

Abstract
The proapoptotic molecule BAX is required for death of sympathetic and motor neurons in the setting of trophic factor deprivation. Furthermore, adult Bax−/− mice have more motor neurons than do their wild-type counterparts. These findings raise the possibility that BAX regulates naturally occurring cell death during development in many neuronal populations. To test this idea, we assessed apoptosis using TUNEL labeling in several well-studied neural systems during embryonic and early postnatal development inBax−/− mice. Remarkably, naturally occurring cell death is virtually eliminated between embryonic day 11.5 (E11.5) and postnatal day 1 (PN1) in most peripheral ganglia, in motor pools in the spinal cord, and in the trigeminal brainstem nuclear complex. Additionally, reduction, although not elimination, of cell death was noted throughout the developing cerebellum, in some layers of the retina, and in the hippocampus. Saving of cells was verified by axon counts of dorsal and ventral roots, as well as facial and optic nerves that revealed 24–35% increases in axon number. Interestingly, many of the supernumerary axons had very small cross-sectional areas, suggesting that the associated neurons are not normal. We conclude that BAX is a critical mediator of naturally occurring death of peripheral and CNS neurons during embryonic life. However, rescue from naturally occurring cell death does not imply that the neurons will develop normal functional capabilities.
Keywords: primary afferent neurons, motor neuron, apoptosis, programmed cell death, Bcl-2 gene family, BAX
Naturally occurring cell death is a widespread phenomenon serving a variety of fundamental functions in multicellular animals. Within the developing nervous system, most (although not all) classes of neurons seem to undergo this process to some degree (Oppenheim, 1991). The traditional view has been that neurons extend axons into target fields and then “compete” for trophic factors, with the “losers” undergoing programmed cell death. The recent introduction of sensitive techniques to detect DNA fragmentation in situ has substantially refined this view. For example, recent histological analyses in mice have revealed naturally occurring cell death present in cells of the peripheral nervous system before the period of target innervation (ElShamy and Ernfors, 1996; Fariñas et al., 1996; White et al., 1996). Further unexpected results have demonstrated that cell death is an ongoing process during the proliferation of neuronal progenitors in the ventricular zone of embryonic telencephalon and during the neuronal migration of cells in the intermediate zone of the developing cortex (Blaschke et al., 1996; see also Schindler et al., 1997). Taken together, these results suggest that cell death occurs during several phases of neural development and that the cell death may be more extensive than appreciated previously. Interestingly, despite these new insights, the function served by naturally occurring cell death in the nervous system remains obscure.
Major advances in understanding naturally occurring cell death have been stimulated by the discovery of the BCL-2 (for review, see Merry and Korsmeyer, 1997) and Caspase (for review, see Martinou and Sadoul, 1996) families of proteins that regulate apoptosis. Analysis of theBcl-2 gene family has revealed a broad range of both promoters and suppressors of apoptosis. Many of these molecules are present and active within the nervous system. For example, elimination of BCL-2, a cell death inhibitor, leads to progressive degeneration of both motoneurons and sensory neurons at early postnatal ages (Michaelidis et al., 1996). Likewise, targeted disruption ofBcl-xL, a Bcl-2 related gene, reveals an early requirement for this molecule to prevent programmed cell death (Motoyama et al., 1995). In addition to the death-inhibiting members of the Bcl-2 gene family, death-promoting members, BAX (Oltvai et al., 1993), BAK (Kiefer et al., 1995), and BAD (Yang et al., 1995), have been characterized. Targeted disruption of theBax gene has shown that BAX is required for at least two populations of neurons to undergo programmed death in the setting of trophic factor or target deprivation (Deckwerth et al., 1996). Furthermore, motoneurons in the facial nucleus are increased in number in these mice, suggesting an effect on naturally occurring neuron death as well (Deckwerth et al., 1996). Finally, introduction of theBax null mutation intoBcl-xL-deficient mice greatly reduces the excessive apoptosis that occurs in the developing nervous system inBcl-xL-deficient mice (Schindler et al., 1997).
The discovery that neuronal apoptosis is powerfully regulated by BAX offers the opportunity to define periods of naturally occurring cell death, determine which neuronal populations are regulated, and determine the fate of supernumary neurons that ordinarily die during nervous system development. We have addressed these issues inBax−/− mice by studying several well-characterized neural systems during embryonic and early postnatal life. We show here that BAX primarily regulates the survival of postmitotic neurons. This effect is seen in most populations of peripheral neurons and many populations of CNS neurons. Perhaps surprisingly, many neurons in BAX-susceptible populations are markedly atrophic, suggesting that these cells may not function normally.
MATERIALS AND METHODS
Animals. Wild-type and Bax−/− mice were obtained from overnight matings [day of vaginal plug = embryonic day 0.5 (E0.5)] of mice heterozygous for Bax (Knudson et al., 1995). Pregnant females were killed by halothane overdose to harvest embryos on E11.5, E12.5, E13.5, E14.5, E15.5, and E17.5. Staging of embryos was verified by crown–rump length and degree of limb development (Kaufman, 1992).
The genotyping of mice was performed by PCR using a set of three primers: Bax exon 5 forward primer (5′-TGATCAGAACCATCATG-3′), Bax intron 5 reverse primer (5′-GTTGACCAGAGTGGCGTAGG3′), and Neo reverse primer (5′-CCGCTTCCATTGCTCAGCGG-3′). Cycling parameters were 5 min at 94°C for one cycle and 1 min at 94°C, 1 min at 55°C, and 1 min at 74°C each for a total of 30–35 cycles. PCR products were resolved on a 1.5% agarose gel.
Staining and analysis of apoptotic figures. Whole staged embryos were immediately frozen on dry ice and stored at −80°C. Before use, embryos were embedded in Tissue-Tek OCT compound (Miles, Elkhart, IN) and sectioned at 18 μm on a cryostat. To detect cell death in the DRG, we stained sections with the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method using the ApopTag fluorescent detection kit (Oncor, Gaithersburg, MD) according to the instructions of the manufacturer. Slides were then washed three times (5 min each) with PBS and visualized with FITC.
The TUNEL method labels cells containing fragmented DNA, a hallmark of apoptosis. Whether a cell exhibited positive labeling (FITC-labeled 3′-OH ends of fragmented DNA) was evaluated by examination of tissue sections using epifluoresence at a magnification of 225×. Cells undergoing apoptosis were recognized by an intensely fluorescent nucleus. For lumbar DRG quantitative analysis, fluorescent cells were counted in at least 20 lumbar DRG or spinal cord sections per animal, and a mean number of TUNEL-positive (TUNEL+) cells per section was determined. All observations reported are based on analysis of multiple tissue sections from three to five Bax−/− mice at each of the ages indicated and from similar numbers of Bax+/+littermates.
Histological analysis of myelinated-axon number and area.After overdose with halothane, adult animals were perfused with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m phosphate buffer (PB), pH 7.4. The dorsal and ventral roots of lumbar levels 4 and 5 (L4/L5) and the optic and facial nerves were removed, soaked in fresh fixative overnight, rinsed in PB, osmicated for 30 min in 1% osmium tetraoxide, rinsed, dehydrated in ascending concentrations of ethyl alcohol, and embedded in Epon resin. Semithin sections were cut at 1 μm. A minimum of three Bax+/+ and threeBax−/− mice were studied.
Cross-sections of each nerve root were photographed, montages were constructed, and myelinated axons were counted manually. For determination of axonal cross-sectional areas, high-power images of nerve roots were scanned into Macintosh Adobe Photoshop 4.0 using a Polaroid Slide scanner. The images were calibrated and transferred into the image analysis program, National Institutes of Health Image, for axon area measurements. Measurements do not include the myelin sheath surrounding the axons.
Analysis of cranial nerve VII soma size. Adult animals of known genotype were deeply anesthetized with a ketamine/xylazine/acepromazine (3:3:1) cocktail and perfused transcardially with phosphate buffer followed by 4% paraformaldehyde. Heads were removed and post-fixed 2 hr before the brainstem and caudal telencephalon were removed. After being rinsed overnight in phosphate-buffered sucrose (5% sucrose), brains were dehydrated via a graded series of alcohols and embedded in Paraplast X-tra (Fisher Scientific, Houston, TX). Twelve micrometer serial sections were cut through the entire facial nucleus and mounted on Superfrost Plus slides (Fisher Scientific). Sections were stained with 0.5% cresyl violet. To determine soma size, we analyzed 400 neurons from eachBax−/− and Bax+/+ animal. Every fourth section was photographed and scanned into the Adobe Photoshop program. After being encoded to blind genotype, every cell with a clear nucleoli was traced and imported into the National Institutes of Health Image analysis program to determine soma area. No attempt was made to account for the tissue shrinkage caused by histological processing.
RESULTS
BAX regulates developmental death of neurons in spinal ganglia and spinal cord
DRGs and spinal cord
In control mice, abundant TUNEL+ cells were detected in lumbar DRGs between E12.5 and E14.5 (Fig. 1). Relatively few apoptotic figures were present before E12.5 or after E14.5 (data not shown). In sharp contrast with control mice, virtually no cell death was detected between E12.5 and E14.5 in theBax−/− mice (Fig. 1). Quantification confirmed the visual impression of virtual elimination of naturally occurring cell death in the Bax−/− animals (Fig.2).
Fig. 1.

Photomicrographs of FITC-labeled cells using the TUNEL method in ventral spinal cord and DRG of wild-type andBax null mutant mice (E12.5–E14.5). Note that a considerable number of FITC-labeled cells are seen throughout wild-type DRG and ventral spinal cord, whereas FITC-labeled cells are absent inBax null mutant littermates. DRG, Dorsal root ganglion; VH, ventral horn of spinal cord. Scale bar, 50 μm.
Fig. 2.

Histograms comparing the number of TUNEL-labeled cells in DRG and motor pools of wild-type and Bax null mutant mice. The total number of TUNEL-labeled cells present in DRG and ventral spinal cords of staged wild-type (gray bars) and Bax null mutant (black bars) mice is shown. Cell number is expressed as a mean number of TUNEL-labeled cells present in each section of DRG and ventral spinal cord. Note that TUNEL-labeled cells were virtually absent on all days examined in Bax null mutants.
To examine whether BAX was necessary for developmental motoneuron death, we quantitated the number of apoptotic figures present on different embryonic days in lumbar spinal cords of Bax−/−and Bax+/+ mice. At E11.5, lumbar spinal cords of both genotypes lacked apoptotic figures. Beginning at E12.5, lumbar spinal cords of Bax+/+ mice exhibited large numbers of apoptotic figures (Fig. 1, top left). In contrast,Bax−/− mice of the same age exhibited few, if any, TUNEL+ cells (Fig. 1, top right). Similar findings were observed in E14.5 Bax−/− and Bax+/+ spinal cords (Fig. 1, bottom). The period of naturally occurring cell death seemed to be primarily over by E15.5. Surprisingly, analysis of lumbar spinal cords in Bax+/+ mice at E13.5 demonstrated few apoptotic figures (Fig. 1, middle). This observation appears to be consistent with the early and late periods of motoneuron cell death present in chick cervical spinal cord (Yaginuma et al., 1996).
Few TUNEL+ cells were observed outside the motor pools between E11.5 and postnatal day 1 (PN1) in either Bax+/+ orBax−/− mice. However, we did find occasional TUNEL+ cells in the ventricular zone of both genotypes. These data suggest that the deletion of BAX does not affect survival of proliferating cells. By PN4, increased cell death outside motor pools was observed in bothBax+/+ and Bax−/− mice. Whether these TUNEL+ cells were interneurons or glia was not determined.
BAX regulates developmental death of cranial ganglia and brain
Trigeminal and cochleovestibular ganglia
Cell death was readily detected in the trigeminal ganglion from E11.5–PN4 in wild-type mice. Thus, naturally occurring death is detected over a longer developmental time frame in this ganglion than in DRGs. Remarkably, almost no TUNEL+ cells were detected in the trigeminal ganglia of Bax−/− mice at any of the ages studied. Representative tissue sections are shown in Figure3.
Fig. 3.
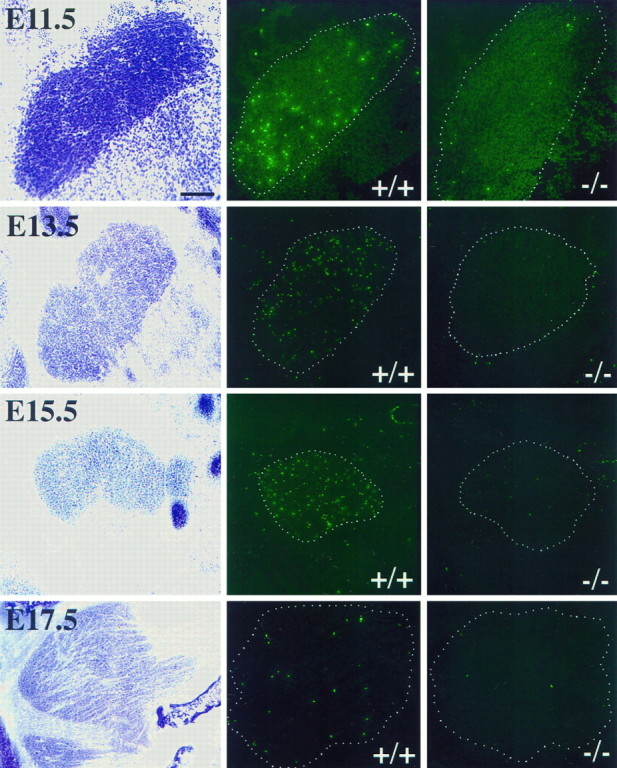
Photomicrographs of trigeminal ganglion (E11.5–E17.5) stained with cresyl violet (left) and corresponding sections labeled using the TUNEL method (center and right) in wild-type andBax null mutant littermates, respectively. Note that a considerable number of FITC-labeled cells are seen throughout wild-type trigeminal ganglion at every age shown (center), whereas FITC-labeled cells are absent in Bax null mutant littermates (right). Scale bar, 50 μm.
We also studied the cochleovestibular ganglia that were assessed at E13.5–E17.5. During that time frame, abundant TUNEL+ cells could be seen in wild-type mice. Similar to the situation in the DRG and trigeminal ganglion, TUNEL+ cells were virtually absent inBax−/− mice (Table 1).
Table 1.
Apoptotic figures in Bax−/− andBax+/+ mice
| E11.5 | E13.5 | E15.5 | E17.5 | PN1 | PN4 | |
|---|---|---|---|---|---|---|
| Brainstem | ↓ | ↓ | ↓ | ↓ | ↓ | |
| Cerebellum | ↓ | ↓ | ↓ | ↓ | ||
| DRG | – | ↓ | ↓ | ↓ | ↓ | ↓ |
| Hippocampus | ↓ | |||||
| (PN5) | ||||||
| Retina | – | – | – | – | – | ↓ |
| Spinal cord | – | ↓ | ↓ | ↓ | ↓ | – |
| Sympathetic ganglia | ↓ | |||||
| Telencephalon | – | – | – | – | – | |
| Trigeminal ganglia | ↓ | ↓ | ↓ | ↓ | ↓ | – |
| Cochleovestibular ganglia | ↓ | ↓ | ↓ | |||
| Optic nerve | – | |||||
| Sciatic nerve | – |
Arrows show changes in the number of apoptotic figures of Bax−/− when compared with wild-type littermates. – indicates no difference in apoptosis between Bax null mutant and wild-type littermates. Blank space indicates that timepoint was not examined.
Trigeminal brainstem nuclear complex and cerebellum
Brainstems were examined at ∼48 hr intervals between E13.5 and PN4. As expected from the results described above for ventral motor pools, the numbers of TUNEL+ cells in the facial nucleus were markedly reduced in Bax−/− mice (data not shown). We next focused on structures in the brainstem associated with sensory processing. Analysis was concentrated on the trigeminal brainstem nuclear complex (TBNC) that is the central target of trigeminal primary afferents. Results at PN1 are shown in Figure 4(top). In control mice, large numbers of TUNEL+ cells were observed in the postnatal period. The developmental cell death in this brainstem structure was virtually eliminated in Bax−/−mice.
Fig. 4.
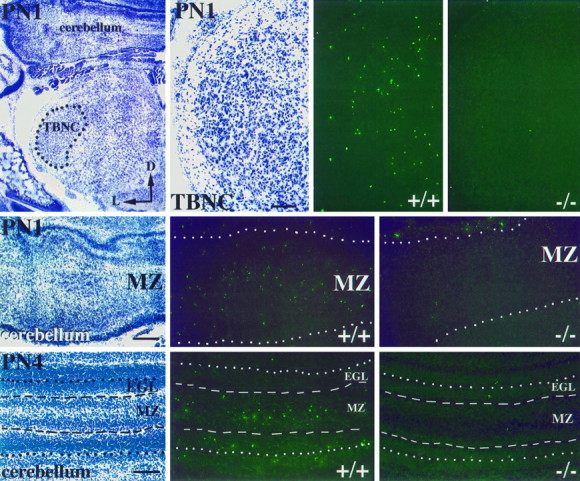
Cresyl violet staining and TUNEL labeling in postnatal TBNC and cerebellum of wild type and Bax null mutants. Low- and high-power photomicrographs of cresyl violet staining in TBNC (top, left) and corresponding high-power TUNEL-labeled sections (top,right) at PN1 are shown. Notice the absence of TUNEL-labeling in Bax−/− TBNC (top,rightmost). Photomicrographs of the cerebellar MZ stained with cresyl violet and the TUNEL method at PN1 (middle) and PN4 (bottom) in wild-type and Bax null mutant mice are shown. Note the extensive labeling at PN1 and PN4 throughout the MZ of wild-type cerebellum.TBNC, Trigeminal brainstem nuclear complex;MZ, marginal zone; EGL, external granule cell layer; D, dorsal; L, lateral. Scale bars, 50 μm.
Apoptotic figures were also readily detected in the cerebellum of wild-type mice at PN1 (Fig. 4, middle). TUNEL+ cells were observed throughout the marginal zone (MZ) of the cerebellum. This is a zone in which immature Purkinje cells are found during migration to their final position. However, no TUNEL+ cells were seen in this region in Bax−/− mice. Three days later at PN4, apoptotic figures could be detected in both the MZ and external granule cell layers in wild-type mice (Fig. 4, bottom). In contrast, inBax−/− mice, only a few TUNEL+ cells were noted in these regions. We repeatedly observed such differences in multiple sections within the developing cerebellum in all pairs of animals examined.
Eye
The developing eye was analyzed in detail (Fig.5). Here, the pattern of naturally occurring cell death is more complex than in the periphery. Abundant cell death was observed as early as E11.5 in the developing layers of both the wild-type and Bax−/− eye (see Fig. 5,top) and continued through PN1. It is clear that many cells (precursors, postmitotic neurons, and non-neuronal cells) in both the developing eye and the associated optic nerve (Fig. 5,middle) are TUNEL+. Thus, cell death during this period seems to be BAX-independent.
Fig. 5.

TUNEL-labeled cells in the developing eye of wild-type and Bax null mutant mice at E11.5 (top), the optic nerve in a Bax null mutant at E17.5 stained with cresyl violet and the corresponding TUNEL-labeled section (middle), and TUNEL-labeled cells in the neural layers of the retina in wild-type and Baxnull mutant mice at PN4 (bottom). Numerous TUNEL-labeled cells were observed in both the developing neuroepithelium of the eye (top) and in the glial cells of the optic nerve (middle, arrows) of wild-type andBax−/− animals. In contrast, TUNEL-positive cells were absent in the RGC layer of the Bax null mutant at PN4, a time at which peak cell death is occurring (bottom).Asterisk, Optic chiasm; RGC, retinal ganglion cell layer; vh, vitreous humor. Scale bars, 50 μm.
Between PN1 and PN4, abundant TUNEL+ cells were also seen throughout the developing retina in both wild-type and Bax null tissue (Fig. 5, bottom). However, in the retinal ganglion cell (RGC) layer, TUNEL+ figures were absent in Bax−/− mice. This time period has been established previously as the period of highest developmental death in the wild-type RGC layer (Young, 1984). Thus, BAX seems to influence the death of RGCs in the postnatal period. Elimination of death in Bax−/− RGCs results in a substantial increase in the size of the optic nerve (see below).
Hippocampus
At PN3, the time of greatest programmed cell death in developing mouse hippocampus (Reznikov, 1982), abundant TUNEL+ figures are apparent in Bax+/+ mice. Apoptosis was particularly prominent in CA2 and CA3. Apoptotic figures were seen in both the granule and molecular cell layers. As we observed in the other regions analyzed, virtually no TUNEL+ figures are seen in Bax−/−mice (n = 3; data not shown).
BAX may not influence survival of non-neuronal cells
Sciatic and optic nerve
The majority of glial cells associated with the optic (Barres et al., 1992, 1993; Raff et al., 1993) and the sciatic (Jessen et al., 1994; Dong et al., 1995; Gavrilovic et al., 1995; Trachtenberg and Thompson, 1996) nerves are dependent on an axon-derived signal for prevention of cell death during embryonic and postnatal periods of development. To determine whether programmed cell death in glial cells is also affected by the deletion of Bax, we examined optic and sciatic nerves at PN4 for the presence of apoptotic figures. In contrast to the elimination of programmed cell death in the DRG and the RGC layer, the mean number of TUNEL+ cells per section (∼5) in at least 10 sections of each Bax null mutant optic and sciatic nerve (n = 4) did not differ from that of wild-type littermates (n = 3; data not shown). These results suggest that the bulk of the naturally occurring cell death in non-neuronal cells is not prevented by deletion of BAX at this age.
Many neurons in adult Bax−/− mice are atrophic
The virtual elimination of apoptosis in many regions of the nervous system during the period of naturally occurring cell death raises the question of mature neuron fate. To begin to address this issue, we examined nerve morphology and myelinated-axon number in the optic nerve and in L4/L5 dorsal and ventral roots. Additionally, we measured both the number and area of myelinated axons in the facial nerve as well as the cross-sectional area of facial motoneurons.
Low-power photomicrographs of typical optic nerves from adult wild-type and Bax null mutant mice are shown in Figure6. Optic nerves in Bax null mutant mice were dramatically enlarged; cross-sectional areas were increased by 30%. The increase in nerve diameter seems to be caused by greater numbers of both small myelinated (Table2) and unmyelinated axons. Whether supernumerary retinal ganglion cell axons innervate normal targets in the lateral geniculate and superior colliculus is unknown. Interestingly, there was not only an increase in the number of very small myelinated axons but also an across-the-board reduction in axon size (see below).
Fig. 6.
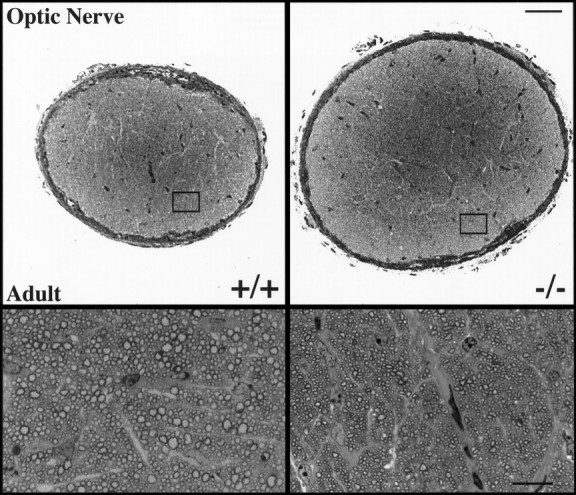
Semithin sections of myelinated axons in the optic nerve of adult wild-type and Bax null mutant mice. The dramatic increase in size of the optic nerve from a Baxnull mutant mouse when compared with that from a wild-type littermate is shown (top, at low magnification). That the increase in optic nerve diameter is attributable to the increased number of axons present in the Bax null mutant when compared with a Bax+/+ mice is shown (bottom, at high magnification). Note also the across-the-board reduction in the size of myelinated axons. Scale bars, 50 μm.
Table 2.
Optic nerve size and myelinated-axon number
| Surface area of optic nerve | Axons per 160 μm2 region of optic nerve | |
|---|---|---|
| Wild type | 59500 ± 7934 μm2* | 162 ± 10* |
| Bax−/− | 83230 ± 3317 μm2 | 233 ± 11 |
*Mean ± SE.
In contrast to the large increase in cross-sectional area of optic nerves, little increase was detected in the area of L4/L5 dorsal or ventral roots. However, counts of myelinated axons in L4/L5 dorsal roots revealed an increase of 24% in Bax null mutant nerves compared with control nerves. Similarly, 34% more axons were detected in the ventral roots of Bax−/− animals (Table3). These findings are thus consistent with our observations of the virtual elimination of apoptosis in retina, DRG, and spinal cord.
Table 3.
Number of myelinated axons in sensory and motor nerves
| No. Axons | % Increase | ||
|---|---|---|---|
| L4/L5 DR | WT | 1737 ± 103* | — |
| KO | 2284 ± 104 | 24 | |
| L4/L5 VR | WT | 932 ± 67 | — |
| KO | 1422 ± 47 | 34 | |
| CN VII | WT | 2156 ± 73 | — |
| KO | 3327 ± 62 | 35 |
*Mean ± SE.
DR, Dorsal root; VR, ventral root; WT, wild type; KO, knockout; and CN VII, cranial nerve VII.
Photomicrographs of semithin sections of facial nerves stained for myelin from Bax−/− and wild-type mice are shown in Figure7. Similar to observations inBax−/− L4/L5 roots, there was no increase in the cross-sectional area of the facial nerve of null mice. However, there were 35% more myelinated axons in the facial nerve ofBax−/− animals versus littermate controls (Table 3). This finding is consistent with the increase in facial neuron numbers demonstrated previously (Deckwerth et al., 1996). Strikingly, however, supernumerary motor axons do not appear to be normal. Most facial motor axons have large cross-sectional areas consistent with a preponderance of α motor neurons projecting axons to the facial muscles. However, in Bax−/− mice, abundant small, thinly myelinated axons were observed (Figs. 7, 8,top). We hypothesize that the smallest axons are from neurons saved from apoptosis. An additional finding, however, was that axons in all size ranges were reduced in size; mean axon caliber in the facial nerve of Bax−/− mice is 14% smaller than that of wild-type littermates.
Fig. 7.
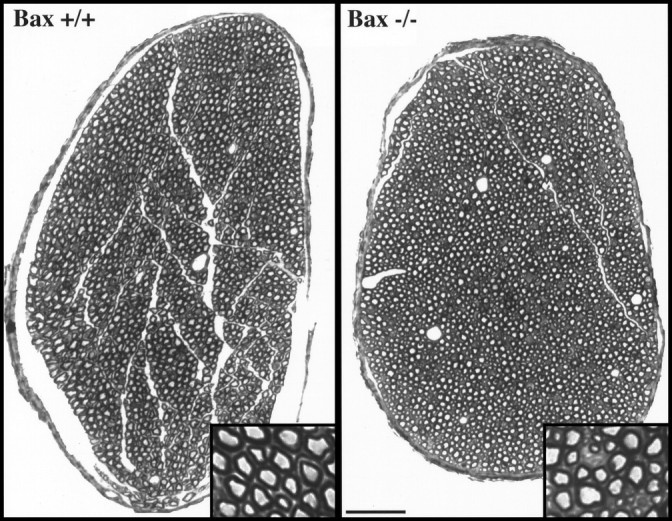
Semithin sections of myelinated axons in the facial nerve of adult wild-type and Bax null mutant mice at low magnification. The insets show, at higher magnification, that many axons in Bax null mutant nerve are markedly atrophic. Scale bar, 50 μm.
Fig. 8.
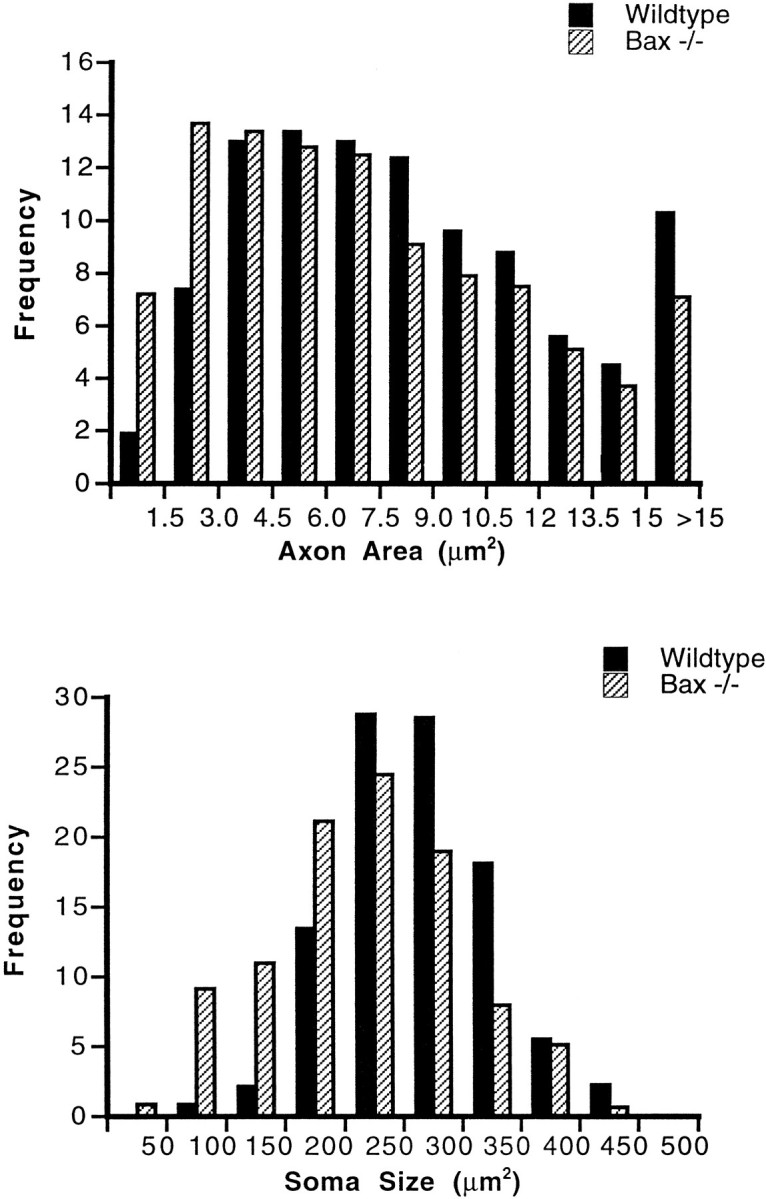
Cross-sectional area of axons and soma ofBax−/− and wild-type facial nucleus. Size-frequency histograms of axon area in adult wild-type and Bax null mutant facial nerves (top) are shown. Size-frequency histograms of motoneuron cross-sectional area in the facial nucleus ofBax null mutant and wild-type littermates (bottom) are shown.
The significant decrease in myelinated axon area in adultBax−/− facial nerves suggested that motoneurons within the facial nucleus would also be atrophic. To test this hypothesis, we measured the cross-sectional area of motoneurons in the facial nucleus of Bax−/− and wild-type littermates. The results are shown in Figure 8 (bottom). Again, two abnormalities were observed, (1) an across-the-board reduction in all soma sizes with a mean somal reduction of 17% and (2) an unexpectedly large increase in the numbers of neurons in the smallest size ranges.
DISCUSSION
The most striking consequence of the Baxdeficiency is the virtual elimination of developmental cell death in many populations of neurons. The uniform reduction in cell death in the neural systems examined suggests that these results will generalize to other regions of the nervous system. A potential objection to our interpretation is that the absence of TUNEL+ staining may not imply that apoptosis has been eliminated. In theory, neurons could die during development without exhibiting the endonucleosomal DNA fragmentation that allows readily detectable nick-end labeling using terminal transferase (see Clarke, 1990). Although this possibility cannot be formally excluded, the fact that supernumerary neurons are present inBax−/− mice make this possibility unlikely.
BAX regulates survival of PNS neurons with differing trophic requirements
In Bax−/− animals, cell death in the DRG and in trigeminal, sympathetic, and cochleovestibular ganglia during midembryonic and early postnatal development was virtually eliminated (see Table 1). Interestingly, these ganglia contain specific subpopulations of neurons that require different neurotrophin family members for survival. As we detected virtually no TUNEL+ cells in any of these ganglia in Bax−/− mice, we conclude that BAX is not linked in a direct way to any specific member of the neurotrophin family. This result is surprising given the large number of BCL-2 family members and in vitro evidence in chick that both BCL-2 (Allsop et al., 1993) and BAX (Middleton et al., 1996) differentially regulate neurotrophin-responsive, as opposed to cytokine-responsive, peripheral neuron populations. The extent to which our results will generalize to cytokine-dependent peripheral ganglia such as the ciliary, or glial-derived neurotrophic factor-dependent (GDNF-dependent) ganglia such as the enteric, remains to be determined.
The powerful effects of BAX on peripheral neuron survival are underscored by the dramatic ability of peripheral neurons fromBax−/− mice to survive in vitro in the absence of exogenous neurotrophic molecules. For example, we (Deckwerth et al., 1996) have shown previously that sympathetic ganglion cells fromBax−/− mice can be cultured in the absence of any exogenous growth factor, including NGF, for more than 21 d. More recently, we have found that DRG neurons from Bax−/− mice can be maintained in vitro for at least 14 d in the absence of any neurotrophin (Lentz et al., 1997). Furthermore, both NGF- and neurotrophin-3-dependent populations survive inBax−/− cultures, as demonstrated by the fact that when either factor is added, a subpopulation of surviving neurons responds. It is important to point out, however, that surviving neurons cultured in the absence of neurotrophins are not normal. In fact,Bax−/− mice may offer a convenient means of separating survival versus growth-promoting effects of neurotrophic factors for many populations of PNS neurons (see below).
BAX regulates survival of several populations of CNS neurons
BAX protein is widely distributed within the CNS, consistent with a role in regulation of CNS neuron survival (Krajewski et al., 1994). Indeed, Bax deficiency decreased neuronal death in several populations of CNS neurons examined. In particular, death of spinal motoneurons between E11.5 and PN1 was virtually eliminated inBax−/− mice. Similarly, there seemed to be a powerful effect on retinal ganglion cells during the postnatal period of naturally occurring cell death. Effects on brainstem, cerebellar, and hippocampal cell death were also observed. These effects are particularly interesting because most populations of CNS neurons are thought to require multiple factors for survival during development.
Reduction of cell death in the CNS results in a 50% increase in motoneuron number (Deckwerth et al., 1996) and a 35% increase in motor axon number (Table 3). Although it is important to point out that very atrophic motoneurons could have been missed in material analyzed at the light microscopic level, the number of supernumerary motoneurons inBax−/− animals approximately corresponds to the number of dying postmitotic motoneurons determined previously in studies of mouse spinal cord and brainstem (Lance-Jones, 1982; Oppenheim, 1991). However, deletion of Bax did not appear to effect the substantial number of proliferative cells that die during neurogenesis (Blaschke et al., 1996; Galli-Resta and Ensini, 1996), because we did not detect differences between Bax−/− and wild-type mice in the number of TUNEL+ cells present in the ventricular zone. Thus, the increased numbers of cells observed in Bax null mice can be explained simply as the result of the elimination of postmitotic cell death.
In this regard, it is interesting to consider differences betweenBax−/− mice and mice with a targeted deletion of CPP32 (Caspase-3). Deletion of CPP32 effectively eliminates death of proliferative cells (Kuida et al., 1996). Moreover, CPP32 may also regulate survival of non-neuronal elements within the CNS (Keane et al., 1997). The end result is a brain that is grossly hypertrophied. In contrast, the brains of Bax−/− mice are not significantly larger than control. The fact that BAX is regulating a period of death that occurs after cell proliferation, combined with the fact that survival of non-neuronal cells may not be increased inBax−/− mice, may in part explain this outcome.
Although the number of TUNEL+ cells is dramatically reduced in the CNS of Bax−/− mice, in vitro data suggest that the consequences of the Bax deficiency may be somewhat different for the PNS and CNS. When disassociated PN0 Bax−/−superior cervical ganglia (SCG) neurons are cultured in the absence of trophic factors, these cells demonstrate an ability to survive for at least 23 d (Deckwerth et al., 1996). In striking contrast, disassociated cultures of PN4 Bax−/− cerebellar granule cells are able to survive only 7 d after potassium deprivation, an apoptosis-inducing event (Miller et al., 1997). These findings suggest that BAX may be a more powerful regulator of apoptosis of peripheral neurons than of central neurons. In fact, differences between PNS and CNS neurons in signal transduction pathways related to survival have recently been demonstrated (Meyer-Frank et al., 1995). It is possible that the variation in the survival-promoting effects of BAX deficiency is related to the differences in signal transduction pathways used by CNS and PNS neurons in vitro.
Neurons saved from naturally occurring cell death may not function normally
It is interesting to consider the fate of neurons saved from apoptosis in the Bax null mutant mice. There are two apparent possibilities. First, supernumerary neurons may be poorly connected with their target structures and may therefore be unable to obtain trophic support. In this scenario, one would expect to find substantial numbers of markedly atrophic neurons and axons, as well as a population of cells of relatively normal size and morphology. Alternatively, supernumerary neurons might form appropriate connections with their peripheral target structures and participate fully in normal functions. This hypothesis would predict increased competition for trophic support, resulting in uniformly smaller soma and axon cross-sectional areas.
In support of the first possibility, a striking finding inBax−/− mice is the presence of markedly atrophic neurons and axons. Furthermore, although the size of all motoneurons is reduced in Bax−/− mice (see below), there was also a unexpected increase in the number of very small caliber axons. In fact, motor axons with cross-sectional areas of less than 3 μm2 were more than twice as frequent inBax−/− mice than in control mice. Indeed, axons in this small size range account for the majority of supernumerary axons encountered in these mice. This latter observation suggests that the supernumerary neurons are unable to acquire target-derived trophic factors that regulate axon caliber (Munson et al., 1997).
During innervation of peripheral structures, target-derived factors influence both the survival and growth of developing neurons. The presence of supernumerary, but markedly atrophic, neurons and axons suggests that these two effects are disassociated inBax−/− mice. Other data are consistent with this view. For example, Bax−/− facial motoneurons survive axotomy in the neonatal period, although the resultant cells, deprived of connection with muscle, are atrophic compared with contralateral motoneurons (Deckwerth et al., 1996). Similarly, Bax−/− DRG neurons, which can survive NGF deprivation in vitro, are markedly atrophic and exhibit vastly reduced neurite outgrowth when compared with Bax−/− DRG neurons grown in the presence of NGF (Lentz et al., 1997). Bax−/− sympathetic ganglia neurons also survive NGF withdrawal in vitro, although the cells undergo reductions of 90% in mRNA and protein synthesis (T. Deckwerth and E. M. Johnson, Jr., personal communication). Thus, deletion of BAX is able to compensate for the survival-promoting but not the growth-promoting effects of target-derived factors.
Complicating the interpretation that atrophic neurons are not fully connected is the fact that motoneurons and associated axons in all size ranges, including the very largest, are smaller in Bax null mutants than in wild-type littermates. Because there is an increase of 35% in facial motor axons of Bax null mutant animals, this finding is consistent with an across-the-board reduction in the amount of trophic support available per neuron. The possibility must also be considered, however, that BAX itself may be involved in regulation of cell and axon size. Although BAX and other BCL-2 family members have traditionally been considered regulators of apoptosis, a recent study suggests that BCL-2 may influence neuronal morphology (Chen et al., 1997).
The phenotype of mice overexpressing the antiapoptotic protein BCL-2 is very similar to the phenotype of mice with a Bax deletion. Both eliminate apoptotic cell death in facial motoneurons, retinal ganglion cells, and other neuronal populations. However, Martinou and colleagues (Martinou et al., 1994) reported that neuron specific enolase-Bcl-2 (NSE-Bcl-2) mice exhibited hypertrophied brains that they attributed to an increase in the overall number of neurons. Unlike brains of NSE-Bcl-2 mice, however, gross observations of brains from Bax null mice do not reveal any discernible differences in size compared with the brains of wild-type littermates. This evidence raises the possibility that neuronal size may be enhanced by overexpression of BCL-2 and reduced by deletion of BAX. Overall, we favor the interpretation that supernumary neurons in Bax−/− mice are not fully connected to their targets and that, in addition, the absence of BAX interferes with the growth of normally connected neurons.
Conclusions
In summary, we have shown that deletion of BAX eliminates much of the postmitotic neuronal death that occurs in both PNS and CNS during development, but the resultant supernumerary neurons and axons are atrophic. BAX mutants therefore constitute a valuable model for understanding the mechanisms as well as the role of cell death in the developing nervous system. Importantly, these mice may represent a valuable tool to test the effects of inhibition of apoptosis in neurodegenerative disorders.
Footnotes
This work was supported by National Institutes of Health Grants NS31768 and P5O-AG05681 to W.D.S. and HD27500 to S.J.K. and by the Muscular Dystrophy Association. We gratefully acknowledge John Harding for his excellent technical assistance and Dr. Judith Mosinger-Ogilvie for valuable discussions of the results.
Correspondence should be addressed to Dr. William D. Snider, Center for the Study of Nervous System Injury, Department of Neurology, Box 8111, Washington University School of Medicine, 600 South Euclid Avenue, St. Louis, MO 63110.
REFERENCES
- 1.Allsop TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Barres BA, Hart IK, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- 3.Barres BA, Jacobson MD, Schmid R, Sendtner M, Raff MC. Does oligodendrocyte survival depend on axons? Curr Biol. 1993;3:489–497. doi: 10.1016/0960-9822(93)90039-q. [DOI] [PubMed] [Google Scholar]
- 4.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 5.Chen DF, Schneider GE, Martinou JC, Tonegawa S. Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature. 1997;385:434–439. doi: 10.1038/385434a0. [DOI] [PubMed] [Google Scholar]
- 6.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 7.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 8.Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulates survival, proliferation and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- 9.ElShamy WM, Ernfors P. Requirement of neurotrophin-3 for the survival of proliferating trigeminal ganglion progenitor cells. Development. 1996;122:2405–2414. doi: 10.1242/dev.122.8.2405. [DOI] [PubMed] [Google Scholar]
- 10.Fariñas I, Yashida CK, Backus C, Reichardt LF. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 1996;17:1065–1078. doi: 10.1016/s0896-6273(00)80240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galli-Resta L, Ensini M. An intrinsic time limit between genesis and death of individual neurons in the developing retinal ganglion cell layer. J Neurosci. 1996;16:2318–2324. doi: 10.1523/JNEUROSCI.16-07-02318.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavrilovic H, Brennan A, Mirsky R, Jessen KR. Fibroblast growth factors and insulin growth factors combine to promote survival of rat Schwann cell precursors without induction of DNA synthesis. Eur J Neurosci. 1995;7:77–85. doi: 10.1111/j.1460-9568.1995.tb01022.x. [DOI] [PubMed] [Google Scholar]
- 13.Jessen KJ, Brennant A, Morgan L, Mirsky R, Kent A, Hasimoto Y, Cavrilovic J. The Schwann cell precursor and its fate: a study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron. 1994;12:509–527. doi: 10.1016/0896-6273(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman MH. The atlas of mouse development. Academic; New York: 1992. [Google Scholar]
- 15.Keane RW, Srinivasan A, Foster LM, Testa M-P, Örd T, Nonner D, Wang H-G, Reed JC, Bredesen DE, Kayalar C. Activation of CPP32 during apoptosis of neurons and astrocytes. J Neurosci Res. 1997;48:168–180. doi: 10.1002/(sici)1097-4547(19970415)48:2<168::aid-jnr9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Kiefer MC, Frauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Barr PJ. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- 17.Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 18.Krajewski S, Krajewski M, Shabaik A, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl-2. Am J Pathol. 1994;145:1323–1334. [PMC free article] [PubMed] [Google Scholar]
- 19.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 20.Lance-Jones C. Motoneuron cell death in the developing lumbar spinal cord of the mouse. Dev Brain Res. 1982;4:473–479. doi: 10.1016/0165-3806(82)90192-4. [DOI] [PubMed] [Google Scholar]
- 21.Lentz SI, Knudson CM, Harding JC, Korsmeyer SJ, Snider WD. Primary sensory neurons are unipolar in the absence of neurotrophins. Soc Neurosci Abstr. 1997;23:1413. [Google Scholar]
- 22.Martinou JC, Sadoul R. ICE-like proteases execute the neuronal death program. Curr Opin Neurobiol. 1996;6:609–14. doi: 10.1016/s0959-4388(96)80092-4. [DOI] [PubMed] [Google Scholar]
- 23.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 24.Merry DE, Korsmeyer SJ. Bcl-2 gene family in the nervous system. Annu Rev Neurosci. 1997;20:245–257. doi: 10.1146/annurev.neuro.20.1.245. [DOI] [PubMed] [Google Scholar]
- 25.Meyer-Frank A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signalling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 26.Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtman B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 27.Middleton G, Nunez G, Davies AM. Bax promotes neuronal survival and anatagonises the survival effects of neurotrophic factors. Development. 1996;122:695–701. doi: 10.1242/dev.122.2.695. [DOI] [PubMed] [Google Scholar]
- 28.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K-I, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 30.Munson JB, Shelton DL, McMahon SB. Adult mammalian sensory and motor neurons: roles of endogenous neurotrophins and rescue by exogenous neurotrophins after axotomy. J Neurosci. 1997;17:470–476. doi: 10.1523/JNEUROSCI.17-01-00470.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 32.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 33.Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 34.Reznikov KY. Cell proliferation and cytogenesis in the mouse hippocampus. Springer; Berlin: 1982. [DOI] [PubMed] [Google Scholar]
- 35.Schindler KS, Latham CB, Roth KA. Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci. 1997;17:3112–3119. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trachtenberg JT, Thompson VJ. Schwann cell apoptosis at developing neuromuscular junctions is regulated by glial growth factor. Nature. 1996;379:174–177. doi: 10.1038/379174a0. [DOI] [PubMed] [Google Scholar]
- 37.White FA, Silos-Santiago I, Molliver DC, Nishimura M, Phillips H, Barbacid M, Snider WD. Synchronous onset of NGF and TrkA survival dependence in developing dorsal root ganglia. J Neurosci. 1996;16:4662–4672. doi: 10.1523/JNEUROSCI.16-15-04662.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yaginuma H, Tomita M, Takashita N, McKay SE, Cardwell C, Yin Q, Oppenheim RW. A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci. 1996;16:3685–3703. doi: 10.1523/JNEUROSCI.16-11-03685.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 40.Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–373. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]