

Abstract
Bcl-xL is a death-inhibiting member of the Bcl-2/Ced9 family of proteins which either promote or inhibit apoptosis. Gene targeting has revealed that Bcl-xL is required for neuronal survival during brain development; however,Bcl-xL knock-out mice do not survive past embryonic day 13.5, precluding an analysis of Bcl-xLfunction at later stages of development. Bcl-xL expression is maintained at a high level postnatally in the CNS, suggesting that it may also regulate neuron survival in the postnatal period. To explore functions of Bcl-xL related to neuron survival in postnatal life, we generated transgenic mice overexpressing humanBcl-xL under the control of a pan-neuronal promoter. A line that showed strong overexpression in brainstem and a line that showed overexpression in hippocampus and cortex were chosen for analysis. We asked whether overexpression of Bcl-xLinfluences neuronal survival in the postnatal period by studying two injury paradigms that result in massive neuronal apoptosis. In the standard neonatal facial axotomy paradigm, Bcl-xLoverexpression had substantial effects, with survival of 65% of the motor neurons 7 d after axotomy, as opposed to only 15% in nontransgenic littermates. To investigate whether Bcl-xLregulates survival of CNS neurons in the forebrain, we used a hypoxia–ischemia paradigm in neonatal mice. We show here that hypoxia–ischemia leads to substantial apoptosis in the hippocampus and cortex of wild-type neonatal mice. Furthermore, we show that overexpression of Bcl-xL is neuroprotective in this paradigm. We conclude that levels of Bcl-xL in postnatal neurons may be a critical determinant of their susceptibility to apoptosis.
Keywords: apoptosis, axotomy, hypoxia-ischemia, Bcl-xL, Ced 9, Bax
Apoptosis is the most common form of physiological cell death and plays an important role in animal development and homeostasis, controlling cell numbers in both vertebrate and invertebrate tissues (Oppenheim, 1991; Raff et al., 1993). Remarkably, apoptosis is also a common response to neuronal injury, particularly in the neonatal period in avians and mammals (for review, see Elliott and Snider, 1998). Recently, several families of molecules that regulate apoptosis in different settings have been identified (for review, see Hengartner and Horvitz, 1994b; White, 1996). The Ced-9/Bcl-2 family of apoptosis regulators is composed of a large number of intracellular proteins with opposing effects in regulating cell death. Some family members, including Bcl-2 (Bakhshi et al., 1985; Hengartner and Horvitz, 1994a) and Bcl-xL (Boise et al., 1993), function to inhibit apoptosis, whereas other members such as Bax (Oltvai et al., 1993), Bcl-xS (Boise et al., 1993), Bad (Yang et al., 1995), and Bak (Chittenden et al., 1995;Farrow et al., 1995; Kiefer et al., 1995) function to promote apoptosis. It appears that homodimerization and heterodimerization between the various death-promoting and death-inhibiting family members regulates the activation of caspases, which execute the cell death program (Korsmeyer, 1995; Yang et al., 1995; Reed, 1997). Although most of the analysis of these molecules has been done in extraneural tissues, there is now compelling evidence that their actions are quite general and include profound influences on neurons. In vitrostudies have demonstrated that Bcl-2 and Bcl-xL can prevent apoptotic neuronal death induced by growth factor deprivation in primary neuronal cultures (Garcia et al., 1992; Allsopp et al., 1993;Frankowski et al., 1995; Gonzalez-Garcia et al., 1995; Greenlund et al., 1995). In contrast, overexpression of Bak and Bax in sympathetic neurons deprived of NGF accelerates apoptosis (Farrow et al., 1995;Easton et al., 1997). Bcl-2, Bcl-xL, Bax, and other members of the Bcl-2 family are expressed temporally and spatially in the nervous system in patterns compatible with a potential role as regulators of neuronal death in vivo (Gonzalez-Garcia et al., 1994; Krajewski et al., 1994; Merry et al., 1994; Frankowski et al., 1995; Parsadanian et al., 1995). Indeed, Bcl-2 overexpression in transgenic animals protects axotomized neonatal motor neurons from death and certain populations of developing neurons from naturally occurring cell death (Dubois-Dauphin et al., 1994; Martinou et al., 1994; Farlie et al., 1995) .
The physiological importance of these molecules for neuronal survival has been demonstrated recently by gene targeting. The phenotype ofBcl-xL null mutants is striking. Mice die at approximately embryonic day 13. Extensive apoptotic cell death is evident in postmitotic immature neurons of developing brain, spinal cord, and dorsal root ganglia (Motoyama et al., 1995). The consequences of the Bcl-2 null mutation are less dramatic, but several populations of neurons in the PNS are partially depleted (Michaelidis et al., 1996). Finally, deletion of the Bax gene also has dramatic but opposite effects. In both sympathetic and motor neuron populations, cell numbers in newborn mice are increased, indicating that naturally occurring cell death is reduced inBax−/− mice (Deckwerth et al., 1996). Furthermore, sympathetic neurons in vitro and facial motor neurons in vivo from Bax knock-out mice survive NGF deprivation and disconnection from their targets by axotomy, respectively.
A striking feature of the antiapoptosis regulator Bcl-xL, which is in contrast to Bcl-2, is that Bcl-xL expression is maintained at high levels throughout the postnatal CNS (Gonzalez-Garcia et al., 1994; Merry et al., 1994;Gonzalez-Garcia et al., 1995; Parsadanian et al., 1995). This persistent expression parallels the time frame when neurons lose their survival dependence on neuronal growth factors. An attractive hypothesis is that relative levels of Bcl-xL, compared with levels of the prodeath regulator Bax, are important in downregulating survival dependence on growth factors. A first step in addressing this hypothesis is to determine whether Bcl-xL can regulate neuronal apoptosis in the postnatal CNS. Studies involvingBcl-xL knock-out mice demonstrate that Bcl-xL is the dominant inhibitor of neuronal apoptosis during embryonic development, but functions of Bcl-xL in the postnatal period cannot be determined in these mice because of early lethality. Currently, there are no data addressing whether Bcl-xL can regulate neuronal apoptosis in the postnatal brain.
To address functions of Bcl-xL in vivo, we generated transgenic mice overexpressing humanBcl-xL in neurons. We found that increased expression of Bcl-xL prevented apoptosis in the standard neonatal facial motor neuron axotomy model. We also found that Bcl-xL overexpression prevented apoptosis of cortical and hippocampal neurons in a hypoxia–ischemia paradigm in neonatal mice. Our results demonstrate that Bcl-xL is a powerful regulator of neuronal apoptosis in the CNS during the early postnatal periodin vivo.
MATERIALS AND METHODS
Generation of transgenic mice. In the first step of making our transgene construct, an 850 bpXbaI–SacI fragment, containing a portion of the SV40 small T antigen with an intron and polyadenylation signal, was cloned into Bluescript SK plasmid (Stratagene, La Jolla, CA), containing an 0.8 kb EcoRI fragment of theBcl-xL cDNA (Boise et al., 1993). This resulted in a plasmid termed pBclxL-pA1. In the second step, the plasmid p253Not (a gift from Dr. F. Miller, Montreal Neurological Institute), containing a 1.1 kbSalI–XhoI fragment of the Tα1 α-tubulin promoter, was digested with XhoI, blunt-ended by Klenow enzyme treatment, further digested withSalI, and cloned into plasmidpBclxL-pA1 digested withSalI and EcoRV. The resulting plasmidpTα1-BclxL-pA6 was digested withNotI and SacI. The 2.75 kb transgene fragment was then eluted from an agarose gel and used for pronuclear injection (strain B6/CBA). Integration of the transgene into the mouse genome was determined by PCR and Southern blot analysis on genomic DNA isolated from mouse tails.
Southern blot and PCR analysis. Eight micrograms of genomic DNA extracted from mouse tail were digested with EcoRI, separated in 0.8% agarose gel, and transferred to a positively charged nylon membrane (Boehringer Mannheim, Indianapolis, IN) by standard capillary blotting. A 0.8 kb digoxigenin (DIG)-labeled PCR product, corresponding to Bcl-xL cDNA, was used as a probe for hybridization in DIG Easy Hyb solution (Boehringer Mannheim). The blots were hybridized at 42°C overnight and then washed for 1 hr in 2× SSC and 0.1% SDS at 65°C and 1 hr in 0.1× SSC and 0.1% SDS. For detection we used the DIG luminescent detection kit (Boehringer Mannheim) following the manufacturer’s instructions. The filters were exposed to Kodak (Rochester, NY) X-OMAT AR film. The films were scanned using a densitometer, and transgene copy number was determined by comparison of the signal intensities between the endogenous and transgene bands.
Integration of the transgene was analyzed routinely by PCR using the 5′-oligo (5′-CTGAATGACCACCTAGAGCCTTGG-3′) and the 3′-oligo (5′-GAATGTTGAGAGTCAGCAGTAGCC-3′). PCR was performed in a 50 μl reaction, containing 1× PCR buffer, 1.5 mmMgCl2, a 150 μm concentration of each dNTP, a 0.3 μm concentration of each primer, and 1.25 U of Taq-polymerase (Life Technologies, Gaithersburg, MD), in the following conditions: 94°C for 1 min, 64°C for 1 min, and 72°C for 1 min, for 35 cycles, followed by a 7 min extension at 72°C.
In situ hybridization. Transcription of the construct shown in Figure 1A produces an mRNA containing the coding regions of both human Bcl-xL and a portion of the SV40 small T antigen. Thus, for in situ hybridization, two different antisense riboprobes were used. The first one corresponds to the coding region of human Bcl-xL. This probe recognizes both endogenous (mouse) and transgenic (human)Bcl-xL mRNA. The second probe corresponds to the SV40 small T antigen and recognizes only transgenicBcl-xL mRNA. F1 offspring from different transgenic lines were used for in situ hybridization. Mice were anesthetized with halothane and quickly decapitated, and the brains and spinal cords were frozen on dry ice. Cryostat sections (12–20 μm) were cut, thaw-mounted onto Super Frost Plus slides (Fisher Scientific, Houston, TX), and stored at −20°. On the day of hybridization, slides were thawed, and hybridization was performed as described previously (Wright et al., 1995) .
Fig. 1.

Generation and characterization ofBcl-xL transgenic mice. A, Schematic presentation of the Bcl-xL transgene construct: RI, EcoRI; N,NotI; S, SacI;Sa, SalI; X,XhoI; Xb, XbaI.B, Southern blot analysis of the different transgenic lines. The 6 kb fragment corresponds to the endogenousBcl-xL gene. The 0.8 kb fragment corresponds toBcl-xL transgene. Lines 7193,7199, and 7194 had the highest copy numbers and were used for further analysis.
Facial nerve axotomy and motor neuron analysis. F1 offspring from matings between Tα1-Bcl-xL transgenic animals and CF1 wild-type animals were anesthetized on postnatal day 2 (P2) with methoxyflurane, and the right facial nerve was transected as it exited the stylomastoid foramen. The contralateral side served as control for the axotomy. Animals were killed 1 week after the axotomy with an overdose of sodium pentobarbital and fixed by intracardiac perfusion with 4% paraformaldehyde. The brains were removed, embedded in paraffin, sectioned at 12 μm, and stained with cresyl violet. Six wild-type and six transgenic animals were studied. The facial nucleus was identified, and every fourth section was evaluated. To ensure the accuracy of cell counts in the facial nucleus, we compared the numbers of cells counted with the physical disector with the counts obtained using standard profile methods. No significant difference in the number of neurons in the facial nucleus was found between profile counts of the nucleoli and the physical disector method. Therefore, counts of nucleoli that came into focus through the plane of the section were used as the primary method of determining cell number. Student’st test was used to compare the mean number of surviving motor neurons between wild-type control andBcl-xL-overexpressing animals.
Immunohistochemistry, terminal deoxynucleotidyl transferase-mediated deoxy-UTP nick end-labeling staining, and electron microscopy. Transgenic and control embryos were perfused in PBS followed by 3% paraformaldehyde with 15% picric acid in 0.1m phosphate buffer, pH 7.4. The brains were immediately removed and cryoprotected in 30% sucrose/PBS. Tissue was then frozen in O.C.T. compound and sectioned coronally on a cryostat at 12 μm. After rehydration in PBS, sections were blocked for 30 min in Superblock buffer (Pierce, Rockford, IL) with 1% porcine gelatin, 2% normal horse serum, and 0.3% Triton X-100. A rabbit polyclonal antibody to the receptor tyrosine kinase Ret (kindly provided by Qiao Yan, Amgen) was diluted 1:1500 in the blocking solution and incubated overnight at 4°C. The following day, sections were rinsed in PBS and incubated with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:150 in the blocking solution for 30 min at room temperature. Slides were incubated subsequently with HRP-labeled streptavidin (ABC kit, Vector). Primary antibodies were visualized with 3–3′-diaminobenzidine as the HRP substrate. Slides were then rinsed, dehydrated through graded alcohols, and coverslipped. Terminal deoxynucleotidyl transferase-mediated deoxy-UTP nick end labeling (TUNEL) was performed on P7 mouse brain tissue according to the manufacturer’s instructions (Oncor, Inc.). Electron microscopy on P7 mouse brain was performed as described previously (Golden et al., 1993).
Hypoxic–ischemic injury. Briefly, P7 mice from transgenic line 7194 and wild-type littermates were anesthetized with 2.5% halothane and balance room air, and the left common carotid artery was exposed and ligated permanently as described previously (Ferriero et al., 1995; Holtzman et al., 1996; Cheng et al., 1997). The incision was sutured, and the pups were returned to the dam for a 2 hr recovery and feeding period. The pups were then placed in containers through which humidified 8% oxygen and balance nitrogen flowed for the next 1 hr. The containers were partially submerged in a 37°C water bath to maintain normothermia during this period. After retrieval from the hypoxic chamber, pups were returned to their dam. Mortality during surgery or the exposure to hypoxia was ∼20%; however, mortality did not differ between the control and transgenic animals. Seven days after treatment, brains were processed, and tissue loss caused by hypoxia–ischemia was determined by calculating the amount of surviving tissue in the damaged versus the undamaged hemisphere in coronal sections exactly as described previously (Holtzman et al., 1996;Cheng et al., 1997).
RESULTS
Generation of transgenic mice
To overexpress human Bcl-xL in neurons we used the α-tubulin (Tα1) promoter, which has been well characterized previously using a lacZ reporter gene in transgenic mice. In most lines, this promoter driveslacZ at high levels in neurons during embryonic development and at lower, but readily detectable, levels postnatally (Gloster et al., 1994; Majdan et al., 1997). After injection of the transgene construct (Fig. 1A), 12 founder mice were obtained of 83 screened. Transgene copy number was determined by Southern blot analysis (Fig. 1B). Transgene copy number differed from line to line and varied from 2 to 60 copies .
Analysis of Bcl-xL expression in different transgenic lines
The expression pattern of the transgene was analyzed in F1 offspring of all transgenic lines using in situhybridization. The cDNA sequences of human and mouseBcl-xL are highly conserved, and theBcl-xL riboprobe that we used detected both endogenous and transgenic Bcl-xL mRNA. To distinguish between these, we designed a riboprobe corresponding to the SV40 small T antigen mRNA, which is included in the transcript derived from the transgene construct and therefore allowed us to detect specific expression of the transgene.
In situ hybridization results showed that most of the transgenic lines expressed Bcl-xL mRNA, but that expression patterns were highly variable. The SV40 small T antigen antisense probe allowed a definitive analysis of transgene expression, because no signal above background was detected by in situhybridization when wild-type neuronal tissue was hybridized (Fig.2C). Lines 7193, 7194, and 7199 were chosen for detailed analysis of Bcl-xLtransgene expression. In line 7193, Bcl-xL was expressed at particularly high levels in cerebellum and in motor neurons of the facial nucleus at P7 (Fig. 2A,B). In line 7194, Bcl-xL was expressed in many neurons of the cerebral cortex, as well as in most neurons in thalamic nuclei at P9 (Fig. 3A,B). The transgene was also expressed at high levels in neurons in the CA1, CA2, and CA4 regions of the hippocampus, with less expression in CA3 and dentate gyrus (Fig. 3B). A minority of neurons in the facial motor nucleus also expressed the Bcl-xL transgene in this line.
Fig. 2.

The Bcl-xL transgene is expressed at high levels in the facial motor nucleus of line 7193.A, Section through the brainstem of a transgenic mouse hybridized with a probe for Bcl-xL. This probe detects endogenous and transgenic Bcl-xL mRNA, both of which are expressed throughout the brainstem and facial nucleus. B, Adjacent section from the same animal as inA, hybridized with the SV40 probe, which detects only the transgene-derived Bcl-xL mRNA. Note the high levels of expression of SV40 in motor neurons of the facial nucleus (arrows). C, Brainstem section of a wild-type littermate hybridized with the SV40 probe. Note that no signal was detected in the facial nucleus. Scale bar, 0.5 mm.
Fig. 3.

The Bcl-xL transgene is expressed at high levels in the forebrain of line 7194.A, Section through the forebrain of a transgenic mouse hybridized with a probe for Bcl-xL. This probe detects both the endogenous and transgene-derivedBcl-xL. B, Adjacent section from the same brain as in A, hybridized with the SV40 probe, which detects only transgene-derived Bcl-xLmRNA. Note the high levels of transgene expression in the cerebral cortex (arrows) and thalamic nuclei. Also note the high expression in CA1, CA2 and CA4 regions of the hippocampus, and the minimal expression in the dentate and CA3 regions. Scale bar, 1 mm.
Bcl-xL overexpression rescues facial motor neurons from axotomy-induced cell death
To address the question of whether Bcl-xLoverexpression may influence motor neuron survival, we transected neonatal facial nerves, which causes massive death of facial motor neurons in wild-type animals. The right facial nerve was transected at P2 in F1 offspring from different Tα1-Bcl-xLtransgenic lines and littermate controls. One week after axotomy, the animals were killed. It was obvious in initial experiments that many cells were present in the facial nucleus on the axotomized side inBcl-xL transgenic mice. To verify that the cells in the axotomized facial motor nucleus were motor neurons, we stained sections with an antibody to the glial cell line-derived neurotrophic factor receptor Ret, which is expressed selectively by motor neurons in spinal cord and brainstem (Trupp et al., 1997). In nonaxotomized wild-type control animals, labeling with this antibody demarcates the facial motor pool clearly (Fig.4A). In axotomized wild-type animals, only occasional cells were labeled in the lateral portion of the nucleus (Fig. 4B). It should be noted that although 15% of facial motor neurons remain when the entire nucleus is counted, virtually all surviving neurons are in the medial subnucleus, which projects axons to the auricular musculature and are unlesioned in this paradigm (Dubois-Dauphin et al., 1994). In striking contrast to controls, in the axotomized transgenic animals, cells throughout the facial nucleus were labeled (Fig. 4C), indicating that the surviving cells are in fact motor neurons and not interneurons or glia.
Fig. 4.
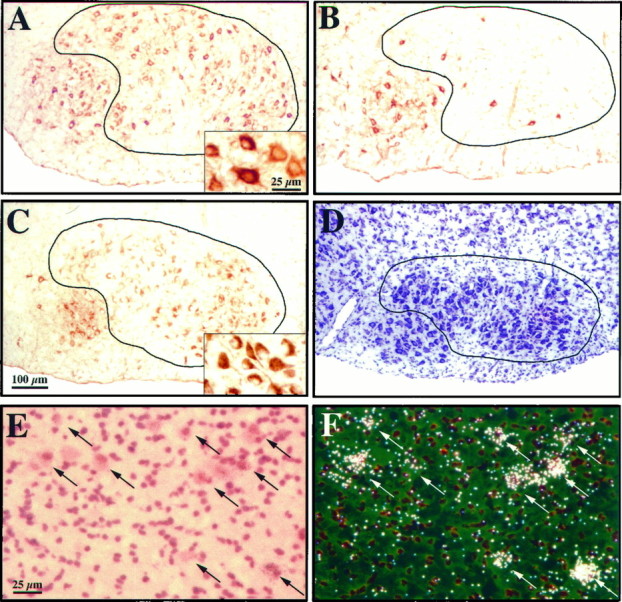
Bcl-xL overexpression protects motor neurons from axotomy-induced cell death. A, Ret-labeled motor neurons in the facial nucleus of a nonaxotomized wild-type mouse. Note the labeling in the medial and lateral (circled) portion of the nucleus. Inset, High-magnification view of Ret labeling in wild-type neurons. B, Ret-labeled motor neurons in the facial nucleus of an axotomized wild-type mouse. Cells in the lateral portion of the nucleus have degenerated (circled), and Ret is no longer detectable except in the medial portion of the nucleus, which is not affected by this lesion paradigm. C, Ret-labeled motor neurons in the facial nucleus of an axotomized transgenic Bcl-xL mouse (line 7193). Many lateral motor neurons survive axotomy (circled), although they are reduced in size.Inset, High-magnification view of Ret labeling in the rescued lateral lateral motor neurons. (Compare the size of motor neurons in the inset in C with the insetin A.) D, Cresyl violet-stained motor neurons in the facial nucleus of an axotomized transgenicBcl-xL mouse. E, High-magnification bright-field view of facial motor neurons 7 d after axotomy in transgenic line 7194. Arrows indicate rescued motor neurons. F, Dark-field view of the same section as in E, labeled with the SV40 probe to detect transgene-derived sequences. Note the high degree of correlation between the rescued motor neurons shown in E and the expression of transgene-derived Bcl-xL shown inF (arrows). Scale bars:A–D, 100 μm; E–F, insets, 25 μm.
Counts of facial motor neurons in Nissl-stained serial sections 1 week after axotomy in line 7193, in which most facial motor neurons expressed the Bcl-xL transgene, demonstrated that an average of 65% of motor neurons survived 1 week after nerve lesion (Figs. 4C,D, 5). The surviving cells were reduced in size (Fig. 4, compareA,C, insets), indicating that motor neurons disconnected from target tissue by axotomy underwent considerable atrophy. Counts of the facial nucleus in unlesioned transgenic animals compared with unlesioned controls revealed no significant difference in the number of motor neurons (Fig. 5). This indicates that although injury-induced apoptosis is suppressed, naturally occurring cell death in the embryonic period was not prevented.
Fig. 5.

Quantification of facial motor neuron rescue in transgenic line 7193. The bar graph shows numbers of facial motor neurons in control and axotomized wild-type andTα1-Bcl-xL transgenic mice. Note the large difference in numbers of facial motor neurons that survive axotomy between wild-type and trangenic animals. Asteriskindicates statistically significant differences (p < 0.05).
In lines 7194 and 7199, fewer facial motor neurons expressed transgenic human Bcl-xL, and fewer motor neurons survived axotomy (e.g., in 7199, ∼33% of the motor neurons survived 1 week after the lesion; n = 3). The correlation between transgene expression and neurons that survive axotomy was studied in line 7194. For example, Figure 4, E and F, shows motor neuron survival (bright field) and transgene expression (dark field) of the same section in the lateral region of the facial nucleus 1 week after axotomy. As indicated by thearrows, each of the surviving facial motor neurons expresses the Bcl-xL transgene.
Neonatal hypoxic–ischemic insult results in apoptosis of forebrain neurons
As opposed to some hypoxia–ischemia paradigms in the adult CNS in which many cells may die via necrosis (Brown and Brierley, 1972), recent data suggest that after hypoxic–ischemic injury to the neonatal brain, many cells die via apoptosis (Ferrer et al., 1994; MacManus et al., 1994; Mehmet et al., 1994; Hill et al., 1995). To examine this issue further, we used a well characterized model of neonatal hypoxia–ischemia (modified Levine procedure), which results in unilateral hypoxic–ischemic brain injury (Rice et al., 1981; Johnston, 1983; Ferriero et al., 1995; Holtzman et al., 1996). We found that unilateral carotid ligation and exposure to 8% O2 for 2.5 hr in P7 rats or 1 hr in P7 mice result in significant damage to the hemisphere ipsilateral to carotid ligation and no damage to the contralateral hemisphere (Ferriero et al., 1995; Holtzman et al., 1996;Cheng et al., 1997). In P7 mice, analysis of the brain ipsilateral to carotid ligation revealed that in cortex, hippocampus, and striatum, some nuclei begin to label with the TUNEL method at 6 hr after the termination of hypoxic exposure (Fig.6B). The number of TUNEL-positive nuclei increased after this time and reached a peak by 18 hr (Fig. 6C,D). Only occasional TUNEL-positive cells were seen contralateral to carotid ligation, but the number of these cells was similar to that seen in normal P7 mice (data not shown). That the TUNEL labeling was nuclear was demonstrated 12 hr after ischemia by staining sections with hematoxylin and eosin (Fig. 6E,F).
Fig. 6.

TUNEL labeling after unilateral carotid ligation and exposure to hypoxia. At different time points after hypoxia–ischemia, the cortex ipsilateral to carotid ligation (ischemic cortex) was assessed for the presence of TUNEL-positive cells in P7 mice. A, There were no TUNEL-positive nuclei at 0 hr. B, Occasional TUNEL-positive nuclei began to appear at 6 hr. C, There was an increase in TUNEL labeling at 12 hr, which reached a peak at ∼18 hr (D). Higher-power photomicrographs counterstained with hematoxylin and eosin demonstrate that TUNEL labeling is nuclear. E–F, Arrows point to shrunken TUNEL-positive nuclei (brown) that are also stained with hematoxylin in the cortex 12 hr after hypoxia–ischemia. The arrowheads point to larger, normal-appearing neuronal nuclei adjacent to the smaller, TUNEL-positive nuclei. Scale bars: A–D, 30 μm; E–F, 7.5 μm.
Whereas the TUNEL method is a sensitive indicator of apoptosis, it is not specific. We therefore performed electron microscopy. We found that there were occasional apoptotic cells in cortex and hippocampus 6 hr after hypoxic treatment ipsilateral to carotid ligation. By 12 hr, there were many cells in both hippocampus and cortex with condensed chromatin and cell shrinkage in every section of ischemic tissue examined (Fig. 7A). Although we cannot rule out that some necrosis occurs, our findings corroborate those of others and suggest that many cells are dying via apoptosis in this model.
Fig. 7.

Electron microscopy reveals evidence of apoptosis after hypoxic–ischemic injury. A, Example of a cell from the cortex of a P7 mouse brain ipsilateral to carotid ligation, 12 hr after exposure to 8% oxygen. There is condensed chromatin (arrows) within the nucleus. B, There is a normal-appearing neuronal nucleus in the P7 mouse cortex contralateral to carotid ligation 12 hr after exposure to 8% oxygen. Scale bar, 5 μm.
Bcl-xL overexpression protects neonatal mouse brain from hypoxic–ischemic insult
We were interested in whether Bcl-xLoverexpression could prevent neuronal cell death in this model that favors apoptosis in the CNS. Our in situ hybridization results showed that in line 7194, the Bcl-xLtransgene is expressed at high levels during the first 2 weeks after birth in the cerebral cortex and hippocampus, regions that are damaged significantly in this hypoxia–ischemia paradigm. Based on thesein situ hybridization results, we tested whether overexpression of Bcl-xL would protect against neonatal hypoxic–ischemic brain injury.
P7 mice from transgenic line 7194 and wild-type littermates received unilateral (left) carotid artery ligation and were exposed to 8% oxygen for 1 hr. Brains were analyzed 1 week later for extent of tissue damage. We found that the amounts of tissue loss (ipsilateral to carotid ligation) in the striatum, cortex, and hippocampus of wild-type animals were 40.1, 30.8, and 52.3%, respectively. In transgenic animals from line 7194, the amounts of tissue loss in striatum, cortex, and hippocampus were 24.6, 11.9, and 31.9%, respectively. Thus, there was 61.5% less damage in the cortex, 39% less damage in the hippocampus, and 38.5% less damage in the striatum of animals overexpressing Bcl-xL (Fig.8). All of these changes were statistically significant.
Fig. 8.

Overexpression of Bcl-xL protects the neonatal mouse brain from hypoxic–ischemic insults. A, Coronal sections of P14 mouse brains 1 week after unilateral (left) carotid ligation and exposure to hypoxia for 1 hr at P7. There was significantly less tissue damage in Bcl-xLtransgenic brains (line 7194) compared with wild-type littermates.B, Quantitative measures of volume loss in transgenic and wild-type animals. The volume of tissue loss in each brain region ipsilateral to carotid ligation (lesioned hemisphere) was compared in each animal with the volume of tissue remaining in the matching brain regions contralateral to carotid ligation (unlesioned hemisphere). The percent volume loss in each structure was determined in each animal, and data are presented as the mean ± SEM.
DISCUSSION
Bcl-xL prevents axotomy-induced apoptosis in the early postnatal period
Using the pan-neuronal promoter Tα1 α-tubulin, we overexpressed human Bcl-xL in neurons. As expected from previous studies, the pattern of transgene expression was highly variable among different lines (Gloster et al., 1994; Majdan et al., 1997). Overexpression of Bcl-xL protected facial motor neurons from axotomy-induced cell death in the neonatal period. The degree of protection varied between the different lines and was correlated with the percentage of facial motor neurons that expressed the transgene. Because all facial motor neurons express endogenousBcl-xL in the postnatal period presumably to some degree, the absolute level of expression must be critical in regulating susceptibility to apoptosis.
It is interesting to compare our results with Bcl-xLoverexpression with other studies that have modulated levels of expression of genes encoding Bcl-2 family members. Overexpression of Bcl-2 under the neuron-specific enolase (NSE) promoter produced results that were similar to those reported here (Dubois-Dauphin et al., 1994). In two of those lines, the majority of motor neurons were protected from axotomy-induced cell death. Thus, an interesting implication of our study is that from a pharmacological standpoint, Bcl-xLand Bcl-2 appear to be interchangeable in regulating motor neuron survival in vivo. In mice that lack the proapoptotic regulator Bax, profound saving of motor neurons was also observed after neonatal facial nerve axotomy (Deckwerth et al., 1996). This latter finding supports the idea that proapoptotic and antiapoptotic Bcl-2 family members interact to regulate susceptibility of postnatal motor neurons to apoptosis.
It is perhaps surprising that there was no reduction in the amount of naturally occurring cell death in the facial motor nucleus. Studies ofBcl-2 overexpression using the NSE promoter showed that lines were highly variable in their ability to prevent naturally occurring cell death related presumably to the developmental age of onset of transgenic Bcl-2 expression (Martinou et al., 1994). The known properties of the Tα1 α-tubulinpromoter suggest that it should be active during early development (Gloster et al., 1994). However, because we were interested in postnatal responses, we did not characterize expression patterns of transgenic Bcl-xL during embryogenesis. Thus, we do not know whether the lack of increase in neuronal numbers is attributable to a relatively late onset of transgenicBcl-xL expression or expression in the embryo that is below the level required to prevent naturally occurring cell death.
Bcl-xL reduces damage caused by neonatal hypoxia-ischemia
Endogenous Bcl-xL is widely expressed in the nervous system in both embryonic and postnatal life (Gonzalez-Garcia et al., 1994; Merry et al., 1994; Gonzalez-Garcia et al., 1995; Parsadanian et al., 1995). To determine whether regulation of apoptosis in vivo by Bcl-xL generalized to cells other than motor neurons, we sought a model of apoptosis of forebrain neurons. Hypoxic–ischemic injury in the adult brain generally leads to necrotic cell death within the “core” of infarcted tissue. However, previous studies have suggested that even severe hypoxic–ischemic brain injury in the neonatal period can lead to apoptosis. We have confirmed and extended these earlier studies (Ferrer et al., 1994; Mehmet et al., 1994; Hill et al., 1995). Carotid ligation in wild-type neonatal mice followed by exposure to hypoxia resulted in a marked amount of DNA damage in cortex, hippocampus, and striatum, as revealed by TUNEL staining. Because TUNEL may not be specific for apoptotic cell death, we performed ultrastructural analysis. Abundant nuclei with changes that resembled apoptosis were seen by EM at 6 and 12 hr after injury. Analogous results have been reported recently that show that injection of glutamate receptor agonists that trigger necrosis in mature rat brain leads to apoptosis in neonatal brain (Portera-Cailliau et al., 1997). Further evidence that this neonatal hypoxic–ischemic injury-induced death has a prominent apoptotic component is provided by the fact that it can be prevented almost completely by injections of BDNF into the lateral ventricle in neonatal rats (Cheng et al., 1997). Interestingly, BDNF is only marginally effective in reducing damage caused by ischemia later in CNS development in the same model (Cheng et al., 1997) .
Overexpression of Bcl-xL had impressive survival-promoting effects in neonatal hypoxia–ischemia-induced brain injury. There was ∼50% reduction in the volume of damage in the hippocampus, cortex, and striatum in mice that overexpress Bcl-xL. It is possible that the protection would have been even greater if Bcl-xL was expressed in all cells (e.g., glia and endothelial cells) and not limited to neurons. Thus, Bcl-xLappears to be effective in regulating death of cells contained completely within the CNS as well as those with peripheral projections (facial motor neurons). This generality of Bcl-xL action is interesting in light of recent demonstrations that PNS and CNS neurons exhibit differing patterns of survival dependence related to neurotrophins and neural activity (Meyer-Franke et al., 1995) .
These observations may have useful clinical implications. This experimental paradigm is a rodent model of hypoxic brain injury, which occurs in the perinatal period in humans and leads to conditions such as cerebral palsy. Once such an insult has occurred, blood supply and oxygen levels usually can return to normal. Thus, if molecules such as Bcl-xL can protect cells against death caused by a “transient” insult, it may provide enough protection for long-term cellular survival and function once the brain environment returns to baseline. An exciting possibility is that factors that augment Bcl-xL actions may be useful as treatments after neonatal hypoxic–ischemic insults.
Potential interactions of Bcl-xL and Bax in regulating postnatal neuron survival
A striking feature of gene expression in Bcl-2 family members is that Bcl-2 is downregulated rapidly during development in all except a few neuronal populations. In contrast, Bcl-xL is either maintained or upregulated during development and into adulthood (Gonzalez-Garcia et al., 1994; Merry et al., 1994; Gonzalez-Garcia et al., 1995; Parsadanian et al., 1995). Similarly, although the precise developmental time frame is less clear, Bax is also expressed at appreciable levels by mature neurons (Krajewski et al., 1994). These expression patterns raise the possibility that Bcl-xL and Bax may be the most important regulators of apoptosis as neurons mature. Our results provide support for this idea. It is clear that overexpression of Bcl-xL in the postnatal period diminishes susceptibility to apoptosis markedly in response to the powerful stimuli of axonal injury and hypoxia–ischemia.
Our results also raise the intriguing possibility that changes in the levels of Bcl-xL and Bax in the postnatal period are related to the profound variations in susceptibility to apoptosis that neurons undergo during development. For example, in the facial axotomy paradigm, cell death is far less extensive in the adult than it is in the postnatal period (for review, see Snider and Thanedar, 1989;Elliott and Snider, 1998). Similarly, after hypoxia–ischemia, although substantial cell death occurs in the adult brain, it occurs by apoptosis less typically than at the early developmental stage studied here (Deshpande et al., 1992). The reasons for such profound changes in neuronal vulnerability to apoptosis associated with these injuries are unknown. One possibility is that an increase in levels of Bcl-xL relative to Bax may lead to a significant change in the threshold for neuronal apoptosis. Indeed, reversal of this ratio, i.e., overexpression of Bax, rapidly leads to death of postnatal sympathetic ganglion neurons in vitro that are normally growth factor-independent (Easton et al., 1997). Taken together, these results support the idea that the Bcl-xL/Bax ratio is critical in setting a threshold for neuronal apoptosis in the postnatal CNS.
Footnotes
This work was supported by a Paul Beeson Physician faculty scholar award from American Federation for Aging Research and National Institutes of Health Grant NS35902 to D.M.H., a Muscular Dystrophy Association Grant and National Institutes of Health grant P50AGO5681 to W.D.S., and the Alan and Edith Wolf Charitable Foundation. We thank J. Harding, R. Gerfen, L. Worley, J. DeMarro, and A. Shah for technical assistance and J. Gidday and M. Jacquin for their advice and expertise.
Correspondence should be addressed to Dr. W. D. Snider, Department Of Neurology, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110.
REFERENCES
- 1.Allsopp TE, Wyatt S, Paterson HF, Davies AM. The proto-oncogene bcl-2 can selectively rescue neurotrophic factor-dependent neurons from apoptosis. Cell. 1993;73:295–307. doi: 10.1016/0092-8674(93)90230-n. [DOI] [PubMed] [Google Scholar]
- 2.Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ. Cloning and chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit of 18. Cell. 1985;41:899–906. doi: 10.1016/s0092-8674(85)80070-2. [DOI] [PubMed] [Google Scholar]
- 3.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2 -related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 4.Brown AW, Brierley JB. Anoxic-ischemic cell change in rat brain: light microscopic and fine-structural observations. J Neurol Sci. 1972;16:59–84. doi: 10.1016/0022-510x(72)90102-5. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM. Marked age-dependent neuroprotection by brain-derived neurotrophic factor against neonatal hypoxic-ischemic brain injury. Ann Neurol. 1997;41:521–529. doi: 10.1002/ana.410410416. [DOI] [PubMed] [Google Scholar]
- 6.Chittenden T, Harrington EA, O’Connor R, Flemington C, Lutz RJ, Evan GI, Guild BC. Induction of apoptosis by the Bcl-2 homologue Bak. Nature. 1995;374:733–736. doi: 10.1038/374733a0. [DOI] [PubMed] [Google Scholar]
- 7.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM J, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 8.Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- 9.Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou J-C. Neonatal motoneurons overexpressing the bcl-2 proto-oncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci USA. 1994;91:3309–3313. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM J. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from BAX deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott JL, Snider WD (1998) Lesion models of motor neuron death. In: Cell death and diseases of the nervous system (Koliatsos V, Ratan R, eds), in press.
- 12.Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard O. bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA. 1995;92:4397–4401. doi: 10.1073/pnas.92.10.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrow SN, White JHM, Martinou I, Raven T, Pun K-T, Grinham CJ, Martinou J-C, Brown R. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature. 1995;374:731–733. doi: 10.1038/374731a0. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I, Tortosa A, Macaya A, Sierra A, Moreno D, Munell F, Bianco R, Squier W. Evidence of nuclear DNA fragmentation following hypoxia-ischemia in the infant rat brain, and transient forebrain ischemia in the adult gerbil. Brain Pathol. 1994;4:115–122. doi: 10.1111/j.1750-3639.1994.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 15.Ferriero DM, Sheldon RA, Holtzman DM, Bredt DS. Mice without neuronal nitric oxide synthase have less injury after perinatal hypoxia-ischemia. Ann Neurol. 1995;38:504A. [Google Scholar]
- 16.Frankowski H, Misotten M, Fernandez P-A, Martinou I, Michel P, Sadoul R, Martinou J-C. Function and expression of the Bcl-x gene in the developing and adult nervous system. NeuroReport. 1995;6:1917–1921. doi: 10.1097/00001756-199510020-00023. [DOI] [PubMed] [Google Scholar]
- 17.Garcia I, Martinou I, Tsujimoto Y, Martinou J-C. Prevention of programmed cell death of sympathetic neurons by the Bcl2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 18.Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The Tα1 α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golden JP, Rana JZ, Davis J, Zahm DS, Jacquin MF. Organization of the proximal, orbital segment of the infraorbital nerve at multiple intervals after axotomy at birth: a quantitative electron microscopic study in rat. J Comp Neurol. 1993;338:159–174. doi: 10.1002/cne.903380203. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, Nunez G. bcl-xL is the major bcl-x mRNA form expressed during murine development and its product localized to mitochondria. Development. 1994;120:3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Garcia M, Garcia I, Ding L, O’Shea S, Boise LH, Thompson CB, Nunez G. bcl-x is expressed in embryonic and postnatal neuronal tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenlund LJS, Korsmeyer SJ, Johnson EM. Role of Bcl-2 in the survival and function of developing and mature sympathetic neurons. Neuron. 1995;15:649–661. doi: 10.1016/0896-6273(95)90153-1. [DOI] [PubMed] [Google Scholar]
- 23.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homologue of the mammalian proto-oncogene bcl-2. Cell. 1994a;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 24.Hengartner MO, Horvitz HR. Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev. 1994b;4:581–586. doi: 10.1016/0959-437x(94)90076-f. [DOI] [PubMed] [Google Scholar]
- 25.Hill IE, MacManus JP, Rasquinha I, Tuor UI. DNA fragmentation indicative of apoptosis following unilateral cerebral hypoxia-ischemia in the neonatal rat. Brain Res. 1995;676:398–403. doi: 10.1016/0006-8993(95)00145-g. [DOI] [PubMed] [Google Scholar]
- 26.Holtzman DM, Sheldon RA, Jaffe W, Cheng Y, Ferriero DM. NGF protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol. 1996;39:114–122. doi: 10.1002/ana.410390117. [DOI] [PubMed] [Google Scholar]
- 27.Johnston MV. Neurotransmitter alterations in a model of perinatal hypoxic-ischemic brain injury. Ann Neurol. 1983;13:511–518. doi: 10.1002/ana.410130507. [DOI] [PubMed] [Google Scholar]
- 28.Kiefer MC, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Barr PJ. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995;374:736–739. doi: 10.1038/374736a0. [DOI] [PubMed] [Google Scholar]
- 29.Korsmeyer SJ. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 30.Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC. Immunohistochemical determination of in vivo distribution of Bax, a dominant inhibitor of Bcl2. Am J Pathol. 1994;145:1323–1336. [PMC free article] [PubMed] [Google Scholar]
- 31.MacManus JP, Hill IE, Huang Z-G, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 32.Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA. Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci. 1997;17:6988–6998. doi: 10.1523/JNEUROSCI.17-18-06988.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinou J-C, Dubois-Dauphin M, Staple JK, Rodriguez, Francowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of Bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 34.Mehmet H, Yue X, Squier MV, Lorek A, Cady E, Penrice J, Sarraf C, Wylezinska M, Kirkbride V, Cooper C, Brown GC, Wyatt JS, Reynolds EOR, Edwards AD. Increased apoptosis in the cingulate sulcus of newborn piglets following transient hypoxia-ischemia is related to the degree of high energy phosphate depletion during the insult. Neurosci Lett. 1994;181:121–125. doi: 10.1016/0304-3940(94)90574-6. [DOI] [PubMed] [Google Scholar]
- 35.Merry DE, Veis DJ, Hickey WF, Korsmeyer SJ. bcl-2 protein expression is widespread in the developing nervous system and retained in the adult PNS. Development. 1994;120:301–311. doi: 10.1242/dev.120.2.301. [DOI] [PubMed] [Google Scholar]
- 36.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 37.Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, Thoenen H. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/s0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 38.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K-I, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x -deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 39.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 40.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 41.Parsadanian AS, Elliott JL, Snider WD. Multiple Ced-3 and Ced-9 homologues are expressed in the murine nervous system. Soc Neurosci Abstr. 1995;21:2019. [Google Scholar]
- 42.Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- 43.Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 44.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 45.Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- 46.Snider WD, Thanedar S. Target deprivation of hypoglossal mo-toneurons during development and during maturity. J Comp Neurol. 1989;279:489–498. doi: 10.1002/cne.902790312. [DOI] [PubMed] [Google Scholar]
- 47.Trupp M, Belluardo N, Funakoshi H, Ibáñez CF. Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-α indicates multiple mechanisms of trophic actions in the adult rat CNS. J Neurosci. 1997;17:3554–3567. doi: 10.1523/JNEUROSCI.17-10-03554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996;10:1–15. doi: 10.1101/gad.10.1.1. [DOI] [PubMed] [Google Scholar]
- 49.Wright DE, White FA, Gerfen RW, Silos-Santiago I, Snider WD. The guidance molecule semaphorin III is expressed in regions of spinal cord and periphery avoided by growing sensory axons. J Comp Neurol. 1995;361:321–333. doi: 10.1002/cne.903610209. [DOI] [PubMed] [Google Scholar]
- 50.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]