

Abstract
Activated protein C (APC), an important inhibitor of the coagulation system, has recently been shown to prevent tissue injury by blocking the activation of leukocytes. To determine whether APC can also prevent post-traumatic spinal cord injury (SCI), a condition in which leukocytes play an important role, we tested the effects of APC on SCI induced in rats by compression trauma. Administration of APC, either before or after the induction of SCI, markedly reduced the motor disturbances in these animals. In contrast, neither an inactive derivative of activated factor X (DEGR-Xa), a selective inhibitor of thrombin generation, nor active site-blocked APC (DIP-APC) reduced the motor disturbances. Histological examination revealed that intramedullary hemorrhages, observed 24 hr after trauma, were significantly reduced in the animals administered APC. The increase in the tissue level of tumor necrosis factor-α (TNF-α) and the accumulation of neutrophils in the damaged segment of the spinal cord were significantly inhibited in the animals that had received APC, but these were not inhibited in those administered DIP-APC or DEGR-Xa. The induction of leukocytopenia had the same effect as APC, in that it significantly reduced motor disturbances, tissue levels of TNF-α, and neutrophil accumulation in the animals subjected to compressive SCI. These findings suggest that in SCI, APC reduces motor disturbances primarily by reducing the amount of TNF-α at the site of injury, thus inhibiting neutrophil accumulation and the resultant damage to the endothelial cells.
Keywords: activated protein C, post-traumatic spinal cord injury, leukocytes, motor disturbances, TNF-α, neutrophils
Spinal cord injury (SCI) is a serious condition that produces lifelong disabilities (Stover and Fine, 1987). Only limited therapeutic measures are currently available for its treatment (Bracken et al., 1990). SCI induced by trauma is a consequence of an initial physical insult that is followed by a progressive injury process that involves various pathochemical events that lead to tissue destruction (Young, 1988; Bracken et al., 1990). Therapeutic intervention for SCI should therefore be directed at reducing or alleviating this secondary process. Although the mechanisms are not fully understood, progressive vascular events, such as ischemia/reperfusion-induced endothelial damage, are involved in this process (Demopoulos et al., 1978; Means and Anderson, 1983; Xu et al., 1990; Blight, 1992). We have demonstrated that activated neutrophils are important in inducing the damage to endothelial cells observed in SCI induced by trauma (Taoka et al., 1997a).
Activated protein C (APC) is an important physiological anticoagulant that is generated from protein C by the action of the thrombin–thrombomodulin complex on the endothelial cells (Walker et al., 1979). APC inactivates factors Va and VIIIa, thereby regulating the coagulation system (Walker et al., 1979; Esmon, 1992). APC is also implicated in the regulation of the inflammatory process by its inhibition of cytokine production by monocytes (Grey et al., 1993,1994). We demonstrated previously that APC prevents the injury to endothelial cells induced by activated leukocytes, primarily by inhibiting the ability of monocytes to produce tumor necrosis factor-α (TNF-α) (Murakami et al., 1996, 1997). Because TNF-α is a potent activator of neutrophils (Klebanoff et al., 1986), it is possible that APC may also prevent the secondary effects of trauma-induced SCI by inhibiting neutrophil activation. We therefore evaluated the effects of APC in a rat model of compression-induced SCI.
MATERIALS AND METHODS
Reagents. APC was obtained from human thrombin-activated protein C and purified by cation-exchange chromatography as described previously (Katsu-ura et al., 1994). Nitrogen mustard was obtained from Sigma (St. Louis, MO). All other reagents used were of analytical grade.
Preparation of dansyl glutamyl-glycyl-arginyl chloromethyl ketone-treated factor Xa. Factor X, purified from human plasma and activated with Russell’s viper venom (Bajaj et al., 1981), was inactivated by incubation with a 20-fold molar excess of dansyl glutamyl-glycyl-arginyl chloromethyl ketone (DEGR) for 30 min at 25°C, after which the mixture was subjected to extensive dialysis against a solution containing 20 mm Tris-HCl, pH 7.4, and 100 mm NaCl. DEGR-treated factor Xa (DEGR-Xa) has been shown to selectively inhibit thrombin generation by competing with intact factor Xa for prothrombinase complex formation (Nesheim et al., 1981).
Preparation of diisopropyl fluorophosphate-treated APC (DIP-APC). APC was inactivated with DIP (Sigma) by incubating APC (1 mg/ml) with 19 mmol/l of DIP in PBS, pH 7.4, for 2 hr and dialyzing it extensively against the same buffer (Grey et al., 1994). The effectiveness of inactivation was monitored amidolytically by measuring the rate of hydrolysis of the chromogenic substrate S-2366 (Chromogenix AB, Stockholm, Sweden) at 405 nm. The amount of APC activity remaining was <1%.
Animal model of spinal cord injury. The study protocol was approved by the Kumamoto University Animal Care and Use Committee. The care and handling of the animals were conducted in accordance with the guidelines of the National Institutes of Health. Under pentobarbital anesthesia (45 mg/kg, i.p.) (Abbott Laboratories, North Chicago, IL), adult pathogen-free male Wistar rats (Nihon SLC, Hamamatsu, Japan), weighing 300–350 gm, were subjected to laminectomy using a surgical airtome at the level of the 12th thoracic vertebra (Th12). Spinal cord injury (SCI) was induced by applying a 20 gm weight extradurally to the spinal cord at Th12 for 20 min as described previously (Taoka et al., 1997a,b). This technique causes paralysis of the lower extremities in a reproducible manner (Taoka et al., 1995; Hamada et al., 1996). Laminectomy alone was performed as a sham operation. APC (100 μg/kg) was administered intravenously to rats 30 min before (pretreatment group) or after (post-treatment group) the compressive trauma. DIP-APC (100 μg/kg) and DEGR-Xa (10 mg/kg) were administered intravenously 30 min before trauma. The control and leukocytopenic animals received saline instead of anticoagulants or other drugs.
Grading of motor disturbance. The motor function of rats was assessed in a blind manner using the the inclined-plane test (Rivlin and Tator, 1977) and footprint analysis (Kunkel-Bagden and Bregman, 1993). In the inclined-plane test, recovery from motor disturbance was assessed before, and again at 1, 7, 14, and 21 d after the compression. We recorded the maximum inclination of the plane on which the rats could maintain themselves for 5 sec without falling.
Footprint analysis was performed before and 3 weeks after the compression injury, as illustrated in Figure1. The hindpaws were wetted, and the animals were made to walk on paper coated with bromophenol blue (Wako Pure Chemical Industries) dissolved in acetone. The base of support was determined by measuring the distance between the central pads of the hindpaws (DBF). The stride lengths of the right and left hindpaws (RSL and LSL) were measured in two consecutive prints.
Fig. 1.

Footprint analysis. The base of support (DBF) and right and left stride lengths (RSL and LSL) were measured from footprints.
Histological examination of the spinal cord. Rats were killed at random 24 hr after compressive SCI. They were perfused transcardially with 10% formaldehyde in a phosphate-buffered solution; ∼ 1 cm of the spinal cord at Th12 was removed immediately and immersed overnight in the same solution. Transverse, semi-serial sections of 5 μm thickness were prepared and embedded in paraffin. These sections were subsequently stained with hematoxylin and eosin. The samples were assessed by a pathologist who had no knowledge of any animal’s group.
Assay of myeloperoxidase activity. The extent of leukocyte infiltration was assessed by measurement of myeloperoxidase (MPO) activity (Lundberg and Arfors, 1983) using a modification of a method described previously (Xu et al., 1990). Within 1 hr after the rats were killed, sections of ∼1 cm were dissected from the Th12 region, removed, and placed in ice-cold 0.9% NaCl bath. A 10% (wt/vol) tissue homogenate was mixed with a 20 mm phosphate buffer, pH 6.0, containing 0.5% hexadecyltrimethyl ammonium bromide (Sigma) and sonicated for 30 sec. After centrifugation (4500 ×g at 4°C for 20 min), 0.1 ml of supernatant was added to 0.6 ml of 0.1 m phosphate buffer, pH 6.0, containing 1.25 mg/ml o-dianisidine and 0.05% H2O2. After 5 min, the change in absorbance at 460 nm was measured spectrophotometrically (DU-54, Beckman, Irvine, CA), and the MPO activity in each sample was calculated using a standard curve for purified MPO (Sigma).
Assay of TNF-α. The level of TNF-α in spinal cord tissue was determined before and 1, 2, 3, 4, 6, 12, and 24 hr after SCI according to the methods described by Murakami et al. (1997). Briefly, within 1 hr after the rats were killed, sections from the Th12 region measuring ∼1 cm were dissected, removed, and placed in ice-cold 0.9% NaCl. A 20% (wt/vol) tissue homogenate was mixed with a 0.1m phosphate buffer, pH 7.4, sonicated for 30 sec, and centrifuged at 4500 × g for 20 min at 4°C. The concentration of TNF-α in the supernatant was determined using an enzyme-linked immunosorbent assay (ELISA) kit for rat TNF-α (Genzyme Corporation, Cambridge, MA). Results are expressed as picograms of TNF-α per gram of tissue.
Induction of leukocytopenia by nitrogen mustard. Rats were made leukocytopenic by the intravenous injection of nitrogen mustard (NM) (Müller-Berghaus and Eckhart, 1975). Because the dose of NM of 1.75 mg/kg administered in an earlier study had caused death in all rats within 10 d of laminectomy, probably as a result of infection (Taoka et al., 1997b), we now administered an intravenous dose of 1.0 mg/kg. This dose caused no deaths for 3 weeks after SCI. NM or 0.9% NaCl was administered intravenously to rats 2 d before induction of SCI. The circulating leukocyte count on day 0 was 9375 ± 1365/μl (n = 10) in controls and 3350 ± 230/μl in NM-treated rats (n = 10) (p < 0.01). In differential leukocyte counts made on peripheral blood smears, the number of neutrophils counts on day 0 was 1298 ± 428/μl in controls and 733 ± 112/μl in NM-treated rats (p < 0.01), and the number of monocytes was 539 ± 286/μl in controls and 182 ± 38/μl in NM-treated animals (p < 0.01).
Statistical analysis. Data are presented as the mean ± SD. Circulating leukocyte counts were compared using Student’st test. Statistical comparisons of the mean degrees in the inclined-plane test, mean distance between feet, mean stride length, mean MPO activity, and the mean TNF-α level between groups used the ANOVA and Scheffe’s post hoc test. A level ofp < 0.05 was defined as statistically significant.
RESULTS
Effect of APC on SCI induced by compression trauma
As reported previously (Hamada et al., 1996), we found that when evaluated by the inclined-plane test, motor disturbances were increased within 24 hr of the compressive SCI. After 24 hr, the neurological scores were significantly higher in the rats treated with APC before the induction of SCI versus that of the controls (Figs.2, 3). From 1 to 21 d after the induction of SCI, the angle of the inclined plane was significantly higher in the rats pretreated with APC than in the controls (Fig. 2).
Fig. 2.

Temporal effect of pretreatment with APC on motor disturbances after compressive SCI. Spinal cord injury was induced by applying a 20 gm weight for 20 min at Th12, and motor disturbances of the hindlimbs were evaluated after SCI using an inclined-plane test. APC (100 μg/kg) or buffer (as a control) was administered intravenously 30 min before injury. Closed circles, Traumatized animals; open circles, APC-treated animals. Mean ± SD of 10 experiments. *p < 0.01 versus traumatized animals.
Fig. 3.

Effects of APC, DEGR-Xa, NM-induced leukocytopenia, and DIP-APC on motor disturbances 1 d after induction of SCI as determined by an inclined-plane test. Motor disturbances of the hindlimbs were evaluated 1 d later using an inclined-plane test. APC (100 μg/kg), DIP-APC (100 μg/kg), DEGR-Xa (10 mg/kg), or buffer (as a control) was administered intravenously 30 min before injury, or APC was administered 30 min after injury. Leukocytes were depleted by administration of nitrogen mustard (NM). Mean ± SD of 10 experiments. *p < 0.05 versusTrauma.
The administration of APC after trauma also significantly improved the motor function of rats. Whether evaluated by the inclined-plane test (Figs. 3, 4A) or footprint analysis (Fig. 4B,C), the neurological scores 1 and 21 d after the induction of SCI were higher in the rats administered APC post-traumatically than in controls.
Fig. 4.
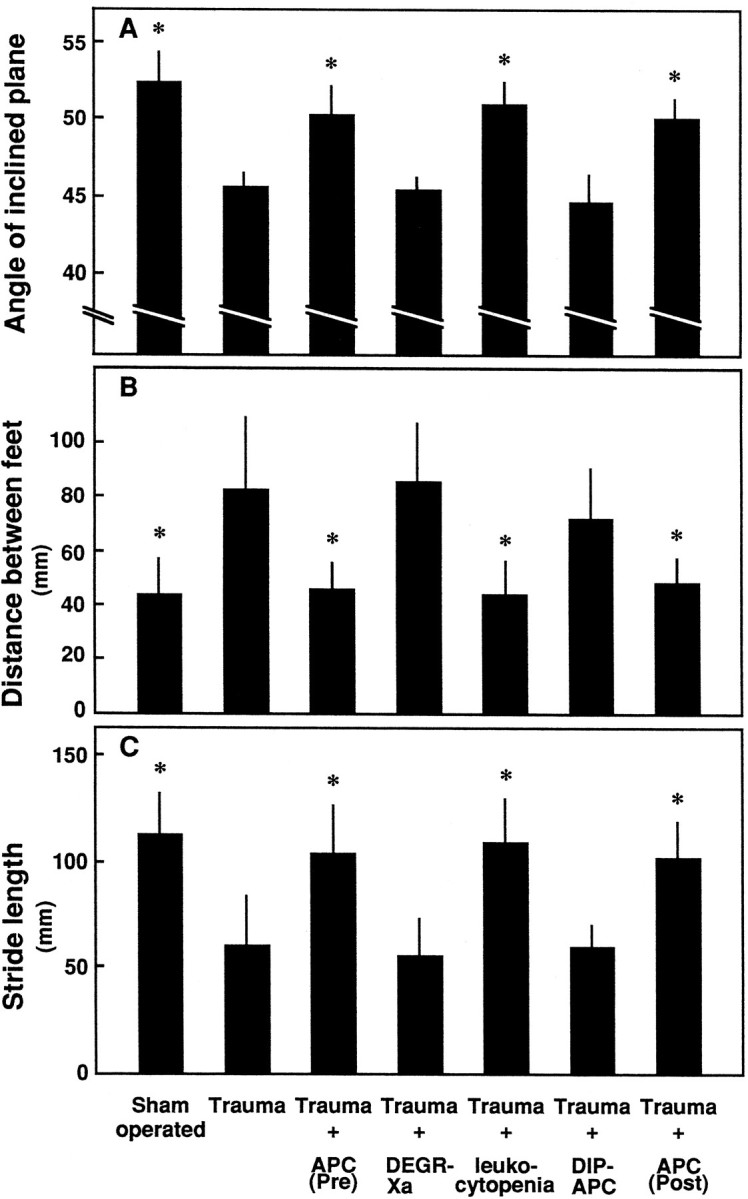
Effects of APC, DEGR-Xa, NM-induced leukocytopenia, and DIP-APC on motor disturbances 21 d after induction of SCI as determined by an inclined-plane test and footprint analysis. Motor disturbances of the hindlimbs were evaluated 21 d after induction of SCI using the inclined-plane test and footprint analysis. Concentrations of APC, DEGR-Xa, NM, and DIP-APC were as in Figure 3. Mean ± SD of 10 experiments. *p < 0.01 versus Trauma.
In contrast to APC, neither DEGR-Xa (a selective inhibitor of thrombin generation) nor DIP-APC (active site-blocked APC) had any effect on the motor function of rats 1 and 21 d after the injury to the spinal cord (Figs. 3, 4).
Histological observations
Histological examination of the traumatized spinal cord 24 hr after the induction of SCI showed the presence of intramedullary hemorrhages in control animals (Fig. 5). These hemorrhages were observed more often in the gray than in the white matter. In contrast, there was markedly less hemorrhage in the animals that had received APC before trauma (Fig. 5). Neither DEGR-Xa nor DIP-APC prevented hemorrhagic changes in the injured spinal cord (data not shown).
Fig. 5.
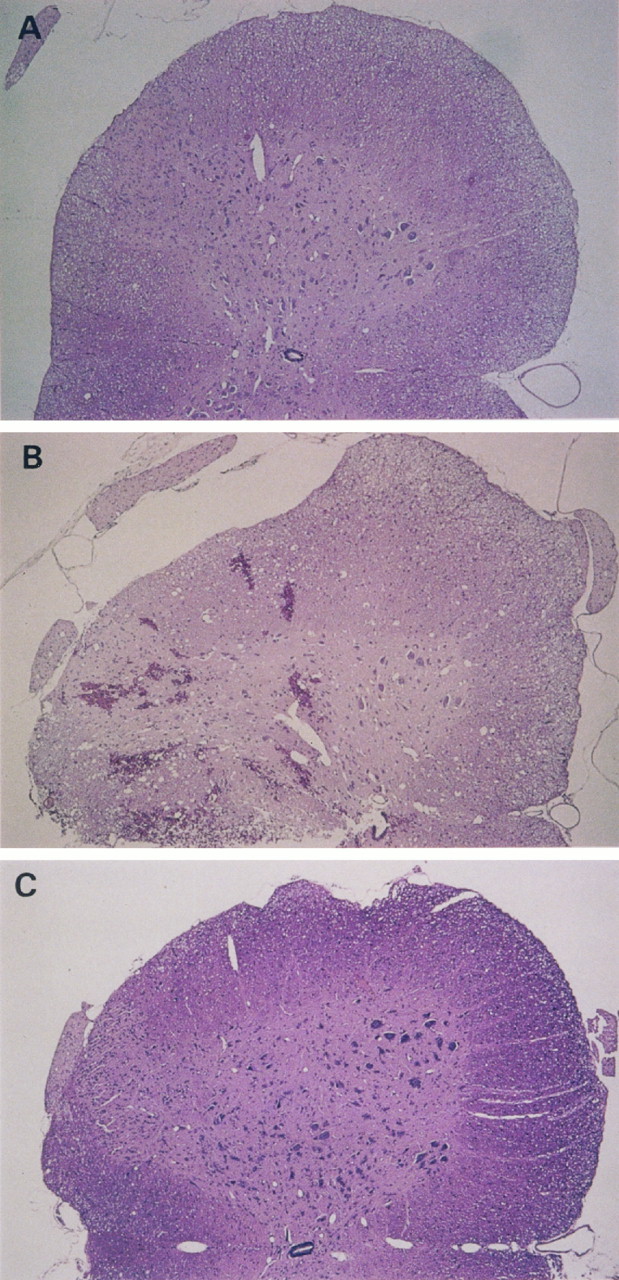
Histology of (A) an intact spinal cord section and in traumatized spinal cord sections from the level of the 12th thoracic vertebra in rats that received (B) saline or (C) APC (150×, hematoxylin and eosin). Five animals in each group were examined; typical results are shown.
Effect of APC on increase in MPO activity induced by trauma
The accumulation of neutrophils at the traumatized spinal cord tissue was evaluated by measuring MPO activity 3 hr after the induction of compressive trauma. The increase in MPO activity, observed in traumatized animals versus sham-operated rats, was inhibited significantly in the spinal cord of animals that received APC 30 min before trauma (Fig.6A). The administration of DEGR-Xa or DIP-APC did not inhibit this increased MPO activity (Fig.6A).
Fig. 6.

Effects of APC, DEGR-Xa, NM-induced leukocytopenia, and DIP-APC on (A) MPO activity and (B) TNF-α levels in traumatized spinal cord. MPO activity and TNF-α level at Th12 were measured 3 or 4 hr after compressive trauma or a sham-operation, respectively. Mean ± SD of five experiments. †p < 0.05 versusTrauma; *p < 0.01 versusTrauma.
Effects of APC and nitrogen mustard-induced leukocytopenia on increased TNF-α in injured segment of spinal cord
After the induction of SCI at Th12, we measured the level of TNF-α in this segment over time. The tissue level of TNF-α increased significantly within 1 hr and peaked at 4 hr (Fig.7). The tissue level of TNF-α 1–6 hr after trauma significantly exceeded that of sham-operated animals (Fig.7).
Fig. 7.

Changes in TNF-α level at Th12 over time after compressive trauma or a sham-operation. TNF-α levels at Th12 were measured before and after the induction of compressive trauma or sham-operation. Closed circles, Traumatized animals;open circles, sham-operated animals. Mean ± SD of five experiments. †p < 0.05 versus sham-operated group; *p < 0.01 versus sham-operated group.
When we evaluated the effect of APC on tissue levels of TNF-α 4 hr after induction of trauma, we found that APC significantly inhibited the compression-induced increases in TNF-α (Fig.6B). The animals administered nitrogen mustard to induce leukocytopenia also showed greatly reduced levels of TNF-α (Fig. 6B). In contrast, neither DEGR-Xa nor DIP-APC had any effect on TNF-α level in animals with SCI (Fig.6B).
DISCUSSION
We observed that APC significantly reduced the deleterious effects of SCI in rats. When administered before or after the induction of trauma, APC reduced the number of intramedullary hemorrhages as well as the severity of motor disturbances. Although the motor disturbances evaluated by using the inclined-plane test were not completely recovered 42 d after trauma, those in animals given APC before or after the trauma were recovered 35 d after trauma (data not shown), suggesting that APC might promote the functional recovery of the motor disturbances in this animal model of spinal cord injury.
Because APC inhibited at the active site (DIP-APC) and a selective inhibitor of thrombin generation (DEGR-Xa) had no effect, our results with APC suggest that the efficacy of this protein may not be mediated by inhibition of thrombin generation. However, this observation does not exclude the possibility that APC may attenuate the spinal cord injury by inactivation of factor Va/VIIIa.
The accumulation of neutrophils in traumatized segments of the spinal cord as reflected by tissue MPO activity was also significantly inhibited in animals treated with APC. The leukocytopenia induced by NM markedly reduced the motor disturbances as well as accumulation of neutrophils in traumatized spinal cord tissue, suggesting that the accumulation of neutrophils may not be an effect but rather may be a cause of the motor disturbances observed after SCI induced by compression trauma. Because DIP-APC did not prevent the accumulation of neutrophils at the injured site, the inhibition of neutrophil accumulation by APC may also be mediated by its serine protease activity. This is consistent with our previous observation that APC prevents endotoxin-induced lung injury by inhibiting the accumulation of neutrophils, and that this effect is also dependent on the serine protease activity of APC (Murakami et al., 1996, 1997).
Although we observed the accumulation of neutrophils at the traumatized site, we found no histological evidence of their infiltration into the tissue of the injured spinal cord (Taoka et al., 1997a). This suggests that neutrophils may accumulate at the endothelial surface, where they may damage the endothelial cells by releasing a wide variety of inflammatory mediators such as granulocyte elastase and oxygen free radicals (Harlan, 1987; Carlos and Harlan, 1994). Indeed, our preliminary study indicates that L-658,758, a specific granulocyte elastase inhibitor (Zimmerman and Granger, 1990), prevents the SCI induced by compressive trauma in this animal model (our unpublished data).
Our results strongly suggest that the production of TNF-α at the site of SCI is implicated in the secondary damage to tissue in SCI. We found that the level of this protein in the traumatized spinal cord tissue was significantly increased after compressive trauma, with a peak seen after 4 hr. These results are consistent with those of other researchers. For example, Wang et al. (1996) showed the presence of TNF-α at the sites of traumatic spinal cord lesions but did not detect this factor in cerebrospinal fluid or in serum. In addition,Yakovlev and Faden (1994) demonstrated that spinal cord impact in rats caused an elevation of TNF-α mRNA levels at the site of trauma 30 min after the injury; the severity of injury was proportional to the level of the TNF-α message. Our additional observation, that leukocytopenic rats in which the level of TNF-α was not increased at the site of trauma exhibited a significant reduction in motor disturbances, indicates that increased levels of TNF-α at the site of injury may be a cause, rather than an effect, of the SCI induced by compressive trauma.
TNF-α contributes to the tissue injury induced by neutrophils by directly activating them (Klebanoff et al., 1986), as well as by increasing the expression of such molecules as E-selectin, which cause the activated neutrophils to adhere to the surface of the endothelial cells (Mulligan et al., 1991). We have also shown that the inhibition of neutrophil adhesion to the endothelial cell surface markedly reduces the severity of the SCI induced by compressive trauma (Taoka et al., 1997a). These observations indicate that the interaction of activated neutrophils with the surface of the endothelial cells is important in the secondary tissue damage that occurs after SCI. Because no increase in the level of TNF-α induced by SCI was found in the animals that had received APC, this suggests that APC may inhibit the accumulation of neutrophils at the site of the traumatic spinal cord injury primarily by inhibiting TNF-α production. This is supported by our earlier findings; i.e., that APC also inhibits the in vivoand in vitro production of TNF-α by monocytes, that this activity depends on the serine protease activity of APC, and that APC does not directly inhibit the neutrophils (Murakami et al., 1997).
Although the precise mechanism(s) by which APC inhibits the production of TNF-α has not been fully elucidated, our finding that DIP-APC did not affect the level of TNF-α at the site of trauma suggests that the serine protease activity of APC may be important in the inhibition of TNF-α production. Grey et al. (1993, 1994) also reported that APC suppresses the production of TNF-α by LPS-stimulated monocytes by inhibiting the coupling of LPS and CD14, but that DIP-APC did not possess this activity. In contrast, however, Grinnell et al. (1994)demonstrated that the inhibition of neutrophil accumulation by APC was not related to the serine protease activity of APC, because the carbohydrate moieties of APC reacted more with E-selectin than with the sialyl Lewis X antigen of neutrophils. This possibility, however, seems less likely in vivo, because we found that DIP-APC did not reduce the accumulation of neutrophils at the traumatized site.
We have demonstrated that APC can lessen the severity of the SCI induced by trauma by inhibiting the accumulation of neutrophils and the production of TNF-α. Because the administration of APC after the injury was as effective as its administration before injury in preventing the secondary effects of SCI, APC may have a potential for clinical use in alleviating the effects of traumatic compression injury to the spinal cord.
Footnotes
Correspondence should be addressed to Dr. Kenji Okajima, Department of Laboratory Medicine, Kumamoto University School of Medicine, Honjo 1-1-1, Kumamoto 860, Japan.
REFERENCES
- 1.Bajaj SP, Rapaport SI, Prodanos CA. A simplified procedure for purification of human prothrombin, factor IX and factor X. Prep Biochem. 1981;11:397–412. doi: 10.1080/00327488108065531. [DOI] [PubMed] [Google Scholar]
- 2.Blight AR. Macrophages and inflammatory damage in spinal cord injury. J Neurotrauma. 1992;9:S83–S91. [PubMed] [Google Scholar]
- 3.Bracken MB, Shepard MJ, Collins WF, Holord TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, Maeshall LF, Perot PL, Piepmeier J, Sonntag KH, Wagner FC, Wilberger JE, Winn HR. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. New Engl J Med. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- 4.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- 5.Demopoulos HB, Yoder M, Gutman EG, Seligman ML, Flamm ES, Ransohoff J. The fine structure of endothelial surfaces in the microcirculation of experimentally injured feline spinal cords. Scan Electron Microsc. 1978;2:677–680. [Google Scholar]
- 6.Esmon CT. The protein C anticoagulant pathway. Arterioscler Thromb. 1992;12:135–145. doi: 10.1161/01.atv.12.2.135. [DOI] [PubMed] [Google Scholar]
- 7.Grey S, Hau H, Salem HH, Hancock WW. Selective effects of protein C on activation of human monocytes by lipopolysaccharide, interferon-g, or PMA: modulation of effects on CD11b and CD14 but not CD25 or CD54 induction. Transplant Proc. 1993;25:2913–2914. [PubMed] [Google Scholar]
- 8.Grey ST, Tsuchida A, Hau H, Orthner CL, Salem HH, Hancock WW. Selective inhibitory effects of the anticoagulant activated protein C on the ponses of human mononuclear phagocytes to LPS, IFN-g, or phorbolester. J Immunol. 1994;153:3664–3672. [PubMed] [Google Scholar]
- 9.Grinnell BW, Hermann RB, Yan SB. Human protein C inhibits selectin-mediated cell adhesion: role of unique fucosylated oligosaccharide. Glycobiology. 1994;4:221–225. doi: 10.1093/glycob/4.2.221. [DOI] [PubMed] [Google Scholar]
- 10.Hamada Y, Ikata T, Katoh S, Nakauchi K, Niwa M, Kawai Y, Fukuzawa K. Involvement of an intracellular adhesion molecule 1-dependent pathway in the pathogenesis of secondary changes after spinal cord injury in rats. J Neurochem. 1996;66:1525–1531. doi: 10.1046/j.1471-4159.1996.66041525.x. [DOI] [PubMed] [Google Scholar]
- 11.Harlan JM. Consequences of leukocytes-vessel wall interactions in inflammatory and immune reactions. Semin Thromb Hemost. 1987;13:425–433. doi: 10.1055/s-2007-1003520. [DOI] [PubMed] [Google Scholar]
- 12.Katsu-ura Y K, Aoki H, Tanabe M, Funatsu A. Characteristic effects of activated human protein C on tissue thromboplastin-induced disseminated intravascular coagulation in rabbits. Thromb Res. 1994;76:353–357. doi: 10.1016/0049-3848(94)90164-3. [DOI] [PubMed] [Google Scholar]
- 13.Klebanoff SJ, Vadas MA, Harlan JM. Stimulation of neutrophils by tumor necrosis factor. J Immunol. 1986;136:4220–4225. [PubMed] [Google Scholar]
- 14.Kunkel-Bagden E, Bregman BS. Methods to assess the development and recovery of locomotor function after spinal cord injury in rats. Exp Neurol. 1993;119:153–164. doi: 10.1006/exnr.1993.1017. [DOI] [PubMed] [Google Scholar]
- 15.Lundberg C, Arfors K. Polymorphonuclear leukocyte accumulation in inflammatory dermal sites as measured by 51Cr-labeled cells and myeloperoxidase. Inflammation. 1983;7:247–255. doi: 10.1007/BF00917262. [DOI] [PubMed] [Google Scholar]
- 16.Means ED, Anderson DK. Neuronophagia by leukocytes in experimental spinal cord injury. J Neuropathol Exp Neurol. 1983;42:707–719. doi: 10.1097/00005072-198311000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Müller-Berghaus G, Eckhart T. The role of granulocytes in the activation of intravascular coagulation and the precipitation of soluble fibrin by endotoxin. Blood. 1975;45:631–641. [PubMed] [Google Scholar]
- 18.Mulligan MS, Varani J, Dame MK. Role of endothelial-leukocyte adhesion molecule 1 (ELAM-1) in neutrophil-mediated lung injury in rats. J Clin Invest. 1991;88:1396–1406. doi: 10.1172/JCI115446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87:642–647. [PubMed] [Google Scholar]
- 20.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K. Activated protein C prevents LPS-induced pulmonary vascular injury by inhibiting cytokine production. Am J Physiol. 1997;272:L197–L202. doi: 10.1152/ajplung.1997.272.2.L197. [DOI] [PubMed] [Google Scholar]
- 21.Nesheim ME, Kettner C, Shaw E, Mann KG. Cofactor dependence of factor Xa incorporation into the prothrombinase complex. J Biol Chem. 1981;256:6537–6540. [PubMed] [Google Scholar]
- 22.Rivlin AS, Tator CH. Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J Neurosurg. 1977;47:577–581. doi: 10.3171/jns.1977.47.4.0577. [DOI] [PubMed] [Google Scholar]
- 23.Stover SL, Fine PR. The epidemiology and economics of spinal cord injury. Paraplegia. 1987;24:225–228. doi: 10.1038/sc.1987.40. [DOI] [PubMed] [Google Scholar]
- 24.Taoka Y, Naruo M, Koyanabi E, Urakado M, Inoue M. Superoxide radicals play important roles in the pathogenesis of spinal cord injury. Paraplegia. 1995;33:450–453. doi: 10.1038/sc.1995.98. [DOI] [PubMed] [Google Scholar]
- 25.Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K. Role of neutrophils in spinal cord injury in the rat. Neuroscience. 1997a;79:1177–1182. doi: 10.1016/s0306-4522(97)00011-0. [DOI] [PubMed] [Google Scholar]
- 26.Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K. Reduction of spinal cord injury by administration of iloprost, a stable prostacyclin analog. J Neurosurg. 1997b;86:1007–1011. doi: 10.3171/jns.1997.86.6.1007. [DOI] [PubMed] [Google Scholar]
- 27.Walker FJ, Sexton PW, Esmon CT. The inhibition of blood coagulation by activated protein C through the selective inactivation of activated factor V. Biochim Biophys Acta. 1979;571:333–342. doi: 10.1016/0005-2744(79)90103-7. [DOI] [PubMed] [Google Scholar]
- 28.Wang CX, Nuttin B, Heremans H, Gybels R. Production of tumor necrosis factor in spinal cord following traumatic injury in rats. J Neuroimmunol. 1996;89:151–156. doi: 10.1016/0165-5728(96)00080-x. [DOI] [PubMed] [Google Scholar]
- 29.Xu J, Hsu CY, Liu TH, Hogan EL, Perot E, Tai H. Leukotriene B4 release and polymorphonuclear cell infiltration in spinal cord injury. J Neurochem. 1990;55:907–912. doi: 10.1111/j.1471-4159.1990.tb04577.x. [DOI] [PubMed] [Google Scholar]
- 30.Yakovlev AG, Faden AI. Sequential expression of c-fos protooncogene, TNF-alpha, and dynorphin genes in spinal cord following experimental traumatic injury. Mol Chem Neuropathol. 1994;23:179–190. doi: 10.1007/BF02815410. [DOI] [PubMed] [Google Scholar]
- 31.Young W. Secondary CNS injury. J Neurotrauma. 1988;5:219–221. doi: 10.1089/neu.1988.5.219. [DOI] [PubMed] [Google Scholar]
- 32.Zimmerman BJ, Granger DN. Reperfusion-induced leukocyte infiltration: role of elastase. Am J Physiol. 1990;259:H390–H394. doi: 10.1152/ajpheart.1990.259.2.H390. [DOI] [PubMed] [Google Scholar]