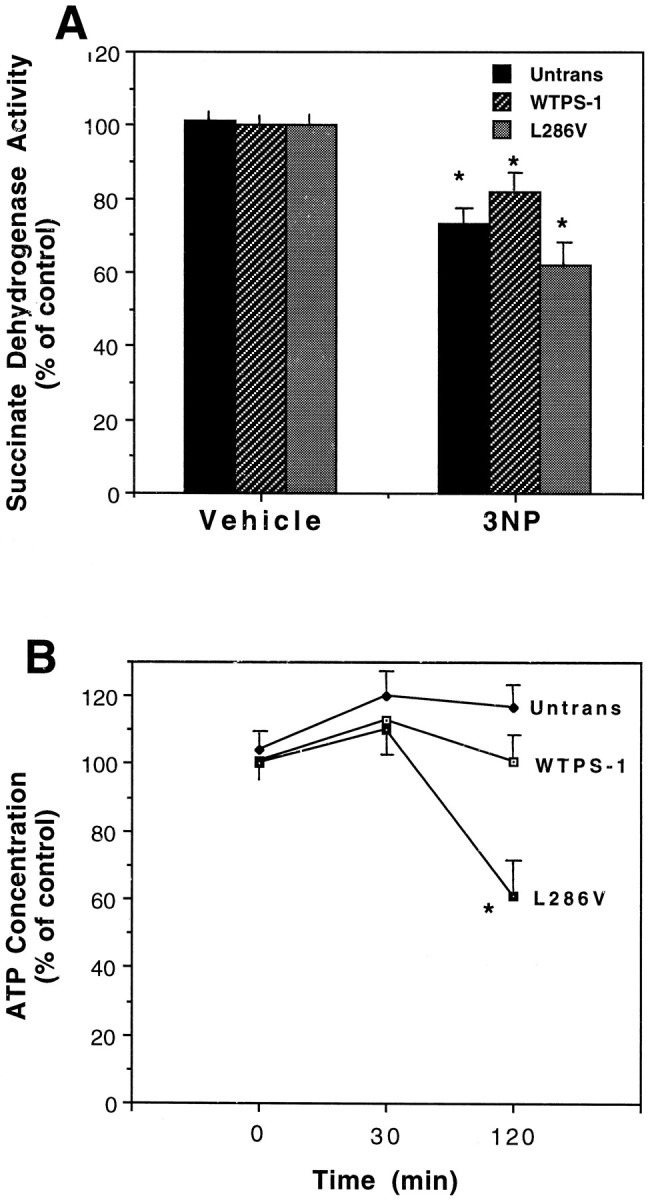
Effects of 3-NP on succinate dehydrogenase activity and ATP levels in PC12 cells expressing mutant PS-1.A, Cultures of untransfected PC12 cells and clones overexpressing either wild-type PS-1 or the L286V PS-1 mutation were exposed to 2 mm 3-NP for 30 min and levels of succinate dehydrogenase activity were measured. Values are the mean and SEM of determinations made in five to seven cultures. *p< 0.02 compared with corresponding value in vehicle-treated cultures.B, Cultures of untransfected PC12 cells and clones overexpressing either wild-type PS-1 or the L286V PS-1 mutation were exposed to 2 mm 3-NP for the indicated time periods and levels of cellular ATP were quantified. Values are expressed as a percentage of basal levels and represent the mean and SEM of determinations made in four to six cultures. Basal ATP levels were: untransfected cells, 19.4 ± 8 nmol/mg protein; cells overexpressing wild-type PS-1, 18.3 ± 6 nmol/mg protein; cells overexpressing mutant PS-1, 19.2 ± 6 nmol/mg protein. *p < 0.01 compared with values for untransfected cells and cells overexpressing wild-type PS-1.
Involvement of elevated [Ca2+]i, oxyradical production, and caspase activation in the apoptosis-enhancing action of mutant PS-1
It was previously reported that PC12 cells expressing mutant PS-1 exhibit enhanced elevation of [Ca2+]iafter exposure to agonists that induce calcium release from IP3-sensitive ER stores (Guo et al., 1996) and after exposure to a concentration of Aβ that induces apoptosis (Guo et al., 1997, 1998). We therefore monitored [Ca2+]i before and after exposure to 3-NP in the different cell lines by fluorescence ratio imaging of the calcium indicator dye fura-2. The basal [Ca2+]i was ∼80–100 nmin each cell line examined. After exposure of cells expressing mutant PS-1 to 2 mm 3-NP, the [Ca2+]i rose rapidly and progressively, with levels reaching 180 nm during the course of a 10 min period (Fig.4A). In control cell lines and the line overexpressing wild-type PS-1, 3-NP did not induce an increase of [Ca2+]i during a 10 min exposure period (Fig. 4A,B).
Fig. 4.
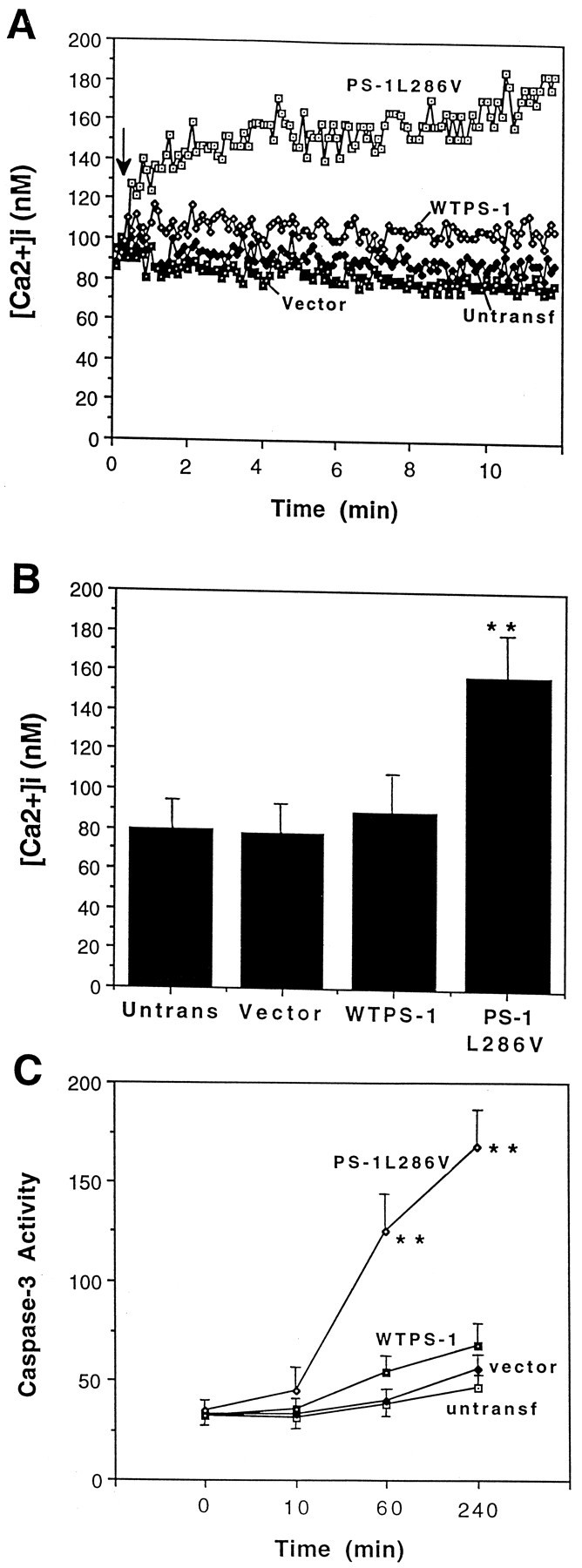
3-NP induces a rapid increase in intracellular free calcium levels in PC12 cells expressing mutant PS-1, which is followed by caspase-3 activation. A, [Ca2+]i was monitored before and after exposure to 2 mm 3-NP in the indicated cell lines; thearrow indicates the time point at which 3-NP was added to the culture medium. Values are the mean of 35–70 cells in three to six separate cultures.B, [Ca2+]i was measured 5 min after exposure of cells of the indicated lines to 2 mm 3-NP. Values are the mean and SEM of determinations made in at least six cultures per condition. **p < 0.01 compared with each of the other values (ANOVA with Scheffe’spost hoc tests). C, Cultures of the indicated lines were exposed for the indicated time periods to 2 mm 3-NP and levels of caspase-3 activity were quantified. Values are expressed as DEVD fluorescence (average pixel intensity per cell) and are the mean and SEM of determinations made in four separate cultures. **p < 0.01 compared with the values for each of the other three cell lines.
To determine whether the elevation of [Ca2+]i and ROS production was causally linked to apoptosis in cells expressing mutant PS-1, we examined the effects of agents that suppress elevations of [Ca2+]i and antioxidants on 3-NP-induced apoptosis. The intracellular calcium chelator BAPTA AM largely prevented apoptosis induced by 3-NP (Table1). Similarly, nifedipine (a blocker of L-type voltage-dependent calcium channels) and dantrolene (a blocker of calcium release from ER stores) significantly attenuated 3-NP-induced apoptosis. GSH and propyl gallate, two antioxidants previously reported to protect cultured neurons against apoptosis induced by Aβ (Mark et al., 1995; Kruman et al., 1997), prevented 3-NP-induced apoptosis in cells expressing mutant PS-1 (Table 1). Because increased [Ca2+]i can induce nitric oxide production and generation of peroxynitrite (Bredt and Snyder, 1992;Kooy and Royall, 1994), and because peroxynitrite can oxidize DHR 123 (Kooy et al., 1994), we ascertained the effects of inhibitors of nitric oxide synthase (NOS) on 3-NP-induced apoptosis in cells expressing mutant PS-1. Levels of apoptosis 24 hr after treatment were untreated control cultures, 2 ± 1%; 2 mm 3-NP, 14 ± 3%; 100 μmNω-nitro-l-arginine plus 2 mm 3-NP, 9 ± 2% (p < 0.05 vs 3-NP alone); and 100 μm 7-nitroindazole plus 2 mm 3-NP, 8 ± 3% (p < 0.05 vs 3-NP alone) (n = 6 separate cultures). In an additional experiment, levels of DHR fluorescence were quantified (in cells expressing mutant PS-1) 2 hr after exposure to 2 mm 3-NP alone or in combination withNω-nitro-l-arginine or 7-nitroindazole. The NOS inhibitors did not block the 3-NP-induced increase in DHR 123 levels (data not shown). Thus, nitric oxide plays an important role in 3-NP-induced apoptosis, but is clearly not the only contributor to the increased ROS accumulation induced by 3-NP.
Table 1.
Evidence for the involvement of calcium influx, ROS production, mitochondrial membrane permeability transition, and caspase activation in 3NP-induced apoptosis in PC12 cells expressing mutant PS-1
| Vehicle | BAPTA | Percentage of cells with apoptotic nuclei | zVAD | |||||
|---|---|---|---|---|---|---|---|---|
| Nif | Dant | Cys A | GSH | PG | ||||
| Control | 6 ± 3.2 | 5 ± 3.5 | 4 ± 1.1 | 5 ± 1.3 | 5 ± 2.4 | 4 ± 0.7 | 7 ± 1.7 | 9 ± 3.4 |
| 3NP | 42 ± 9.3* | 13 ± 4.2** | 11 ± 3.6** | 12 ± 5.3** | 12 ± 6.4** | 11 ± 5.4** | 11 ± 7.5** | 15 ± 4.7** |
Cultures were exposed to 2 mM 3NP for 48 hr and levels of apoptosis were quantified. Vehicle refers to saline. BAPTA, 5 μm BAPTA-AM; Nif, 10 μm nifedipine; Dant, 1 μm dantrolene; Cys A, 1 μm cyclosporin A; GSH, 1 mm glutathione ethyl ester; PG, 10 μmpropyl gallate; zVAD, 10 μm zVAD-fmk. Cultures were pretreated for 1 hr with the indicated agents before exposure to 3NP, and the agents were present during the exposure to 3NP. Cultures were exposed to 2 mm 3NP for 48 hr. Values are the mean and SEM of determinations made in six to eight cultures.
*p < 0.01 compared to value for vehicle-treated cultures.
**p < 0.01 compared to value for 3-NP-treated cultures. ANOVA with Scheffe’s post hoc tests.
Caspases are believed to play important roles in the effector phase of apoptosis in many paradigms (Miller, 1997; Yuan, 1997). Measurements of caspase-3 activation after exposure of cells to 3-NP revealed only a small and transient activation of this caspase at the 1 and 4 hr time points in control PC12 cell lines (untransfected and vector-transfected) and lines overexpressing wild-type PS-1 (Fig.4C). In contrast, a marked and sustained activation of caspase-3 occurred after exposure to 3-NP in PC12 cells expressing mutant PS-1 (Fig. 4C). The broad-spectrum caspase inhibitor zVAD-fmk prevented apoptosis induced by 3-NP in cells expressing mutant PS-1 (Table 1), indicating a causal role for caspase activation in the cell death process.
Evidence for the involvement of decreases in mitochondrial transmembrane potential and reducing potential and permeability transition in the apoptosis-enhancing action of mutant PS-1
A decrease in mitochondrial transmembrane potential has been shown to be associated with the effector phase of apoptosis in many different systems (Zamzami et al., 1995; Kroemer et al., 1997). We used the dye JC-1 to examine the effects of 3-NP on relative mitochondrial transmembrane potential in the different lines of PC12 cells. Levels of JC-1 fluorescence were essentially unchanged 18 hr after exposure to 2 mm 3-NP in control cell lines and the line overexpressing wild-type PS-1 (Fig. 5A). In contrast, the level of JC-1 fluorescence was decreased to <40% of the control level 18 hr after exposure to 3-NP in cells expressing mutant PS-1 (Fig. 5A). The decrease of JC-1 fluorescence in cells expressing mutant PS-1 caused by 3-NP was delayed because no decrease in JC-1 fluorescence occurred during the first 2 hr of exposure (Fig.5B). Levels of MTT reduction, a measure of mitochondrial reducing potential, were decreased in a concentration-dependent manner by 3-NP in PC12 cells expressing mutant PS-1 (Fig. 5C). In contrast, the level of MTT reduction was not significantly changed after exposure to 3-NP in control PC12 cell lines and in cells expressing wild-type PS-1 (Fig. 5C; Table2). Malonate also caused a significant decrease in MTT reduction in cells expressing mutant PS-1 but not in control cell lines (Fig. 5D).
Fig. 5.

Mitochondrial toxins cause decreases in mitochondrial transmembrane potential and energy charge in PC12 cells expressing mutant PS-1, but not in control cell lines. A, B, Indicated cell lines were exposed for 18 hr (A) or the indicated time periods (B) to 2 mm 3-NP, and levels of JC-1 fluorescence were quantified. Values are the mean and SEM of determinations made in six cultures. **p < 0.01 compared with each of the other values (ANOVA with Scheffe’spost hoc test). C, Indicated cell lines were exposed to increasing concentrations of 3-NP for 24 hr and levels of MTT reduction were quantified. Values are the mean and SEM of determinations made in six cultures. *p < 0.05, **p < 0.01 compared with each of the other values at the corresponding 3-NP concentration (ANOVA with Scheffe’spost hoc test). D, Indicated cell lines were exposed to 100 mm malonate for 24 hr and levels of MTT reduction were quantified. Values are the mean and SEM of determinations made in six cultures. **p < 0.01 compared with each of the other values (ANOVA with Scheffe’spost hoc test).
Table 2.
Evidence for the involvement of calcium influx and ROS production in mitochondrial dysfunction following exposure of PC12 cells expressing mutant PS-1 to 3NP
| Vehicle | BAPTA | MTT Reduction (% of control) | PG | Z-VAD | ||||
|---|---|---|---|---|---|---|---|---|
| Nif | Dant | Cys A | GSH | |||||
| Control | 98 ± 3.2 | 96 ± 4.5 | 93 ± 5.3 | 91 ± 8.6 | 89 ± 9.5 | 98 ± 7.4 | 96 ± 3.2 | 97 ± 6.3 |
| 3NP | 51 ± 8.5* | 94 ± 5.8** | 92 ± 10** | 89 ± 6.9** | 92 ± 4.5** | 90 ± 7.2** | 86 ± 7.4** | 96 ± 3.6** |
See Table 1 legend for treatment concentrations and exposure conditions. Values are the mean and SEM of determinations made in 8–12 cultures. The basal (control) level of MTT reduction was 250 ± 8 absorbance units per culture.
*p < 0.01 compared to value for vehicle treated cultures.
**p < 0.01 compared to value for 3-NP treated cultures. ANOVA with Scheffe’s post hoc tests.
Cyclosporin A is an immunosuppressive compound that has been shown to block the MPT (Marchetti et al., 1996; Zamzami et al., 1996). When PC12 cells expressing mutant PS-1 were cotreated with cyclosporin A and 3-NP, apoptosis and impairment of mitochondrial function were largely prevented (Tables 1, 2) suggesting links between the MPT, mitochondrial damage, and nuclear apoptosis. The effect of cyclosporin A was apparently attributable to specific blockade of the MPT because FK 506, an inhibitor of calcineurin had no effect on 3-NP-induced apoptosis (data not shown).
Elevations of [Ca2+]i are causally linked to ROS production and mitochondrial alterations and precede caspase activation in the apoptotic pathway
To better understand the mechanism whereby mutant PS-1 sensitizes PC12 cells to apoptosis induced by 3-NP and to reveal the ordering of events in this apoptotic paradigm, we examined the effects of various pharmacological agents on ROS production, mitochondrial dysfunction, and apoptosis induced by 3-NP in cells expressing mutant PS-1. BAPTA AM, nifedipine, and dantrolene each significantly attenuated the decrease in MTT reduction otherwise induced by 3-NP (Table 2), indicating a key role for elevation of intracellular calcium levels in 3-NP-induced mitochondrial dysfunction. The antioxidants GSH and propyl gallate also prevented the decrease in levels of MTT reduction after exposure of cells to 3-NP (Table 2), indicating a contribution of oxyradicals to the mitochondrial dysfunction induced by 3-NP. Interestingly, levels of MTT reduction were unchanged after 3-NP exposure in cells incubated in the presence of zVAD-fmk, indicating a role for caspases in 3-NP-induced mitochondrial impairment.
We next addressed the question of whether elevation of [Ca2+]i, caspase activation, and/or MPT contribute to ROS production. Removal of extracellular calcium, BAPTA AM, nifedipine, and dantrolene each prevented the increase in DCF fluorescence and DHR 123 fluorescence otherwise induced by 3-NP (Table 3). As expected, the antioxidants GSH and propyl gallate also suppressed ROS accumulation. In contrast, zVAD-fmk was ineffective in preventing the 3-NP-induced increases of DCF and DHR 123 fluorescence (Table 3), indicating that caspase activation was not necessary for generation of ROS. zVAD-fmk also had no effect on the rapid elevation of [Ca2+]i induced by 3-NP, indicating that caspases did not play a role in the initial disruption of calcium homeostasis caused by 3-NP ([Ca2+]i 10 min after exposure to 2 mm 3-NP were control, 75 ± 5 nm; 3-NP, 169 ± 5 nm; zVAD, 83 ± 6 nm; and zVAD plus 3-NP, 162 ± 4 nm (mean + SEM; n = 5–6 cultures). Cyclosporin A attenuated 3-NP-induced increases of DCF and DHR 123 fluorescence (Table 3) but had no effect on the rapid elevation of [Ca2+]i induced by 3-NP (data not shown).
Table 3.
Pharmacological characterization of the involvement of calcium, mitochondrial permeability transition, and caspase activation in 3NP-induced generation of ROS in PC12 cells expressing mutant PS-1
| Vehicle | 0 [Ca]o | BAPTA | Nif | Dant | GSH | PG | Cys A | |
|---|---|---|---|---|---|---|---|---|
| DCF Fluorescence (average pixel intensity per cell) | ||||||||
| Control | 10 ± 1.3 | 11 ± 1.5 | 9 ± 2.1 | 12 ± 1.6 | 12 ± 2.2 | 11 ± 1.9 | 11 ± 2.4 | 9 ± 1.7 |
| 3-NP | 25 ± 4.3* | 11 ± 1.5** | 12 ± 2.5** | 11 ± 1.3** | 14 ± 3.5** | 12 ± 1.6** | 11 ± 2.1** | 13 ± 2.1* |
| DHR 123 Fluorescence | ||||||||
| Control | 11 ± 1.6 | 10 ± 1.5 | 10 ± 2.3 | 11 ± 2.1 | 11 ± 2.4 | 13 ± 2.9 | 12 ± 1.5 | 12 ± 1.9 |
| 3-NP | 29 ± 4.1* | 10 ± 2.7** | 13 ± 2.1** | 10 ± 1.4** | 19 ± 2.6** | 14 ± 2.2** | 13 ± 3.4** | 12 ± 3.6** |
Cultures were pretreated for 1 hr with the indicated agents and were then exposed to 2 mm 3NP for 2 hr, at which time levels of DCF fluorescence and DHR 123 fluorescence were quantified. Values are the mean and SEM of determinations made in at least six cultures.
*p < 0.01 compared to value for vehicle treated-control cultures.
**p < 0.01 compared to value for 3-NP-treated cultures. (ANOVA with Scheffe’s post hoc tests). 3NP, 2 mm 3NP; Vehicle, 0.2% dimethylsulfoxide; 0 [Ca]o, Locke’s solution lacking added calcium and containing 0.5 mm EGTA; BAPTA, 5 μm BAPTA-AM; Nif, 10 μm nifedipine; Dant, 1 μm dantrolene; GSH, 1 mm glutathione ethyl ester; PG, 10 μmpropyl gallate; Cys A, 1 μm cyclosporine A; zVAD, 10 μm zVAD-fmk.
DISCUSSION
Expression of a PS-1 mutation in cultured neural cells rendered the cells vulnerable to apoptosis induced by mitochondrial toxins. The mechanism underlying the endangering action of mutant PS-1 involves destabilization of cellular calcium homeostasis and increased ROS production. Increases of [Ca2+]i and ROS occurred within several to 30 min of exposure to 3-NP and were followed several hours later by mitochondrial failure, as indicated by membrane depolarization and decreased MTT reduction. Untransfected and vector-transfected control PC12 cell lines and a line overexpressing wild-type PS-1 were resistant to apoptosis induced by 3-NP and did not exhibit increased [Ca2+]i and ROS or mitochondrial membrane depolarization. Pharmacological blockade of calcium influx and treatment of cells with the calcium chelator BAPTA AM prevented 3-NP-induced ROS accumulation, decreased mitochondrial energy charge, and significantly decreased apoptosis in cells expressing mutant PS-1, indicating that elevation of [Ca2+]i was causally linked to each subsequent event.
Studies of cultured primary neurons and cell lines showed LD50s for 3-NP and malonate in the ranges of 0.1–5 and 10–100 mm, respectively (Behrens et al., 1995; Zeevalk et al., 1995; Fink et al., 1996; Pang et al., 1997). These concentrations are similar to those effective in inducing apoptosis in PC12 cells expressing mutant PS-1 in the present study. Previous studies of cultured rat hippocampal neurons (Mattson et al., 1993) and PC12 cells (Luo et al., 1997) showed that cyanide and other mitochondrial uncouplers can induce increases of [Ca2+]i that are causally linked to cell death. Our data suggest that the initial [Ca2+]i increase after exposure of cells expressing mutant PS-1 to 3-NP is not the result of mitochondrial membrane depolarization because JC-1 fluorescence was not decreased during the first 2 hr of exposure. One possible explanation is that an early increase in ROS induces calcium release from mitochondria with preservation of mitochondrial transmembrane potential (Richter, 1997). Although 3-NP inhibited succinate dehydrogenase to a similar extent in cells expressing mutant PS-1 as compared with control cell lines, ATP depletion was exacerbated in cells expressing mutant PS-1. Depletion of ATP levels after exposure of cultured cells to cyanide or 3-NP lagged behind the elevation of [Ca2+]i, consistent with previous studies (Mattson et al., 1993; Erecinska and Nelson, 1994; Luo et al., 1997; Pang and Geddes, 1997), and therefore is unlikely to play a major role in the rapid increase of [Ca2+]i induced by 3-NP. However, ATP depletion may contribute to delayed calcium release from ER and influx through plasma membrane voltage-dependent channels as the result of impaired activity of the ER Ca2+-ATPase, and the plasma membrane Na+–K+-ATPase and Ca2+-ATPase. The latter scenario is consistent with data showing that dantrolene and nifedipine protect PC12 cells expressing mutant PS-1 against apoptosis induced by 3-NP. Indeed, PC12 cells expressing mutant PS-1 (Guo et al., 1996) and fibroblasts from human carriers of PS-1 mutations (Ito et al., 1994) exhibit enhanced calcium release from ER stores in response to a variety of stimuli including cholinergic agonists and Aβ. Such enhanced Ca2+ release from ER may be sufficient to render the cells vulnerable to a level of mitochondrial compromise that would not kill normal cells.
The oxidation of the probes DCF and DHR 123 in intact cells may result from the actions of peroxides and peroxynitrite, respectively (Page et al., 1993; Goodman and Mattson, 1994; Kooy et al., 1994; Mattson et al., 1997), although these fluorescent probes can also be oxidized by other ROS. We found that two inhibitors of NOS significantly attenuated apoptosis induced by 3-NP in cells expressing mutant PS-1, suggesting a contribution of nitric oxide to the apoptotic process. The finding that elevation of [Ca2+]i preceded ROS production after exposure to 3-NP is consistent with roles for nitric oxide and peroxynitrite in 3-NP-induced apoptosis, because Ca2+ is a potent activator of NOS (Bredt and Snyder, 1992). Indeed, staurosporine-induced apoptosis in PC12 cells apparently involves peroxynitrite production secondary to elevation of [Ca2+]i (Kruman et al., 1998). Elevations of [Ca2+]i in neurons can result in generation of ROS, including superoxide anion radical (Lafon-Cazal et al., 1993), hydrogen peroxide (Mattson et al., 1995b), and peroxynitrite (Deliconstantinos and Villiotou, 1996). Our data therefore suggest that PS-1 mutations may promote nitric oxide production and ROS accumulation in a calcium-dependent manner. The ROS, in turn, appear to play a central role in subsequent impairment of mitochondrial function and nuclear apoptosis induced by 3-NP, because the antioxidants GSH and propyl gallate prevented the decrease in MTT reduction and nuclear condensation and fragmentation otherwise induced by 3-NP. Indeed, hydrogen peroxide (Whittemore et al., 1994; Hoyt et al., 1997) and peroxynitrite (Estevez et al., 1995; Mattson et al., 1997) were previously shown to impair mitochondrial function and induce apoptosis in cultured neurons. ROS are implicated in the effector phase of apoptosis in many different physiological and pathophysiological settings (Hockenberry et al., 1993; Kane et al., 1993; Kruman et al., 1997); ROS can disrupt mitochondrial calcium homeostasis and promote MPT (Richter, 1993; Packer and Murphy, 1995; Brorson et al., 1997).
Cyclosporin A, an inhibitor of MPT (Petronilli et al., 1994), prevented 3-NP-induced apoptosis in PC12 cells expressing mutant PS-1. The ability of cyclosporin A a to attenuate 3-NP-induced ROS production and preserve mitochondrial reducing potential in cells expressing mutant PS-1 suggests that mutant PS-1 perpetuates a vicious cycle in which perturbed calcium homeostasis promotes MPT and ROS production resulting in further disruption of calcium homeostasis and nuclear apoptosis (Fig. 6). Our data are consistent with previous work showing that cyclosporin A prevents mitochondrial depolarization in cultured cortical neurons exposed to NMDA (Niemenin et al., 1996). The finding that cyclosporin A attenuates accumulation of ROS is at first approximation in contradiction to the data showing that ROS accumulation occurs before loss of transmembrane potential as measured by the JC-1 method. One possible explanation of these data is that there is a time lag between MPT and loss of transmembrane potential as measured by the JC-1. A second possibility is that cyclosporin A suppresses ROS production by a mechanism other than blocking MPT. Inhibition of calcineurin is a well documented action of cyclosporin A, and recent findings suggest a role for calcineurin in several different models of apoptosis (Shibasaki and Mckeon, 1995). However, 3-NP-induced apoptosis in the present study was not blocked by FK 506, another inhibitor of calcineurin.
Fig. 6.

Working model of the mechanisms whereby PS-1 mutations render neurons vulnerable to mitochondrial dysfunction and apoptosis. PS-1 is localized in the ER. PS-1 mutations perturb ER calcium homeostasis in a manner that enhances calcium release when cells are exposed to various potentially toxic environmental signals (e.g., glutamate, amyloid β-peptide, or agonists that activate the IP3 pathway). When mitochondrial electron transport is uncoupled, as in cells exposed to the irreversible succinate dehydrogenase inhibitor 3-NP, calcium efflux from the mitochondria occurs followed by ATP depletion. The elevation of [Ca2+]i in response to mitochondrial impairment is exacerbated in cells expressing mutant PS-1. High cytoplasmic calcium levels promote the production of intramitochondrial and extramitochondrial ROS that damage mitochondrial membranes and proteins resulting in membrane depolarization (Δ¥) and permeability transition (MPT). These mitochondrial alterations result in the release of apoptotic factors (AFs) from mitochondria that then promote nuclear apoptosis. The increased [Ca2+]i and oxidative stress in cells expressing mutant PS-1 may also promote aberrant processing of APP resulting in increased production of amyloidogenic forms of Aβ. Aβ, in turn, may perpetuate a vicious neurodegenerative cascade by inducing oxidative stress, disrupting cellular ion homeostasis and further impairing mitochondrial function. See Discussion for further details and references supporting different aspects of this model.
The caspase inhibitor zVAD-fmk prevented loss of mitochondrial transmembrane potential and energy charge and nuclear apoptosis after exposure of cells expressing mutant PS-1 to 3-NP. However, the increase of [Ca2+]i and ROS production induced by 3-NP was unaffected by zVAD-fmk. These findings suggest that caspases are involved in 3-NP-induced apoptosis and may act at a point subsequent to elevation of [Ca2+]i and ROS production but before mitochondrial failure and nuclear apoptosis. These findings are consistent with previous studies in other cell types implicating caspases as upstream effectors of mitochondrial alterations linked to apoptosis (Harvey et al., 1997; Posmantur et al., 1997). On the other hand, mitochondrial membrane depolarization and MPT have been linked to release of molecules that activate caspases and induce nuclear apoptosis (Anel et al., 1996; Susin et al., 1996; Yang et al., 1997; Kluck et al., 1997; Shidoji et al., 1997). In neural cells, CPP32-like caspases may play a prominent role in the late, postmitochondrial, phase of apoptosis (Kroemer et al., 1997; Yuan, 1997). Because the caspase inhibitor used in the present study (zVAD-fmk) inhibits a broad range of caspases (including CPP32), the identity of the caspases that mediate 3-NP-induced apoptosis in cells expressing mutant PS-1 remains to be established. However, our analysis of caspase-3 activation does suggest that CPP32 is activated relatively early after exposure to 3-NP, during a time period when ROS accumulation and disruption of calcium homeostasis occurs. Interestingly, PS-1 and PS-2 are cleaved by type 3 caspases in cultured cells undergoing apoptosis; the cleavage site is in the cytosolic loop domain just adjacent to the normal cleavage site (Kim et al., 1997;Loetscher et al., 1997). Presenilin mutations result in increased cleavage by caspase-3, suggesting a role for this cleavage in the apoptotic process (Kim et al., 1997).
Apoptosis is believed to occur in several prominent neurodegenerative conditions associated with mitochondrial dysfunction, including AD (Cotman and Anderson, 1995; Smale et al., 1995), cerebral ischemia (Linnik et al., 1993; Nitatori et al., 1995; Du et al., 1996), and Huntington’s disease (Portera-Cailliau et al., 1995). The ability of mutant PS-1 (Guo et al., 1996, 1997; present study) and PS-2 (Wolozin et al., 1996), and perhaps wild-type PS-2 (Deng et al., 1996; Wolozin et al., 1996), to enhance apoptosis is consistent with an action of presenilins at a critical point in the apoptotic pathway. Our findings suggest that PS-1 mutations affect apoptosis by perturbing ER and mitochondrial calcium regulation. Recent studies have shown that mitochondria and ER interact in the regulation of cellular calcium homeostasis. For example, in lymphocytes, mitochondria control calcium release from ER stores (Hoth et al., 1997). PC12 cells expressing mutant PS-1 exhibit altered responses to signals, such as acetylcholine and bradykinin, that activate receptors linked to inositol phospholipid hydrolysis and release of calcium from ER stores (Guo et al., 1996). ER and mitochondrial calcium-regulating systems are increasingly recognized as playing important roles in fundamental neurophysiological events, including neurotransmitter release and synaptic plasticity. The present findings therefore suggest that altered subcellular calcium regulation may contribute to neuronal dysfunction before cell death in AD.
Footnotes
This work was supported by National Institutes of Health Grants AG14554, NS35253, AG05119, and AG05144 to M.P.M. We thank J. G. Begley, W. Fu, H. Luo, J. Partin, and J. Xie for technical assistance and J. W. Geddes, Z. Pang, and W. A. Pedersen for helpful discussions.
Correspondence should be addressed to Mark P. Mattson, 211 Sanders Brown Building, University of Kentucky, Lexington, KY 40536-0230.
REFERENCES
- 1.Anel A, Gamen S, Alava MA, Schmitt-Verhulst AM, Pineiro A, Naval J. Role of oxidative damage and IL-1 β-converting enzyme-like proteases in Fas-based cytotoxicity exerted by effector T cells. Int Immunol. 1996;8:1173–1183. doi: 10.1093/intimm/8.7.1173. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins B, Ferrante R, Kowall NW, Miller J, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histological characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens MI, Koh J, Canzoniero L, Sensi SL, Csernansky CA, Choi DW. 3-Nitropropionic acid induces apoptosis in cultured striatal and cortical neurons. NeuroReport. 1995;6:545–548. doi: 10.1097/00001756-199502000-00034. [DOI] [PubMed] [Google Scholar]
- 6.Benzi G, Moretti A. Contribution of mitochondrial alterations to brain aging. Adv Cell Aging Gerontol. 1997;2:129–160. [Google Scholar]
- 7.Blass JP. Metabolic alterations common to neural and non-neural cells in Alzheimer’s disease. Hippocampus. 1993;3:45–53. [PubMed] [Google Scholar]
- 8.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 9.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 10.Bredt DS, Snyder SH. Nitric oxide, a novel neuronal messenger. Neuron. 1992;8:3–11. doi: 10.1016/0896-6273(92)90104-l. [DOI] [PubMed] [Google Scholar]
- 11.Brorson JR, Sulit RA, Zhang H. Nitric oxide disrupts Ca2+ homeostasis in hippocampal neurons. J Neurochem. 1997;68:95–105. doi: 10.1046/j.1471-4159.1997.68010095.x. [DOI] [PubMed] [Google Scholar]
- 12.Cook DB, Sung JC, Golde TE, Felsenstein KM, Wojczyk BS, Tanzi RE, Trojanowski JQ, Lee VMY, Doms RW. Expression and analysis of presenilin 1 in a human neuronal system: localization in cell bodies and dendrites. Proc Natl Acad Sci USA. 1996;93:9223–9228. doi: 10.1073/pnas.93.17.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotman CW, Anderson AJ. A potential role for apoptosis in neurodegeneration in Alzheimer’s disease. Mol Neurobiol. 1995;10:19–45. doi: 10.1007/BF02740836. [DOI] [PubMed] [Google Scholar]
- 14.Cribbs DH, Chen L, Bendle SM, La Ferla FM. Widespread neuronal expression of the presenilin-1 early onset Alzheimer’s disease in the murine brain. Am J Pathol. 1996;148:1797–1806. [PMC free article] [PubMed] [Google Scholar]
- 15.Deliconstantinos G, Villiotou V. NO synthase and xanthine oxidase activities of rabbit brain synaptosomes: peroxynitrite formation as a causative factor of neurotoxicity. Neurochem Res. 1996;21:51–56. doi: 10.1007/BF02527672. [DOI] [PubMed] [Google Scholar]
- 16.Deng G, Pike CJ, Cotman CW. Alzheimer-associated presenilin-2 confers increased sensitivity to apoptosis in PC12 cells. FEBS Lett. 1996;397:50–54. doi: 10.1016/s0014-5793(96)01142-8. [DOI] [PubMed] [Google Scholar]
- 17.De Strooper B, Beullens M, Contreras B, Levesque L, Craessaerts K, Cordell B, Moechars D, Bollen M, Fraser P, George-Hyslop PS, Van Leuven F. Phosphorylation, subcellular localization, and membrane orientation of the Alzheimer’s disease-associated presenilins. J Biol Chem. 1997;272:3590–3598. doi: 10.1074/jbc.272.6.3590. [DOI] [PubMed] [Google Scholar]
- 18.Doan A, Thinakaran G, Borchelt D, Slunt H, Ratovistsky T, Podlisny M, Selkoe D, Seeger M, Gandy S, Price D, Sisodia S. Protein topology of presenilin 1. Neuron. 1996;17:1023–1030. doi: 10.1016/s0896-6273(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 19.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Duff K, Eckman C, Zehr C, Yu X, Prada C-M, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon M, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 21.Erecinska M, Nelson D. Effects of 3-nitropropionic acid on synaptosomal energy and transmitter metabolism: relevance to neurodegenerative diseases. J Neurochem. 1994;63:1033–1041. doi: 10.1046/j.1471-4159.1994.63031033.x. [DOI] [PubMed] [Google Scholar]
- 22.Estevez AG, Radi R, Barbeito L, Shin JT, Thompson JA, Beckman JS. Peroxynitrite-induced cytotoxicity in PC12 cells: evidence for an apoptotic mechanism differentially modulated by neurotrophic factors. J Neurochem. 1995;65:1543–1550. doi: 10.1046/j.1471-4159.1995.65041543.x. [DOI] [PubMed] [Google Scholar]
- 23.Fink SL, Ho DY, Sapolsky RM. Energy and glutamate dependency of 3-Nitropropionic acid neurotoxicity in culture. Exp Neurol. 1996;138:298–304. doi: 10.1006/exnr.1996.0068. [DOI] [PubMed] [Google Scholar]
- 24.Geddes JW, Pang Z, Wiley DH. Hippocampal damage and cytoskeletal disruption resulting from impaired energy metabolism. implications for Alzheimer’s disease. Mol Chem Neuropathol. 1996;28:65–74. doi: 10.1007/BF02815206. [DOI] [PubMed] [Google Scholar]
- 25.Giannakopoulos P, Bouras C, Kovari E, Shioi J, Tezapsidis N, Hof PR, Robakis NK. Presenilin-1 immunoreactive neurons are preserved in late-onset Alzheimer’s disease. Am J Pathol. 1997;150:429–436. [PMC free article] [PubMed] [Google Scholar]
- 26.Goodman Y, Mattson MP. Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induce oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 27.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochormocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo Q, Furukawa K, Sopher BL, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer’s PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid β-peptide. NeuroReport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- 29.Guo Q, Sopher B, Furukawa K, Pham D, Robinson N, Martin G, Mattson MP. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid β-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–4222. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo Q, Christakos S, Robinson N, Mattson MP (1998) Calbindin blocks the pro-apoptotic actions of mutant presenilin-1: reduced oxidative stress and preserved mitochondrial function. Proc Natl Acad Sci USA, in press. [DOI] [PMC free article] [PubMed]
- 31.Hardy J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 32.Harvey NL, Butt AJ, Kumar S. Functional activation of Nedd2/ICH-1 (Caspase-2) is an early process in apoptosis. J Biol Chem. 1997;272:13134–13139. doi: 10.1074/jbc.272.20.13134. [DOI] [PubMed] [Google Scholar]
- 33.Hockenbery D, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SH. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 34.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoyt KR, Gallagher AJ, Hastings TG, Reynolds IJ. Characterization of hydrogen peroxide toxicity in cultured rat forebrain neurons. Neurochem Res. 1997;22:333–340. doi: 10.1023/a:1022403224901. [DOI] [PubMed] [Google Scholar]
- 36.Ito E, Oka K, Etcheberrigaray R, Nelson TJ, McPhie DL, Tofel-Grehl B, Gibson GE, Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 38.Kaneko I, Yamada N, Sakuraba Y, Kamenosono M, Tutumi S. Suppression of mitochondrial succinate dehydrogenase, a primary target of β-amyloid, and its derivative racemized at Ser residue. J Neurochem. 1995;65:2585–2593. doi: 10.1046/j.1471-4159.1995.65062585.x. [DOI] [PubMed] [Google Scholar]
- 39.Kim T-W, Pettingell WH, Jung Y-K, Kovacs D, Tanzi R. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science. 1997;277:373–376. doi: 10.1126/science.277.5324.373. [DOI] [PubMed] [Google Scholar]
- 40.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 41.Kooy NW, Royall JA. Agonist-induced peroxynitrite production from endothelial cells. Arch Biochem Biophys. 1994;310:352–359. doi: 10.1006/abbi.1994.1178. [DOI] [PubMed] [Google Scholar]
- 42.Kooy NW, Royall JA, Ischoropoulos H, Beckman JS. Peroxynitrite-mediated oxidation of dihydrorhodamine 123. Free Radical Biol Med. 1994;16:149–156. doi: 10.1016/0891-5849(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 43.Kovacs DM, Fausett HJ, Page KJ, Kim T-W, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 44.Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- 45.Kruman I, Bruce-Keller AJ, Bredesen DE, Waeg G, Mattson MP. Evidence that 4-hydroxy nonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruman I, Guo Q, Mattson MP (1998) Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res, in press. [DOI] [PubMed]
- 47.Lafon-Cazal M, Pietri S, Cuicasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 48.Lehmann S, Chiesa R, Harris DA. Evidence for a six-transmembrane domain structure of presenilin 1. J Biol Chem. 1997;272:12047–12051. doi: 10.1074/jbc.272.18.12047. [DOI] [PubMed] [Google Scholar]
- 49.Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- 50.Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002–2008. doi: 10.1161/01.str.24.12.2002. [DOI] [PubMed] [Google Scholar]
- 51.Lippold HJ. Succinic dehydrogenase activity in liver, kidney and brain of rat. Histochemistry. 1982;75:287–291. doi: 10.1007/BF00496018. [DOI] [PubMed] [Google Scholar]
- 52.Loetscher H, Deuschle U, Brockhaus M, Reinhardt D, Nelboeck P, Mous J, Grunberg J, Haass C, Jacobsen H. Presenilins are processed by caspase-type proteases. J Biol Chem. 1997;272:20655–20659. doi: 10.1074/jbc.272.33.20655. [DOI] [PubMed] [Google Scholar]
- 53.Ludolph AC, Seelig M, Ludolph AG, Sabri MI, Spencer PS. ATP deficits and neuronal degeneration induced by 3-nitropropionic acid. Ann NY Acad Sci. 1992;648:300–302. doi: 10.1111/j.1749-6632.1992.tb24562.x. [DOI] [PubMed] [Google Scholar]
- 54.Luo Y, Bond JD, Ingram VM. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci USA. 1997;94:9705–9710. doi: 10.1073/pnas.94.18.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchetti P, Hirsch T, Zamzami N, Castedo M, Decaudin D, Susin SA, Masse B, Kroemer G. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996;157:4830–4836. [PubMed] [Google Scholar]
- 56.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid β-peptide impairs ion- motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Markesbery WR. Oxidative stress hypothesis of Alzheimer’s disease. Free Radical Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 58.Mattson MP. Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997a;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- 59.Mattson MP. Mother’s legacy: mitochondrial DNA mutations and Alzheimer’s disease. Trends Neurosci. 1997b;20:373–375. doi: 10.1016/s0166-2236(97)01114-4. [DOI] [PubMed] [Google Scholar]
- 60.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 61.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995a;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 62.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of [Ca2+]i and neurotoxicity, and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995b;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 63.Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K. Activation of NF-κB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of Mn-SOD and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res. 1997;49:681–697. doi: 10.1002/(SICI)1097-4547(19970915)49:6<681::AID-JNR3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 64.Miller DK. The role of the caspase family of cysteine proteases in apoptosis. Semin Immunol. 1997;9:35–49. doi: 10.1006/smim.1996.0058. [DOI] [PubMed] [Google Scholar]
- 65.Ming LJ. Moldy sugarcane poisoning–a case report with a brief review. Toxicol Clin Toxicol. 1995;33:363–367. doi: 10.3109/15563659509028924. [DOI] [PubMed] [Google Scholar]
- 66.Moudy AM, Handran SD, Goldberg MP, Ruffin N, Karl I, Kranz-Eble P, DeVivo DC, Rothman SM. Abnormal calcium homeostasis and mitochondrial polarization in a human encephalomyopathy. Proc Natl Acad Sci USA. 1995;92:729–733. doi: 10.1073/pnas.92.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Musser DA, Oseroff AR. The use of tetrazolium salts to determine sites of damage to the mitochondrial electron transport chain in intact cells following in vitro photometric therapy with Photofrin 2. J Immunol. 1994;59:621–626. doi: 10.1111/j.1751-1097.1994.tb09666.x. [DOI] [PubMed] [Google Scholar]
- 68.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-d-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 69.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Packer MA, Murphy MP. Peroxynitrite formed by simultaneous nitric oxide and superoxide generation causes cyclosporin-A-sensitive mitochondrial calcium efflux and depolarisation. Eur J Biochem. 1995;234:231–239. doi: 10.1111/j.1432-1033.1995.231_c.x. [DOI] [PubMed] [Google Scholar]
- 71.Page B, Page M, Noel C. A new fluorometric assay for cytotoxicity measurements in vitro. Int J Oncol. 1993;3:473–476. [PubMed] [Google Scholar]
- 72.Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage- dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta. 1994;1187:255–259. doi: 10.1016/0005-2728(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 74.Portera-Cailliau C, Hedreen JC, Price DL, Koliatos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Posmantur R, McGinnis K, Nadimpalli R, Gilbertson RB, Wang KKW. Characterization of CPP32-like protease activity following apoptotic challenge in SH-SY5Y neuroblastoma cells. J Neurochem. 1997;68:2328–2337. doi: 10.1046/j.1471-4159.1997.68062328.x. [DOI] [PubMed] [Google Scholar]
- 76.Richter C. Pro-oxidants and mitochondrial calcium: their relationship to apoptosis and oncogenesis. FEBS Lett. 1993;325:104–107. doi: 10.1016/0014-5793(93)81423-w. [DOI] [PubMed] [Google Scholar]
- 77.Richter C. Reactive oxygen and nitrogen species regulate mitochondrial Ca2+ homeostasis and respiration. Biosci Rep. 1997;17:53–66. doi: 10.1023/a:1027387301845. [DOI] [PubMed] [Google Scholar]
- 78.Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin JC, Chan PH. Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res. 1997;745:343–347. doi: 10.1016/s0006-8993(96)01231-0. [DOI] [PubMed] [Google Scholar]
- 79.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. The amyloid β protein deposited in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 80.Schulz JB, Henshaw DR, MacGarvey U, Beal MF. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem Int. 1996;29:167–171. doi: 10.1016/0197-0186(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 81.Shearman MS, Ragan CI, Iversen LL. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of β-amyloid-mediated cell death. Proc Natl Acad Sci USA. 1994;91:1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shibasaki F, McKeon F. Calcineurin functions in Ca2+-activated cell death in mammalian cells. J Cell Biol. 1995;131:735–743. doi: 10.1083/jcb.131.3.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shidoji Y, Nakamura N, Moriwaki H, Muto Y. Rapid loss in the mitochondrial membrane potential during geranylgeranoic acid-induced apoptosis. Biochem Biophys Res Commun. 1997;230:58–63. doi: 10.1006/bbrc.1996.5883. [DOI] [PubMed] [Google Scholar]
- 84.Smale G, Nichols NR, Brady DR. Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 85.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Kroemer G. Bcl-2 inhibits the mitochondrial release of apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 87.Walter J, Capell A, Grunberg J, Pesold B, Schindzielorz A, Prior R, Podlinsy MB, Fraser P, St. George-Hyslop P, Selkoe DJ, Haass C. The Alzheimer’s disease-associated presenilins are differentially phosphorylated proteins located predominantly within the endoplasmic reticulum. Mol Med. 1996;2:673–691. [PMC free article] [PubMed] [Google Scholar]
- 88.White JR, Reynolds IJ. Mitochondrial depolarization in glutamate stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Whittemore ER, Loo DT, Cotman CW. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. NeuroReport. 1994;5:1485–1488. doi: 10.1097/00001756-199407000-00019. [DOI] [PubMed] [Google Scholar]
- 90.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D’Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 91.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome C from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 93.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 94.Yuan J. Transducing signals of life and death. Curr Opin Cell Biol. 1997;9:247–251. doi: 10.1016/s0955-0674(97)80069-5. [DOI] [PubMed] [Google Scholar]
- 95.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin S, Petit P, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996;384:53–57. doi: 10.1016/0014-5793(96)00280-3. [DOI] [PubMed] [Google Scholar]
- 97.Zeevalk G, Derr-Yellin E, Nicklas W. Relative vulnerability of dopamine and GABA neurons in mesencephalic culture to inhibition of succinate dehydrogenase by malonate and 3-nitropropionic acid and protection by NMDA receptor blockade. J Pharmacol Exp Ther. 1995;275:1124–1130. [PubMed] [Google Scholar]