

Abstract
Specific isoforms of laminin (LN) are concentrated at neuromuscular junctions (NMJs) where they may participate in synaptic organization or function. In myotubes from C2 cells, LN is concentrated within the majority of spontaneous acetylcholine receptor (AChR) aggregates. Neural agrin substantially increases this colocalization, suggesting that agrin can recruit LN into AChR aggregates. Addition of LN to C2 myotubes induces a more than twofold increase in the number of AChR aggregates. These aggregates have a larger size and are more dense than are those induced by agrin, suggesting that LN is involved in the growth and/or stabilization of AChR aggregates. Consistent with this hypothesis, an antiserum to LN reduces the size of individual AChR aggregates but increases their number. In C2 myotubes, extracellular matrix receptors containing the integrin β1 subunit are poorly colocalized with AChR aggregates, suggesting that integrins may not be involved in LN-induced aggregation. In contrast, almost all AChR aggregates are associated with dystroglycan immunoreactivity, and monoclonal antibody (mAb) IIH6 against α-dystroglycan (α-DG), a LN and agrin receptor, causes a concentration-dependent inhibition of LN-induced aggregation. Moreover, S27 cells, which lack a functional α-DG, and two C2-derived cell lines expressing antisense DG mRNA fail to aggregate AChRs in response to LN. Finally, LN-induced AChR aggregation does not involve the phosphorylation of the muscle-specific tyrosine kinase receptor (MuSK) or the AChR β subunit. We hypothesize that the interaction of LN with α-DG contributes to the growth and/or stabilization of AChR microaggregates into macroaggregates at the developing NMJ via a MuSK-independent mechanism.
Keywords: laminin, α-dystroglycan, acetylcholine receptor aggregation, neuromuscular synapse, agrin, MuSK
At the adult neuromuscular junction (NMJ), sites of transmitter release are aligned precisely opposite postjunctional folds in the muscle membrane. At the crests of the folds, acetylcholine receptors (AChRs) are packed at a density of ∼10,000/μm2 (Fertuck and Salpeter, 1974), whereas in adjacent regions of the membrane, the receptor density is ∼1000-fold less. Before innervation, AChRs are diffusely distributed, but within hours of the first contact of the nerve terminal and muscle fiber, AChRs in the postsynaptic membrane begin to aggregate (Bevan and Steinbach, 1977). These early synapses are composed of loose assemblies of small receptor patches (Anderson and Cohen, 1977; Olek et al., 1986) that subsequently coalesce into more tightly organized receptor “plaques” that further increase in size and density until they attain the proportions found in the adult postsynaptic membrane (Steinbach, 1981).
Agrin released from the nerve terminal has been shown to activate the muscle-specific tyrosine kinase receptor MuSK leading to the aggregation of AChRs in the postsynaptic membrane (Valenzuela et al., 1995; DeChiara et al., 1996; Glass et al., 1996). Several studies have shown that agrin, in addition to triggering the aggregation of AChRs, also induces the aggregation of other synaptic proteins including laminin (LN), the heparan sulfate proteoglycan perlecan, acetylcholinesterase, muscle agrin, rapsyn, α-actinin, filamin, vinculin, and α- and β-dystroglycan (β-DG) (Daniels et al., 1984;Wallace, 1989; Nitkin and Rothschild, 1990; Shadiack and Nitkin, 1991;Lieth et al., 1992; Cohen et al., 1995). Furthermore, in skeletal muscle from MuSK knock-out mice, there is a complete failure in synapse formation, indicating that as for AChRs, agrin-induced aggregation of extracellular matrix (ECM), membrane, and cytoskeletal proteins requires the activation of MuSK. Therefore, agrin activation of MuSK initiates the creation of a separate compartment within the muscle fiber containing a specialized synaptic basal lamina (BL), as well as membrane and cytoskeletal proteins.
Several previous results indicate that LN may assist agrin in the formation and consolidation of postjunctional AChR aggregates. For example, LN is concentrated at agrin-induced AChR aggregates in culture, and isoforms of LN containing the β2 chain are restricted to the NMJ (Hunter et al., 1989). Furthermore, LN has been reported to induce AChR aggregation on cultured myotubes and to enhance the aggregating activity of a neuroblastoma × glioma hybrid cell-conditioned medium with agrin-like activity (Vogel et al., 1983). Recently, Denzer et al. (1997) reported that agrin isoforms containing a 15 amino acid N-terminal insertion bind with different affinities to LN, showing a preference for heterotrimers containing the synapse-specific β2 LN chain. AChR aggregates induced by this agrin splice variant are considerably smaller than are those induced by C-terminal fragments of agrin, suggesting that the interaction of agrin with LN can modulate the size of AChR aggregates.
Several LN receptors have been shown to be expressed and developmentally regulated in skeletal muscle and the C2 mouse muscle cell line. These include members of the integrin family of heterodimeric ECM receptors (Song et al., 1992; Collo et al., 1993;Belkin et al., 1996) as well as the dual LN and agrin receptor α-dystroglycan (α-DG), which is an extracellular peripheral membrane protein and a member of the dystrophin/utrophin-associated protein complex (Smalheiser and Schwartz, 1987; Douville et al., 1988;Ervasti and Campbell, 1991, 1993; Ibraghimov-Beskrovnaya et al., 1992;Matsumura et al., 1992; Gee et al., 1993). Most integrins that bind LN contain the β1 subunit that is found at NMJs (Bozyczko et al., 1989;Belkin et al., 1996) and at agrin- and nerve-induced AChR aggregates in cultured myotubes (Bozyczko et al., 1989; Anderson and Qiao Shi, 1996). LN also binds with high affinity to α-DG, and this interaction is thought to play a role in maintaining the structural integrity of the sarcolemma (Henry and Campbell, 1996). α-DG is concentrated at the NMJ in vivo (Matsumura et al., 1992) and at AChR aggregates in cultured myotubes (Cohen et al., 1995) and has been implicated in AChR aggregation (Campanelli et al., 1994; Gee et al., 1994, Cohen et al., 1995; but see Sugiyama et al., 1994). In the present study, we sought to determine the role of LN in agrin-induced AChR aggregation and to identify the receptor involved. We find that agrin-induced aggregation of AChRs is accompanied by a recruitment of LN at these aggregates and that exogenous LN alone induces the aggregation of AChRs on C2 myotubes in addition to potentiating the aggregating activity of agrin. We further show that LN-mediated AChR aggregation does not involve the phosphorylation of MuSK or the AChR β subunit. Finally, we used three mutant cell lines derived from the C2 cell line that express reduced levels of α-DG to demonstrate that this receptor is responsible for the observed effects of LN on AChR aggregation. Our results suggest a role for LN and α-DG in the growth and/or stabilization of AChRs into compact aggregates during synapse formation at the NMJ.
MATERIALS AND METHODS
Materials. LN was purified from Engelbreth–Holm–Schwarm (EHS) tumor by the method of Timpl et al. (1982). Coomassie blue- and silver-stained gels revealed only two bands at ∼215 kDa (β/γ chains) and 400 kDa (α chain) that did not cross-react with a monoclonal antibody to the LN α2. Polyclonal anti-LN antiserum was produced by immunizing rabbits with EHS tumor LN. This antiserum has been characterized previously (Morissette and Carbonetto, 1995) and recognizes all three subunits of LN 1 (α1, β1, and γ1) with a very faint band at 150 kDa corresponding to entactin. There is no cross-reactivity with agrin (F. Montanaro and S. Carbonetto, unpublished observations) or LN α2 chain (Morissette and Carbonetto, 1995). Anti-LN antiserum was purified by chromatography on Affi-Gel Blue (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions and was subsequently dialyzed into PBS, pH 7.2. Recombinant rat agrin(12,4,8) was purified from the conditioned medium of COS cells expressing a truncated C-terminal fragment (amino acids 1145–1940) (Ferns et al., 1993). Monoclonal antibody (mAb) IIH6 ascites fluid was prepared as described previously (Ervasti and Campbell, 1991). Monoclonal antibodies to β-DG and to the core protein of mouse perlecan were purchased from Novo Castra Laboratories (Newcastle upon Tyne, UK) and Chemicon (Temecula, CA), respectively. Monoclonal antibody 3A3 is directed against the rat integrin α1 subunit and was prepared as described previously (Turner et al., 1989). The polyclonal antiserum to the integrin β1 subunit was generated by immunizing rabbits with purified native rat integrin β1 subunit (Tawil et al., 1990). The polyclonal antiserum to mouse collagen IV was purchased from Chemicon. Polyclonal anti-MuSK antiserum was prepared by immunizing rabbits with a synthetic peptide corresponding to the C-terminal 20 amino acids of the rat MuSK protein (Glass et al., 1996).
Cell culture. C2 and S27 myotubes were cultured as described previously (Gee et al., 1994) on collagen-coated 18-mm-round glass coverslips in scored wells on 100 mm Petri plates. For experiments involving the DG-antisense cell lines, 11F, 11E, and control C2 cells were cultured on Falcon tissue culture plastic dishes coated with 0.15% gelatin. Cells were kept in growth medium (DMEM high glucose plus 10% fetal bovine serum; plus 70 μg/ml G-418 for 11F and 11E cells only) until confluent. Cultures were then switched to fusion medium (DMEM high glucose plus 1% horse serum) and allowed to differentiate for an additional 4 d. All cells were treated with LN, agrin, or anti-LN IgG on the third day of fusion.
Immunofluorescence microscopy. To visualize surface AChRs, we incubated myotubes with BODIPY- or rhodamine-conjugated α-bungarotoxin (Molecular Probes, Eugene, OR) for 30 min at room temperature. Myotubes were fixed with 2% paraformaldehyde in 0.1m phosphate buffer, pH 7.2, for 20 min and rinsed three times with PBS. For antibody staining, the myotubes were fixed as above and then blocked for 1 hr with PBS plus 1% horse serum. Cells were incubated with primary antibody in blocking buffer for 1 hr and then with the appropriate biotin-conjugated secondary antibody for an additional hour, followed by fluorescein-conjugated streptavidin for 20 min.
Quantification of AChR colocalization and number. All quantifications were performed by visual inspection of a minimum of 10 random fields using a 40× objective. For colocalization studies, AChR aggregates were scored as partially overlapping with the antigen of interest if at least a quarter of their surface colocalized with the antigen. AChR aggregates were scored as completely overlapping if the antigen immunoreactivity was coextensive with α-bungarotoxin staining in both size and shape. Determination of the number of AChR aggregates per myotube segment was performed as described previously (Gee et al., 1994). A myotube segment corresponds to 5.6 × 103 μm2.
Quantification of AChR number, size, and intensity for Table2. Thirty-five millimeter negatives were scanned into Adobe Photoshop for Windows (version 2.5.1) using an Agfa Arcus II scanner and FotoLook (version 2.05) with TWAIN acquisition. Brightness and contrast were adjusted to −25 and +25, respectively. Files were saved in TIFF format (for MacIntosh) and imported into NIH Image (version 1.59b). Images were processed with Invert Filter; then the background was subtracted using the two-dimensional rolling ball method (radius, 5–15). Background-subtracted images were analyzed for particle size and mean pixel intensity after thresholding the image. The mean pixel intensity is the average gray value for all of the pixels in an aggregate. The possible values for each pixel range from 0 (white) to 255 (black). Thresholded images were carefully compared with their unprocessed counterparts to ensure that AChR aggregate size and number were accurate. Measurements were imported into Excel (version 5.0; Microsoft) and SigmaPlot (version 5.0; Jandel Scientific, Corte Madera, CA) for analysis. AChR aggregates of < 1.0 μm2 were not included because image thresholding of low intensity or out-of-focus aggregates accounted for the majority of particles of this size.
Table 2.
Quantitation of AChR aggregate number, size, and intensity in C2 myotubes
| Treatment | Mean number per field | Mean area (μm2) | Mean average intensity (min = 0, max = 255) | Total area (μm2) |
|---|---|---|---|---|
| Control | 9 ± 1 (n = 11) | 16.4 ± 1.8 | 133 ± 6 | 150 |
| Agrin (200 pM) | 34 ± 4 (n = 13) | 7.5 ± 0.9 | 95 ± 9 | 306 |
| LN (60 nM) | 21 ± 2 (n = 10) | 17.4 ± 1.6 | 163 ± 7 | 356 |
| Agrin (200 pM) + anti-LN IgG (100 μg/ml) | 101 ± 14 (n = 10) | 3.8 ± 0.2 | 73 ± 6 | 385 |
Cultures were treated overnight with the indicated concentrations of neural agrin (Ferns et al., 1993), LN, and/or purified anti-laminin IgG. AChR aggregate number, size, and intensity were quantitated as described in Materials and Methods. The means for the indicated number of fields (n) containing at least one AChR aggregate are shown ± SEM. Total area refers to the area of the myotube occupied by AChR aggregates and is the product of the mean number of aggregates per field and the mean area of each aggregate.
Immunoprecipitation. For immunoprecipitations, cultures of 100 mm dishes of C2 myotubes were treated with agrin or LN for 15 min, rinsed with Ca2+- and Mg2+-free PBS containing 1 mm sodium vanadate, then harvested by scraping, and pelleted. Cell pellets were extracted for 15 min on ice with 25 mm Tris-glycine, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 1 mm sodium vanadate, 50 mm sodium fluoride, 1 μmaprotinin, 1 μm leupeptin, 1 μm pepstatin A, 1 mm benzamidine, 1 mm iodoacetamide, and 1 mm PMSF. Insoluble material was pelleted by centrifugation at 15,000 × g for 10 min at 4°C. Extracts of C2 myotubes were incubated with anti-MuSK antisera for 1 hr at 4°C with agitation; then immune complexes were precipitated by addition of protein G–Sepharose beads. The beads were pelleted at 1500 ×g and were washed three times with 50 mmTris-HCl, pH 8.0, 0.5 m NaCl, 50 mm sodium vanadate, and 1% Triton X-100. Bound proteins were eluted with 30 μl of reducing SDS-PAGE sample buffer. To isolate AChRs, we incubated C2 myotube cultures with 0.1 μg/ml biotin-conjugated α-bungarotoxin for 1 hr at 25°C, harvested and extracted the cells as described above, then added 100 μl of streptavidin–Sepharose beads, and incubated the mixture at 4°C for 1 hr. The beads were washed and prepared for SDS-PAGE as described above.
Western blot analysis. Samples were electrophoretically separated on 8% SDS-PAGE gels and transferred onto nitrocellulose membranes. The proteins were probed with a monoclonal anti-phosphotyrosine antibody (mAb 4G10) (Upstate Biotechnology, Saranac Lake, NY) in buffer containing 5% BSA, 10 mmTris-HCl, pH 7.4, 0.15 m NaCl, 0.5% Nonidet P-40, and 0.1% Tween 20. The blots were then incubated with a horseradish peroxidase-conjugated anti-mouse Ig secondary antibody (Amersham, Arlington Heights, IL). Bound antibody was visualized by chemiluminescence (ECL) (Amersham).
RESULTS
LN colocalizes with AChR aggregates in C2 cells
Previous studies have shown that LN colocalizes with AChR aggregates in cultured myotubes (Vogel et al., 1983; Bayne et al., 1984; Olwin and Hall, 1985; Nitkin and Rothschild, 1990; Gordon et al., 1993) and that it is concentrated at synapses in vivo (Sanes et al., 1990). In addition, agrin treatment of cultured chick myotubes was also shown to promote a rearrangement of LN on the cell surface (Nitkin and Rothschild, 1990). We therefore sought to confirm and quantify the presence of LN at both spontaneous and agrin-induced AChR aggregates in C2 myotubes. Cultures were double labeled with a polyclonal antiserum to LN and with rhodamine-conjugated α-bungarotoxin to visualize AChRs. LN was found to be concentrated at most spontaneous and agrin-induced AChR aggregates (Table1, Fig. 1). In agreement with a previous report (Gordon et al., 1993), LN immunoreactivity was also found in a punctate pattern on the myotube surface and in a few patches devoid of AChRs (Fig. 1). The amount of overlap of LN with AChR aggregates was categorized as partial or complete. Overlap was considered partial when ≥25% of the surface of the AChR aggregate was associated with LN immunoreactivity and complete when the two proteins were coextensive. In untreated C2 myotubes, ∼81% of spontaneous AChR aggregates overlapped partially with LN, and in 38%, the overlap was complete (Table 1). Overnight treatment of C2 myotubes with 200 pm of a recombinant neural agrin fragment [C-Agrin(4,8), hereafter referred to as agrin] increased the overlap of LN in almost all AChR aggregates, with a more than twofold increase in the number of AChR aggregates that overlap completely with LN (Table 1). To determine the specificity of this recruitment of LN to AChR aggregates in C2 cells, we mapped the distribution of other proteins found concentrated at the neuromuscular junction in vivo (Sanes et al., 1990; Belkin et al., 1996) and at spontaneous and agrin-induced AChR aggregates: collagen IV, the heparan sulfate proteoglycan perlecan, and the integrin β1 subunit. We found that only perlecan shows a high degree of overlap with spontaneous AChR aggregates, and this overlap is increased by agrin treatment. In contrast, collagen IV showed a poor colocalization with spontaneous AChR aggregates and was often concentrated in large patches devoid of AChRs on the cell surface (Fig. 1, Table 1). Similar poor colocalization was found with antisera for fibronectin (Montanaro and Carbonetto, unpublished observations). Agrin did not greatly increase localization of collagen IV at AChR aggregates, nor did it seem to alter its distribution on the cell surface. Only a subset of collagen IV chains have been shown to be specifically concentrated at synapses (Sanes et al., 1990), and we cannot exclude the possibility that the low level of colocalization of collagen IV with AChR aggregates is attributable to the inability of our antibody to recognize the synapse-specific collagen IV chains. However, our data indicate that agrin does not cause a reorganization of all BL components but selectively recruits LN and perlecan to AChR aggregates in C2 myotubes. Similar results have been reported with cultures of primary myotubes (Nitkin and Rothschild, 1990).
Table 1.
Overlap of LN and other proteins with AChR aggregates
| AChR Aggregates | Spontaneous | Agrin-induced | ||
|---|---|---|---|---|
| 100% Overlap | 25–100% Overlap | 100% Overlap | 25–100% Overlap | |
| LN | 38% (n = 489) | 81% (n = 139) | 83% (n = 129) | 95% (n = 129) |
| Perlecan | 46% (n = 91) | 66% (n = 91) | 95% (n = 156) | 99% (n = 156) |
| Collagen IV | N.D. | 56% (n = 247) | N.D. | 68% (n = 285) |
| Integrin β1 subunit | 0% (n = 117) | 23% (n = 117) | 2% (n = 115) | 12% (n = 115) |
Untreated C2 myotubes (spontaneous) and C2 myotubes that had been treated overnight with 200 pM agrin (agrin-induced) were double labeled for AChR aggregates using rhodamine-conjugated α-bungarotoxin and for the proteins indicated using indirect immunofluorescence with specific antibodies. The extent of overlap of these proteins with AChR aggregates was quantified by visual inspection. Overlap was considered complete if 100% of an aggregate was coextensive with immunoreactivity for the protein and partial if <100% but >25% was coextensive. Aggregates with <25% overlap with the protein were considered nonoverlapping. Data indicate the percentage of AChR aggregates that scored positive for overlap. The total number of AChR aggregates visualized is given in parenthesis. N.D., Not determined.
Fig. 1.
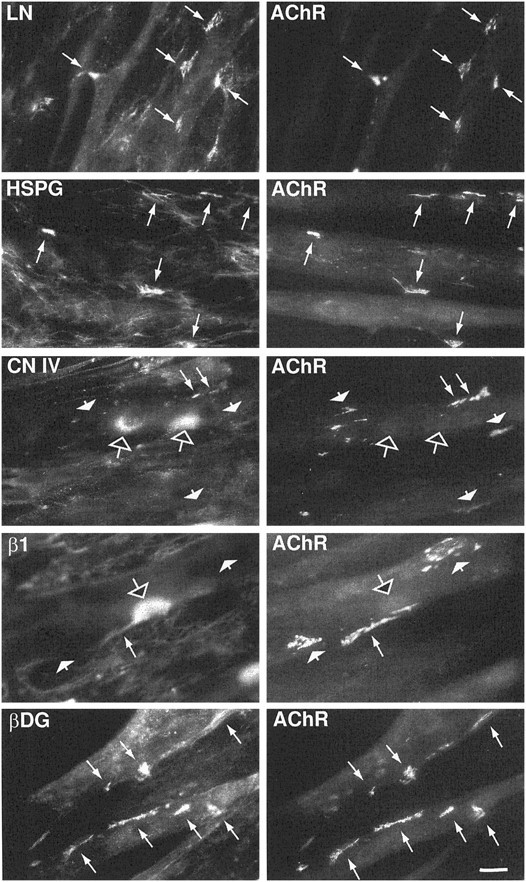
Colocalization of some synaptic proteins with agrin-induced AChR aggregates. C2 myotubes were incubated with 100 pm agrin for 18 hr and then double labeled with rhodamine-conjugated α-bungarotoxin and with antibodies specific forLN, perlecan (HSPG), collagen IV (CN IV), integrin β1 subunit (β1), or β-DG. Both LN and perlecan are distributed diffusely on the surface of the myotubes and in the surrounding matrix but are highly concentrated at and fully coextensive with most AChR aggregates (long arrows). Collagen IV and the integrin β1 subunit were present in diffuse patches devoid of AChRs (open arrows) and were found to colocalize with only a few AChR aggregates (long arrows). The vast majority of AChR aggregates had no detectable collagen IV or integrin β1 immunoreactivity associated with them (short arrows). β-DG immunoreactivity was present over the entire myotube and highly concentrated at both large and small AChR aggregates (long arrows). Scale bar, 20 μm.
Two classes of LN receptors are expressed in skeletal muscle and C2 cells, integrins and α-DG (Collo et al., 1993; Ervasti and Campbell, 1993; Song et al., 1992; Gee et al., 1994; Belkin et al., 1996). Integrins comprise a family of cell adhesion molecules composed of heterodimers of α and β subunits (for review, see Hynes, 1992). LN-binding integrins found in skeletal muscle contain the β1 subunit, and their expression is developmentally regulated in both muscle and C2 cells (Collo et al., 1993; Belkin et al., 1996). Antibodies to the β1 subunit label NMJs (Belkin et al., 1996) as well as AChR aggregates inXenopus myocyte and chick myotube cultures (Bozyczko et al., 1987; Anderson et al., 1996). In our studies, we used an antiserum directed against the extracellular domain of the integrin β1 subunit that should recognize all cytoplasmic splice variants expressed in these cells. We found a very poor correlation with both spontaneous and agrin-induced AChR aggregates in C2 cells (Fig. 1, Table 1). Agrin did not seem to change the distribution of this integrin subunit that remained concentrated in large plaques on the cell surface (Fig. 1,open arrow), its distribution resembling more that of collagen IV than that of LN. By contrast, both α- (data not shown) and β-DG (Fig. 1) are concentrated at and overlap completely with essentially all AChR aggregates (>95%). Taken together, our results show that agrin specifically recruits LN and perlecan at AChR aggregates in C2 cells. Furthermore, the low level of colocalization of the integrin β1 subunit with AChR aggregates in C2 cells suggests that this receptor is unlikely to be responsible for the recruitment of LN into aggregates. Rather, α-DG, which is consistently concentrated at both spontaneous and agrin-induced AChR aggregates, seems a better candidate.
LN induces large AChR aggregates
The possibility that LN is involved in later stages of AChR aggregate formation, such as the maturation of aggregates, was suggested by the observation that agrin-induced reorganization of the ECM lags behind AChR aggregation (Nitkin and Rothschild, 1990). To test this possibility, we treated C2 myotubes with either agrin or LN, measured the number, size, and density of AChR aggregates, and compared these values with those of control cultures. In the absence of added agrin or LN, C2 myotubes had an average of 9 ± 1 spontaneous aggregates per field with a mean size of 16.4 ± 1.8 μm2 (n = 10 fields; Table2, Fig.2A,E). Spontaneous aggregates had a mean pixel intensity of 133 ± 6 (out of a possible 255), reflecting the density of AChRs within an aggregate. Treatment of C2 myotubes with 200 pm agrin induced a more than threefold increase in the number of AChR aggregates compared with control cultures (Table 2, Fig. 2B,F); however this increase in number was accompanied by a twofold decrease in size and a 1.4-fold decrease in apparent density. In spite of the small size of the AChR aggregates, their large number resulted in an increase from 150 to 306 μm2 in the total area per field occupied by AChR aggregates (Table 2).
Fig. 2.

LN affects the size of AChR aggregates. Cultures of C2 myotubes were incubated overnight in fusion medium alone (A, E) or in fusion medium containing 200 pm agrin (B, F), 60 nm LN (C, G), or 200 pm agrin plus 100 μg/ml anti-LN IgG (D, H). A–D, Representative fields of C2 myotubes labeled with rhodamine-conjugated α-bungarotoxin for each treatment.E–H, Representative frequency distribution histograms of AChR aggregate areas. Note that the scale of they-axis of H (agrin + anti-LN) is four times that of E–G. The addition of LN to C2 myotube cultures results in an increase in the area of AChR aggregates, whereas the addition of anti-LN IgG results in an dramatic increase in the number of small AChR aggregates (<5 μm2). Scale bar, 10 μm.
Treatment of C2 cells with 60 nm LN resulted in an approximately twofold increase in the number of AChR aggregates compared with control cultures (Table 2, Fig. 2C,G). The average size of LN-induced AChR aggregates was more than double that of agrin-induced aggregates, resulting in a greater total area occupied by AChR aggregates (Table 2). Furthermore, the AChR density of LN-induced aggregates was higher than that of either spontaneous or agrin-induced aggregates. Thus LN alone induces the formation of large, dense AChR aggregates. To explore the function of LN in agrin-induced AChR aggregation, we incubated C2 myotubes with 200 pm agrin in the presence of 100 μg/ml anti-LN antiserum. This antiserum was raised against purified EHS LN and does not cross-react with the recombinant agrin used in these experiments. As a control for nonspecific antibody effects, an equal concentration of a polyclonal antibody to collagen IV was added to identical cultures of C2 myotubes, and no obvious effect on the size or number of AChR aggregates was observed (data not shown). Only in cultures treated with the anti-LN antiserum was there a dramatic increase in the number of AChR aggregates (Table 2, Fig. 2D,H). These AChR aggregates were half the size of the aggregates observed in cultures treated with agrin alone and had a slightly lower AChR density. A large proportion (>80%) of the myotubes cultured in the presence of anti-LN antiserum showed only microaggregates on their surface (see Fig.2D,H). These microaggregate-rich myotubes were sometimes seen in control and agrin-treated cultures. However they were absent in cultures treated with LN alone or with LN and agrin together. Because the total area covered by AChR aggregates in the presence of the anti-LN antiserum is comparable with that for agrin alone, this antiserum did not seem to completely disperse aggregates into individual AChRs. From these results, we hypothesize that agrin increases the number of AChR microaggregates, while recruiting LN to promote the formation of large, high density aggregates from microaggregates or possibly to inhibit the breakup of larger aggregates into microaggregates.
LN-induced AChR aggregation is mediated by α-DG
Our immunocytochemical data on integrin and α-DG localization favored the latter as a receptor mediating the effects of LN on AChR aggregation. To test this further, we quantified the induction of AChR aggregates by LN on C2 myotubes and on a genetic variant of the C2 muscle cell line S27, which is deficient in the synthesis of glycosaminoglycans (Gordon and Hall, 1989). We and others (Gee et al., 1994; Sugiyama et al., 1994) have shown previously that S27 cells have reduced expression of α-DG as well as reduced binding of the residual α-DG to agrin and LN. These cells also have no spontaneous AChR aggregates and respond poorly to agrin (Gordon et al., 1993). In C2 cells incubated for 18 hr with various concentrations of LN, there was a maximal increase (twofold) in AChR aggregates at 60 nm LN (Fig. 3). In contrast, LN failed to cause aggregation of AChRs on S27 myotubes, even at concentrations as high as 120 nm. Like spontaneous and agrin-induced aggregation, LN-induced aggregation seems to require a functional α-DG and the normal synthesis of glycosaminoglycans.
Fig. 3.

LN induces AChR aggregation in C2 but not S27 cells. Cultures were treated for 18 hr with 0, 12, 24, 60, or 120 nm LN, labeled with rhodamine-conjugated α-bungarotoxin, and the number of AChR aggregates per myotube segment was quantified. Data points represent the mean ± SEM of four experiments. For each experiment, 10 fields taken at random from each of four coverslips were quantified. LN induces the first significant increase in the number of AChR aggregates in C2 cells (open circles) at a concentration of 24 nm. The response saturates at 60 nm with a twofold increase in the number of AChR aggregates over control. An asterisk indicates numbers of LN-induced AChR clusters that are statistically significant from control as assessed by ANOVA and Fisher’s test (p < 0.05). In contrast, no AChR aggregates were present in S27 cultures (filled circles) at any concentration of LN tested.
Because α-DG is not the only protein affected by the mutation in S27 cells, we assessed the involvement of α-DG in LN-induced AChR aggregation with mAb IIH6. mAb IIH6 recognizes a unique carbohydrate epitope on α-DG and inhibits binding of both LN and agrin to α-DG on blots (Gee et al., 1994; Sugiyama et al., 1994). A control monoclonal antibody to the rat integrin α1 subunit, mAb 3A3, had no effect on LN-induced aggregation (data not shown). Treatment of C2 cells with LN in the presence of increasing concentrations of mAb IIH6 resulted in a dose-dependent inhibition of LN-induced AChR aggregation (Fig. 4). A significant effect was observed at mAb IIH6 concentrations as low as 1:200 dilution. Higher concentrations of mAb IIH6 decreased the number of AChR aggregates to that occurring spontaneously in C2 cells. No obvious change in the morphology of the remaining aggregates was observed. Although others (Campanelli et al., 1994; Cohen et al., 1995; but see Sugiyama et al., 1994) have reported effects of mAb IIH6 similar to those we saw with the anti-LN antiserum, in our hands these antibodies had different effects possibly because the functions of α-DG in AChR aggregation are not limited to those of LN.
Fig. 4.

mAb IIH6 against α-DG inhibits LN-induced AChR aggregation. Cultures of C2 myotubes were incubated for 18 hr in medium alone (control) or in medium containing 48 nm LN and increasing concentrations of mAb IIH6 against α-DG. For each condition, the number of AChR aggregates per myotube segment was determined for three experiments. For each experiment, 10 fields taken at random from each of three to four coverslips were quantified. At this concentration, LN induced an ∼30% increase in the number of AChR aggregates per myotube segment compared with control (filled diamond, p < 0.01). mAb IIH6 significantly decreased the number of AChR aggregates compared with LN treatment in a dose-dependent manner at dilutions as high as 1:200 (*, p < 0.01; **, p < 0.001). Although treatment with high concentrations of mAb IIH6 decreased the number of AChR aggregates below spontaneous levels, this decrease was not statistically significant. Values represent means ± SEM, and significant differences from control (filled diamond) or from LN-treated (*) cultures were determined by ANOVA followed by Fisher’s test.
To confirm that α-DG is involved in LN-induced AChR aggregation, we generated C2 clonal cell lines that stably express antisense cDNA to DG. Two such cell lines, 11F and 11E, show reductions of 50 and 80%, respectively, in α-DG expression relative to C2 cells. These cells fuse to form myotubes and express normal levels of other proteins including adhalin, LN, and AChRs (M. Lindenbaum, F. Montanaro, and S. Carbonetto, unpublished observations). C2, 11F, and 11E myotubes were incubated in the presence of 60 nm LN for 18 hr and then assayed for AChR aggregation. Figure5 shows that LN induced a 1.8–2-fold increase in the number of AChR aggregates in C2 cells regardless of the level of spontaneous AChR aggregates. In contrast, there was little change in the number of AChR aggregates in 11F and 11E cells treated with LN (Fig. 5). In other experiments (see below; C. Jacobson, F. Montanaro, M. Lindenbaum, S. Carbonetto, and M. Ferns, unpublished observations), we have shown that 11F and 11E cells produce a significant, although reduced, response to agrin when compared with C2 cells, indicating that they are not inherently incapable of responding to agents known to induce AChR aggregation. Taken together, our results suggest that α-DG is required for LN-induced AChR aggregation in C2 cells.
Fig. 5.

DG-antisense clones fail to aggregate AChRs in response to LN. 11F and 11E cells show reductions in α-DG expression of 50 and 80%, respectively, compared with parental C2 cells. Cells were treated for 18 hr with 60 nm LN and labeled with rhodamine-conjugated α-bungarotoxin, and the number of AChR aggregates per myotube segment was quantified. LN (hatched bars) induced a twofold increase in AChR aggregates in C2 cells compared with control nontreated cultures (filled bars). However, both 11F and 11E cells were unresponsive to LN, indicating that α-DG is necessary for LN-induced AChR aggregation. Values represent means ± SD of two coverslips from one representative experiment; 15–20 fields for each of the two coverslips per treatment and per cell type were quantified. Data from two separate experiments were not pooled because of variability in the level of spontaneous AChR aggregates in C2 cells. An asteriskindicates a statistically significant difference between control and LN treatment within each cell type (p < 0.05,t test).
LN interaction with α-DG enhances agrin-induced AChR aggregation
Vogel et al. (1983) reported that LN can enhance the AChR aggregating activity of a factor in neuroblastoma-conditioned medium. Because agrin is produced by a variety of neural cells, we tested whether recombinant agrin might also act cooperatively with LN in this regard and further whether this was mediated by α-DG. To optimize the sensitivity of this assay, we used a subthreshold concentration of LN (12 nm) that alone did not cause any increase in the number of AChR aggregates in C2, 11F, or 11E cells (Fig.6). Addition of agrin alone to C2, IIF, and IIE cultures caused a significant increase in the number of AChR aggregates. This increase was enhanced significantly by simultaneous treatment with agrin and LN (12 nm) in C2 and to a lesser extent in IIF cells, but there was no effect in IIE cells. These results show that LN can potentiate the activity of agrin and that α-DG is involved in this process.
Fig. 6.

LN potentiates the AChR aggregating activity of agrin via its interaction with α-DG. Cells were treated with 12 nm LN (wide hatched bars), 100 pm agrin (fine hatched bars), or both (filled bars) for 18 hr, and the number of AChR aggregates per myotube segment was quantified as described in Materials and Methods. The concentration of LN used is subthreshold and does not affect the number of AChR aggregates compared with control cultures (open bars) for all cell types. LN enhances the effect of agrin by twofold and 1.7-fold in C2 and 11F cells, respectively. However, in 11E cells, where the α-DG level is only 20% of that in C2 cells, LN does not significantly enhance the effect of agrin. Note that in both DG-antisense clones agrin alone induces AChR aggregation. Values represent means ± SD for two experiments. For each experiment, 15–20 fields from each of two coverslips were quantified. An asterisk indicates a statistically significant difference between agrin and agrin + LN treatment within each cell type (p < 0.05, t test).
LN does not induce the phosphorylation of MuSK or the AChR β subunit
Neural agrin induces a rapid, but transient, phosphorylation of the receptor tyrosine kinase MuSK that subsequently leads to the phosphorylation of the AChR β subunit (Wallace et al., 1991; Qu and Huganir, 1994; DeChiara et al., 1996; Ferns et al., 1996;Glass et al., 1996). We therefore sought to determine whether LN-induced AChR aggregation is similarly dependent on the phosphorylation of MuSK and leads to the phosphorylation of the AChR β subunit. AChRs were isolated using α-bungarotoxin conjugated to Sepharose beads, and MuSK was immunoprecipitated. Isolates were then analyzed by SDS-PAGE and immunoblotting with a monoclonal anti-phosphotyrosine antibody (4G10). Blots were then stripped and reprobed with mAb 124 to the AChR β subunit or with anti-MuSK antiserum to verify the identity of the phosphorylated bands (data not shown). Treatment of C2 myotubes with 200 pm neural agrin leads to a rapid phosphorylation of MuSK (∼110 kDa; Fig.7A). However, treatment with LN did not induce MuSK phosphorylation, even at concentrations that induce maximal aggregation (Fig. 3B). Agrin also induced a robust increase in tyrosine phosphorylation of an ∼50 kDa band that corresponds in size to the AChR β subunit (Fig. 7B). In contrast, concentrations of LN that are sufficient to induce maximal aggregation do not phosphorylate the AChR β subunit above endogenous levels seen in untreated cultures (Fig. 7B). We conclude that the observed effects of LN on the size and number of AChR aggregates do not involve phosphorylation of either MuSK or the AChR β subunit.
Fig. 7.
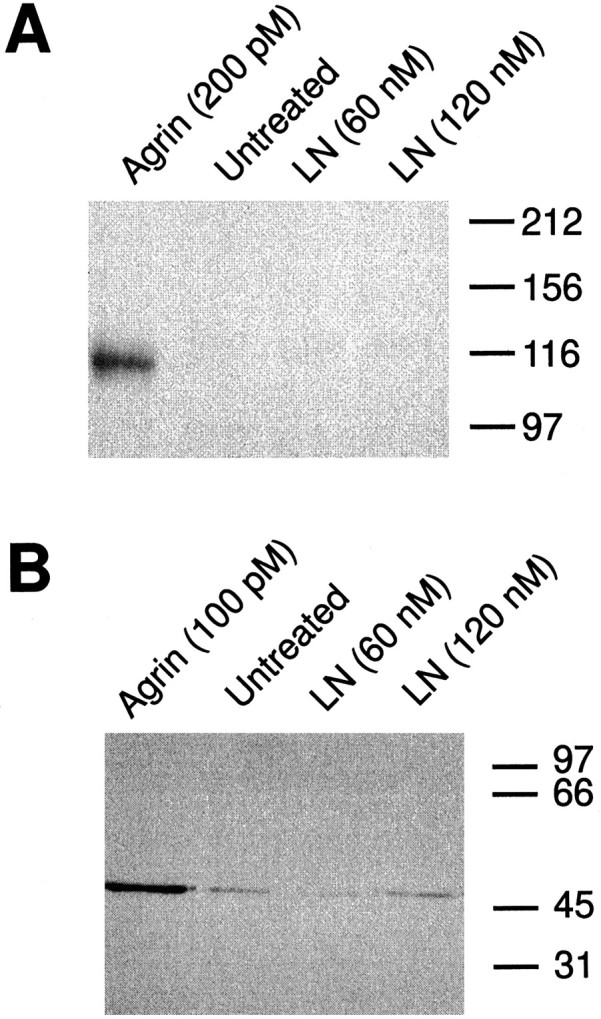
LN does not induce the phosphorylation of MuSK and the AChR β subunit. C2 myotubes were treated for 15 min with the indicated concentrations of neural agrin or LN; then AChRs or MuSK were purified from the cell extracts (see Materials and Methods). The isolates were analyzed by SDS-PAGE, transferred to nitrocellulose, and then assayed for phosphotyrosine content by immunoblotting with mAb 4G10. A, Anti-phosphotyrosine immunoblot of MuSK immunoprecipitates. Treatment of C2 myotubes with 200 pmagrin produced a large increase in MuSK phosphorylation compared with endogenous levels. LN, on the other hand, failed to induce MuSK phosphorylation at either of the two concentrations tested.B, Anti-phosphotyrosine immunoblot of purified AChR subunits. A prominent band at ∼50 kDa, corresponding in size to the AChR β subunit, is evident in AChR isolates from cells treated with 100 pm agrin. In contrast, treatment of C2 myotubes with either 60 or 120 nm LN did not induce the phosphorylation of the AChR β subunit above the level seen in untreated cells. Molecular mass standards are indicated at the right of each panel (in kDa).
DISCUSSION
The formation of the postsynaptic apparatus of the skeletal neuromuscular junction is a complex process involving ECM, membrane, and cytoskeletal elements (for review, see Hall and Sanes, 1993;Carbonetto and Lindenbaum, 1995). Agrin released from nerve terminals activates MuSK to trigger events within muscle that lead to the aggregation of AChRs in the postsynaptic membrane. Diffusely distributed AChRs on the muscle cell surface first form loose assemblies of microaggregates beneath the nerve terminal that later coalesce into large aggregates (Anderson and Cohen, 1977; Steinbach, 1981). The results presented here suggest that LN is important in the later stages of this process and that it acts through α-DG via a MuSK-independent mechanism.
Our results extend previous data (Vogel et al., 1983) on LN stimulation of AChR aggregation by demonstrating a role for LN in determining the size and density of AChR aggregates. LN-rich regions are coextensive with a majority, but not all, of spontaneous and agrin-induced AChR aggregates. In untreated C2 myotubes, 81% of spontaneous aggregates are closely associated with LN. This value agrees well with results obtained in an earlier study in which 70–90% of spontaneous receptor aggregates in chick myotube cultures colocalized with LN (Bayne et al., 1984). Agrin causes a dramatic change in the distribution of LN on the cell surface, reflected by the twofold increase in AChR aggregates that overlap completely with LN. In addition, we show that treatment of cultures with agrin and an anti-LN antiserum leads to the formation of AChR microaggregates with an apparent lower AChR density, consistent with the notion that endogenous LN is necessary for their cohesion. These observations could suggest that the recruitment of LN at the nascent neuromuscular synapse by agrin might assist the latter in forming a continuous densely packed AChR aggregate under the nerve terminal. Indeed, microaggregates formed in the presence of the anti-LN antiserum resemble those seen in the early stages of AChR aggregation at neuromuscular synapses forming in vivo (Steinbach, 1981) and at synapses between neurites and myotubes in vitro(Anderson and Cohen, 1977; Kidokoro et al., 1980; Role et al., 1985). Microaggregates are also observed at initial stages of AChR aggregation induced by bath-applied agrin, where AChR microaggregates (<4 μm2) begin forming within 1–2 hr of adding agrin to chick myotubes in culture (Godfrey et al., 1984; Wallace, 1988). Larger aggregates (>4 μm2) are seen at later times (12–20 hr) and are formed in part by lateral migration and fusion of pre-existing microaggregates (Wallace, 1988). During this time, the total area occupied by AChR aggregates and their density increase. Similarly, we find that AChR aggregates induced by LN exhibit an increased size and density compared with their agrin-induced counterparts. These observations support a model in which LN promotes the fusion of microaggregates into larger, more densely packed aggregates. However, they do not eliminate the possibility that LN stabilizes large aggregates once they have formed and that the anti-LN antiserum leads to the dispersal of these preformed macroaggregates.
In C2 cells, LN induces AChR aggregation in a dose-dependent manner with a maximal effect at 60 nm. In our hands, LN first shows a significant effect on AChR aggregation at 24 nm; however Vogel et al. (1983) reported an effect with only 1.2 nm LN. One possible explanation for this difference could be the presence of small amounts of agrin in the LN preparation used byVogel et al. (1983). Coomassie blue and silver staining of our purified LN revealed only a broad band at ∼200 kDa and a second band at ∼400 kDa corresponding to the β/γ and α chains of LN, respectively (Montanaro and Carbonetto, unpublished observations). We further tested the purity of our LN by immunoblot with antibodies to agrin and LN α2 chain (merosin) and found that neither of these proteins was present (Montanaro and Carbonetto, unpublished observations). Alternatively, this observed difference may derive from the different types of cells used and from a relative weak response of C2 cells to exogenous LN compared with primary myotube cultures and G8-1 cells.
Agrin-induced AChR aggregation has been shown to require activation of the tyrosine kinase receptor MuSK and to lead to the phosphorylation of the AChR β subunit. Subsequent identification, cloning, and functional studies of MuSK (Valenzuela et al., 1995; DeChiara et al., 1996; Glass et al., 1996) indicated that its activation is responsible for initiating virtually all events in presynaptic and postsynaptic differentiation (DeChiara et al., 1996). Our studies indicate that LN-induced AChR aggregation does not involve the phosphorylation of either MuSK or the AChR β subunit, even at concentrations that induce maximal receptor aggregation. We would therefore predict that phosphorylation inhibitors or a lack of MuSK would have no effect on this process in vitro. The complete lack of AChR aggregates in MuSK null mice indicates that in vivo LN and agrin do not induce the aggregation of AChRs via parallel pathways. Instead we envisage a model in which LN acts downstream of agrin-induced MuSK activation to potentiate the action of agrin and to consolidate AChR microaggregates into larger aggregates or to stabilize microaggregates within the forming subsynaptic receptor “plaque.”
While this paper was being reviewed, Sugiyama et al. (1997) published a paper showing that LN 1, but not LN 2 or LN 11, induces AChR clustering in a muscle cell line derived from MuSK knock-out mice, although at a 20-fold lower level than that in C2 cells (DeChiara et al., 1996). In addition, they demonstrated that LN-induced AChR aggregation occurs at a much slower rate than does agrin-induced AChR aggregation. Like us, they concluded that LN induces the aggregation of AChRs via a pathway that does not involve MuSK signaling but that in vivoactivation of MuSK is required to initiate AChR aggregation. Furthermore, they find that the aggregating activity of LN is determined by the identity of its α chain. Our results complement and extend those of Sugiyama et al. (1997) by implicating α-DG as the receptor that mediates this MuSK-independent aggregation. α-DG is known to bind specifically to G domains in the LN α chain (Gee et al., 1993) but is known to bind with similar affinity to LN 1 and LN 2 (Yamada et al., 1994, 1996). The unique ability of LN 1 to induce AChR aggregation could result from modulation of LN binding to α-DG by heparan sulfate proteoglycans. Indeed heparin has been shown to modulate differentially the interaction of LN 1 and LN 2 to α-DG (Pall et al., 1996). Alternatively, the LN α2 chain is much more prone to proteolysis than is the α1 chain during purification (Leivo and Engvall, 1988; Ehrig et al., 1990), and this can result in loss of the G domains and a loss of binding to α-DG. This could explain the reported inability of purified LN 2 to induce AChR aggregation and would further support our observations that α-DG mediates this aggregation.
Our data indicate that the effects of agrin and LN on C2 cells may not be mediated by integrins but rather involve the dual agrin and LN receptor α-DG. First, α-DG is immunocytochemically localized at AChR aggregates where LN appears concentrated, and it is the only protein detected in overlays of C2 myotube extracts probed with LN (Gee et al., 1994). Second, the Kd for LN binding to α-DG (∼90 nm; Gee et al., 1993) is close to the concentration of LN that induces half-maximal aggregation on C2 myotubes (∼30 nm). Third, S27 cells do not aggregate AChRs in response to LN. Although these cells synthesize normal amounts of LN and deposit it into the ECM, they do not retain LN on their surfaces (Gordon et al., 1993). Consistent with this observation, the binding of LN to α-DG is dramatically decreased in S27 myotube extracts compared with C2 extracts (Campanelli et al., 1994; Gee et al., 1994; Sugiyama et al., 1994). Moreover, the amount of full-size α-DG is greatly reduced in S27 cells (Sugiyama et al., 1994). Thus, a reduction in α-DG levels or in its affinity for LN may be responsible for the inability of S27 cells to retain LN on their surfaces and to aggregate AChRs in response to exogenous LN. Fourth, mAb IIH6 against α-DG blocks LN-induced receptor aggregation in a dose-dependent manner. This antibody inhibits the binding of both LN and agrin to α-DG and has been reported previously to interfere with agrin-induced AChR aggregation (Campanelli et al., 1994; Gee et al., 1994; Cohen et al., 1995; but see Sugiyama et al., 1994). Interestingly, in our hands, mAb IIH6 and the anti-LN antiserum had different effects on AChR aggregation, the former affecting the number of aggregates and the latter their size and number. This difference could be attributable to the fact that in C2 myotubes mAb IIH6 interferes with α-DG binding to LN and muscle agrin, which could also be involved in AChR aggregation. Finally, we have found that two C2-derived cell lines transfected with antisense DG cDNA and expressing reduced levels of α-DG do not show significant AChR aggregation in response to LN. 11F and 11E cells show a specific decrease in α-DG expression with levels 50 and 20% of parental C2 cells, respectively. Like S27 cells, both 11F and 11E cells show an abnormal LN deposition on their surface (Lindenbaum, Montanaro, and Carbonetto, unpublished observations) and a decreased response to agrin (Jacobson, Montanaro, Lindenbaum, Carbonetto, and Ferns, unpublished observations). Taken together, these data suggest a role of α-DG in LN-induced AChR aggregation. In addition, we have shown that a subthreshold concentration of LN can significantly potentiate the AChR aggregating activity of recombinant neural agrin, providing further evidence of a cooperation between LN and agrin in AChR aggregation. As for LN-induced AChR aggregation, this synergistic effect of LN on agrin also seems to be mediated by α-DG because it was not observed in 11E cells that have the lowest level of α-DG expression.
Experiments of others have emphasized the presence of integrins at the NMJ in vivo and at AChR aggregates in culture (Bozyczko et al., 1987; Anderson et al., 1996; Belkin et al., 1996). Although we are unaware of any perturbation experiments with the function-blocking antibodies used for localization (Bozyczko et al., 1987) this precise colocalization suggests that integrins are involved in aggregation and makes them a potential candidate for mediating the effects of LN on muscle cells. Somewhat surprisingly however, we have found a poor localization of integrins in agrin-induced AChR aggregates on C2 myotubes. The antibody used in these experiments is directed against the β1 subunit that is common to most, if not all, integrin LN receptors. This antiserum recognizes the extracellular domain of the β1 subunit and should therefore cross-react with the two cytoplasmic splice variants β1A and β1D recently identified in skeletal muscle and C2 cells. In particular, β1D was localized to the NMJ, where it is believed to act as a LN receptor by dimerizing with the α7 subunit. We have no definite explanation for the low colocalization of integrins with agrin-induced AChR aggregates in C2 cells. However, in all previous studies, AChR aggregation in culture was induced by either cocultured neurons or extracts containing full-length agrin. It is therefore possible that recruitment of integrins to AChR aggregates requires additional nerve-derived factors not present in our culture system or agrin domains not present in our recombinant agrin fragment. Further experiments with full-length agrin and function-blocking anti-integrin antibodies are required to address this issue.
How does LN stimulate the formation of large densely packed AChR aggregates? Previous studies have shown that LN can self-polymerize in solution via domains at the end of each of its short arms (Yurchenco et al., 1990; Colognato-Pyke et al., 1995) and forms an independent network in basement membranes (Yurchenco et al., 1992). Thus, one possibility is that LN self-associates into a multimeric structure and in so doing “traps” AChR microaggregates at sites of assembly. A similar mechanism has been proposed by Cohen et al. (1997) to mediate the aggregation of α-DG, β-DG, and dystrophin by exogenous LN inXenopus myocytes. Interestingly, this aggregation is not blocked by inhibitors of tyrosine phosphorylation. Similarly, exogenous LN causes the aggregation of α-DG on C2 myotubes (Lindenbaum, Montanaro, and Carbonetto, unpublished observations). Although there is no evidence of a direct interaction of LN with the AChR, expression of α- and β-DG together with rapsyn in nonmuscle cells results in a coclustering of these proteins (Apel et al., 1995). β-DG could therefore interact with rapsyn that is able to aggregate AChRs when expressed in nonmuscle cells (Froehner et al., 1990; Phillips et al., 1991; Maimone and Merlie, 1993; Scotland et al., 1993). Hence, the aggregation of α/β-DG complexes by LN in muscle cells may cause aggregation of AChRs via rapsyn by a MuSK-independent mechanism.
During synapse formation, the binding of agrin to MuSK would activate rapsyn and initiate aggregation of AChRs, MuSK, and α-DG as well as LN bound to α-DG. The local increase in the concentration of LN may potentiate direct interactions between LN–LN as well as LN–agrin (Denzer et al., 1997) molecules and lead to the coalescence of microaggregates of AChRs into large dense aggregates similar to those found at mature synapses.
Footnotes
This work was supported by grants to S.C. from the Medical Research Council of Canada (MA 9000 and MA 10182), the National Centres of Excellence, and the Muscular Dystrophy Association (USA) and to S.C.F from the National Institutes of Health. F.M. and C.J. were supported by studentships from the National Centres of Excellence. During part of this research, S.H.G. was supported by a postdoctoral fellowship from the Medical Research Council of Canada. We thank Drs. Steven Roberds and Kevin Campbell (University of Iowa) for supplying antibodies to α-DG, Dr. John Lindstrom (University of Pennsylvania) for supplying monoclonal antibody to the AChR β subunit, and Dr. James E. Faber for the use of his image capture apparatus. Neural agrin was a gift from Drs. James Campanelli and Richard Scheller (Stanford University) and Dr. Michael Ferns (McGill University).
F.M. and S.H.G. contributed equally to this study.
Correspondence should be addressed to Dr. Salvatore Carbonetto, Montreal General Hospital Research Institute, 1650 Cedar Avenue, Montreal, Quebec H3G 1A4, Canada.
REFERENCES
- 1.Anderson MJ, Cohen MW. Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol (Lond) 1977;268:757–773. doi: 10.1113/jphysiol.1977.sp011880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson MJ, Qiao Shi Z. Proteolytic disruption of laminin-integrin complexes on muscle cells during synapse formation. Mol Cell Biol. 1996;16:4972–4984. doi: 10.1128/mcb.16.9.4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apel ED, Roberds SL, Campbell KP, Merlie JP. Rapsyn may function as a link between the acetylcholine receptor and the agrin-binding dystrophin-associated glycoprotein complex. Neuron. 1995;15:115–126. doi: 10.1016/0896-6273(95)90069-1. [DOI] [PubMed] [Google Scholar]
- 4.Bayne EK, Anderson MJ, Fambrough DM. Extracellular matrix organization in developing muscle: correlation with acetylcholine receptor aggregates. J Cell Biol. 1984;99:1486–1501. doi: 10.1083/jcb.99.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belkin AM, Zhidkova NI, Balzac F, Altruda F, Tomatis D, Maier A, Tarone G, Koteliansky VE, Burridge K. β1D integrin displaces the β1A isoform in striated muscles: localization at junctional structures and signalling potential in nonmuscle cells. J Cell Biol. 1996;132:211–226. doi: 10.1083/jcb.132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bevan S, Steinbach JH. The distribution of alpha-bungarotoxin binding sites of mammalian skeletal muscle developing in vivo. J Physiol (Lond) 1977;267:195–213. doi: 10.1113/jphysiol.1977.sp011808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bozyczko D, Decker C, Muschler J, Horwitz AF. Integrin on developing and adult skeletal muscle. Exp Cell Res. 1989;183:72–91. doi: 10.1016/0014-4827(89)90419-9. [DOI] [PubMed] [Google Scholar]
- 8.Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 9.Carbonetto S, Lindenbaum M. The basement membrane at the neuromuscular junction: a synaptic mediatrix. Curr Opin Neurobiol. 1995;5:596–605. doi: 10.1016/0959-4388(95)80064-6. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MW, Jacobson C, Godfrey EW, Campbell KP, Carbonetto S. Distribution of alpha-dystroglycan during embryonic nerve-muscle synaptogenesis. J Cell Biol. 1995;129:1093–1101. doi: 10.1083/jcb.129.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen MW, Jacobson C, Yurchenco PD, Morris GE, Carbonetto S. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. J Cell Biol. 1997;136:1047–1058. doi: 10.1083/jcb.136.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collo G, Starr L, Quaranta V. A new isoform of the laminin receptor integrin α7β1 is developmentally regulated in skeletal muscle. J Biol Chem. 1993;268:19019–19024. [PubMed] [Google Scholar]
- 13.Colognato-Pyke H, O’Rear JJ, Yamada Y, Carbonetto S, Cheng YS, Yurchenco PD. Mapping of network-forming, heparin-binding, and alpha 1 beta 1 integrin-recognition sites within the alpha-chain short arm of laminin-1. J Biol Chem. 1995;270:9398–9406. doi: 10.1074/jbc.270.16.9398. [DOI] [PubMed] [Google Scholar]
- 14.Daniels MP, Vigny M, Sonderegger P, Bauer HC, Vogel Z. Association of laminin and other basement membrane components with regions of high acetylcholine receptor density of cultured myotubes. Int J Dev Neurosci. 1984;2:87–99. doi: 10.1016/0736-5748(84)90063-7. [DOI] [PubMed] [Google Scholar]
- 15.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 16.Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137:671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douville PJ, Harvey WJ, Carbonetto S. Isolation and partial characterization of high affinity laminin receptors in neural cells. J Biol Chem. 1988;263:14964–14969. [PubMed] [Google Scholar]
- 18.Ehrig K, Leivo I, Argraves WS, Ruoslahti E, Engvall E. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci USA. 1990;87:3264–3268. doi: 10.1073/pnas.87.9.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 20.Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- 22.Ferns M, Deiner M, Hall Z. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphorylation. J Cell Biol. 1996;132:937–944. doi: 10.1083/jcb.132.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fertuck HC, Salpeter MM. Localization of acetylcholine receptor by 125I-labeled alpha bungarotoxin binding at mouse motor endplates. Proc Natl Acad Sci USA. 1974;71:1376–1378. doi: 10.1073/pnas.71.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Froehner SC, Luetje CW, Scotland PB, Patrick J. The postsynaptic 43K protein clusters muscle nicotinic acetylcholine receptors in Xenopus oocytes. Neuron. 1990;5:403–410. doi: 10.1016/0896-6273(90)90079-u. [DOI] [PubMed] [Google Scholar]
- 25.Gee SH, Blacher RW, Douville PJ, Provost PR, Yurchenco PD, Carbonetto S. Laminin-binding protein 120 from brain is closely related to the dystrophin-associated glycoprotein, dystroglycan, and binds with high affinity to the major heparin-binding domain of laminin. J Biol Chem. 1993;268:14972–14980. [PubMed] [Google Scholar]
- 26.Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 27.Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- 28.Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J Cell Biol. 1984;99:615–627. doi: 10.1083/jcb.99.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon H, Hall ZW. Glycosaminoglycan variants in the C2 muscle cell line. Dev Biol. 1989;135:1–11. doi: 10.1016/0012-1606(89)90152-8. [DOI] [PubMed] [Google Scholar]
- 30.Gordon H, Lupa M, Bowen D, Hall Z. A muscle cell variant defective in glycosaminoglycan biosynthesis forms nerve-induced but not spontaneous clusters of the acetylcholine receptor and the 43 kDa protein. J Neurosci. 1993;13:586–595. doi: 10.1523/JNEUROSCI.13-02-00586.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall ZW, Sanes JR. Synaptic structure and development: the neuromuscular junction. Cell [Suppl] 1993;72:99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 32.Henry MD, Campbell KP. Dystroglycan: an extracellular matrix receptor linked to the cytoskeleton. Curr Opin Cell Biol. 1996;8:625–631. doi: 10.1016/s0955-0674(96)80103-7. [DOI] [PubMed] [Google Scholar]
- 33.Hunter DD, Shah V, Merlie JP, Sanes JR. A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature. 1989;338:229–234. doi: 10.1038/338229a0. [DOI] [PubMed] [Google Scholar]
- 34.Hynes RO. Integrins: versatility, modulation and signalling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 35.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveillle CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 36.Kidokoro Y, Anderson MJ, Gruener R. Changes in synaptic potential properties during acetylcholine receptor accumulation and neurospecific interactions in Xenopus nerve-muscle cell culture. Dev Biol. 1980;78:464–483. doi: 10.1016/0012-1606(80)90347-4. [DOI] [PubMed] [Google Scholar]
- 37.Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, striated muscle, and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA. 1988;85:1544–1548. doi: 10.1073/pnas.85.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieth E, Cardasis CA, Fallon JR. Muscle-derived agrin in cultured myotubes: expression in the basal lamina and at induced acetylcholine receptor clusters. Dev Biol. 1992;149:41–54. doi: 10.1016/0012-1606(92)90262-f. [DOI] [PubMed] [Google Scholar]
- 39.Maimone MM, Merlie JP. Interaction of the 43 kDa postsynaptic protein with all subunits of the muscle nicotinic acetylcholine receptor. Neuron. 1993;11:53–66. doi: 10.1016/0896-6273(93)90270-2. [DOI] [PubMed] [Google Scholar]
- 40.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 41.Morissette N, Carbonetto S. Laminin α2 chain (M chain) is found within the pathway of avian and murine retinal projections. J Neurosci. 1995;15:8067–8082. doi: 10.1523/JNEUROSCI.15-12-08067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nitkin RM, Rothschild TC. Agrin-induced reorganization of extracellular matrix components on cultured myotubes: relationship to AChR aggregation. J Cell Biol. 1990;111:1161–1170. doi: 10.1083/jcb.111.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olek AJ, Ling A, Daniels MP. Development of ultrastructural specializations during the formation of acetylcholine receptor aggregates on cultured myotubes. J Neurosci. 1986;6:487–497. doi: 10.1523/JNEUROSCI.06-02-00487.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olwin BB, Hall ZW. Developmental regulation of laminin accumulation in the extracellular matrix of a mouse muscle cell line. Dev Biol. 1985;112:359–367. doi: 10.1016/0012-1606(85)90407-5. [DOI] [PubMed] [Google Scholar]
- 45.Pall EA, Bolton KM, Ervasti JM. Differential heparin inhibition of skeletal muscle α-dystroglycan binding to laminins. J Biol Chem. 1996;271:3817–3821. doi: 10.1074/jbc.271.7.3817. [DOI] [PubMed] [Google Scholar]
- 46.Phillips WD, Kopta C, Blount P, Gardner PD, Steinbach JH, Merlie JP. ACh receptor-rich membrane domains organized in fibroblasts by recombinant 43-kilodalton protein. Science. 1991;251:568–570. doi: 10.1126/science.1703661. [DOI] [PubMed] [Google Scholar]
- 47.Qu Z, Huganir RL. Comparison of innervation and agrin-induced tyrosine phosphorylation of the nicotinic acetylcholine receptor. J Neurosci. 1994;14:6834–6841. doi: 10.1523/JNEUROSCI.14-11-06834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Role LW, Matossian VR, O’Brien RJ, Fischbach GD. On the mechanism of acetylcholine receptor accumulation at newly formed synapses on chick myotubes. J Neurosci. 1985;5:2197–2204. doi: 10.1523/JNEUROSCI.05-08-02197.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanes JR, Engvall E, Butkowski R, Hunter DD. Molecular heterogeneity of basal laminae: isoforms of laminin and collagen IV at the neuromuscular junction and elsewhere. J Cell Biol. 1990;111:1685–1699. doi: 10.1083/jcb.111.4.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scotland PB, Colledge M, Melnikova I, Dai Z, Froehner SC. Clustering of the acetylcholine receptor by the 43 kDa protein: involvement of the zinc finger domain. J Cell Biol. 1993;123:719–728. doi: 10.1083/jcb.123.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shadiack AM, Nitkin RM. Agrin induces alpha-actinin, filamin, and vinculin to co-localize with AChR clusters on cultured chick myotubes. J Neurobiol. 1991;22:617–628. doi: 10.1002/neu.480220607. [DOI] [PubMed] [Google Scholar]
- 52.Smalheiser NR, Schwartz NB. Cranin, a laminin-binding protein of cell membranes. Proc Natl Acad Sci USA. 1987;84:6457–6461. doi: 10.1073/pnas.84.18.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ. H36-α7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J Cell Biol. 1992;117:643–657. doi: 10.1083/jcb.117.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinbach JH. Developmental changes in acetylcholine receptor aggregates at rat skeletal neuromuscular junctions. Dev Biol. 1981;84:267–276. doi: 10.1016/0012-1606(81)90394-8. [DOI] [PubMed] [Google Scholar]
- 55.Sugiyama JE, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 56.Sugiyama JE, Glass DJ, Yancopoulos GD, Hall ZW. Laminin-induced acetylcholine receptor clustering: an alternative pathway. J Cell Biol. 1997;139:181–191. doi: 10.1083/jcb.139.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tawil NJ, Houde M, Blacher R, Esch F, Reichardt LF, Turner DC, Carbonetto S. α1β1 integrin heterodimer functions as a dual laminin/collagen receptor in neural cells. Biochemistry. 1990;29:6540–6544. doi: 10.1021/bi00479a028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Timpl R, Rhode H, Risteli L, Ott U, Gehron Robey P, Martin GR. Laminin. Methods Enzymol. 1982;82:831–839. doi: 10.1016/0076-6879(82)82104-6. [DOI] [PubMed] [Google Scholar]
- 59.Turner DC, Flier LA, Carbonetto S. Identification of a cell-surface protein involved in PC-12 cell-substratum adhesion and neurite growth on laminin and collagen. J Neurosci. 1989;9:3287–3296. doi: 10.1523/JNEUROSCI.09-09-03287.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, Le Beau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 61.Vogel Z, Christian CN, Vigny M, Bauer HC, Sonderegger P, Daniels MP. Laminin induces acetylcholine receptor aggregation on cultured myotubes and enhances the receptor aggregation activity of a neuronal factor. J Neurosci. 1983;3:1058–1068. doi: 10.1523/JNEUROSCI.03-05-01058.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wallace BG. Regulation of agrin-induced acetylcholine receptor aggregation by Ca++ and phorbol ester. J Cell Biol. 1988;107:267–278. doi: 10.1083/jcb.107.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wallace BG. Agrin-induced specializations contain cytoplasmic, membrane, and extracellular matrix-associated components of the postsynaptic apparatus. J Neurosci. 1989;9:1294–1302. doi: 10.1523/JNEUROSCI.09-04-01294.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wallace BG, Qu Z, Huganir RL. Agrin induces phosphorylation of the nicotinic acetylcholine receptor. Neuron. 1991;6:869–878. doi: 10.1016/0896-6273(91)90227-q. [DOI] [PubMed] [Google Scholar]
- 65.Yamada H, Shimizu T, Tanaka T, Campbell KP, Matsumura K. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 1994;352:49–53. doi: 10.1016/0014-5793(94)00917-1. [DOI] [PubMed] [Google Scholar]
- 66.Yamada H, Chiba A, Endo T, Kobata A, Anderson LVB, Hori H, Fukuta-Ohi H, Kanazawa I, Campbell KP, Shimizu T, Matsumura K. Characterization of dystroglycan-laminin interaction in peripheral nerve. J Neurochem. 1996;66:1518–1524. doi: 10.1046/j.1471-4159.1996.66041518.x. [DOI] [PubMed] [Google Scholar]
- 67.Yurchenco PD, Cheng YS, Schittny JC. Heparin modulation of laminin polymerization. J Biol Chem. 1990;265:3981–3991. [PubMed] [Google Scholar]
- 68.Yurchenco PD, Cheng YS, Colognato H (1992) Laminin forms an independent network in basement membranes. J Cell Biol [Erratum (1992) 118:493] 117:1119–1133. [DOI] [PMC free article] [PubMed]