

Abstract
It has been shown that abnormalities in axonal transport occur in several mouse models with motoneuron degeneration and also in the human disease amyotrophic lateral sclerosis. In this report, we have examined the potential of neurotrophic factors to act on axonal transport properties in a mouse mutant, progressive motor neuronopathy (pmn). This mouse mutant has been characterized as a “dying-back” motoneuronopathy, with a loss of motoneuron cell bodies and motor fibers. Retrograde transport to the spinal cord motoneurons was determined using fluorescent tracers either injected into the gastrocnemius muscle or applied directly onto the cut sciatic nerve. Because the rate of retrograde labeling was significantly reduced in the pmn, we examined the potential of neurotrophic factors to compensate for the impairment. Ciliary neurotrophic factor (CNTF), brain-derived neurotrophic factor (BDNF), and neurotrophin-3 (NT-3) but not glial-derived neurotrophic factor (GDNF) or nerve growth factor (NGF) were capable of significantly improving the rate of labeling. The differential effects of these factors agree with previous studies showing that molecules that promote cell survival do not necessarily compensate for axonal deficiency. Because impairment of axonal properties appears as an early event in motoneuron pathology, our results may have important clinical implications in the treatment of motoneuron diseases.
Keywords: progressive motor neuronopathy, pmn, neurotrophic factors, retrograde transport, motoneuron disease, ALS, motoneuron cell death
Previous work has shown that certain neurotrophic factors have an important effect on alleviating the pathological symptoms in animal models of motoneuron disease (for review, see Sagot et al., 1997). For example, in progressive motor neuronopathy (pmn) mice both ciliary neurotrophic factor (CNTF) and neurotrophin 3 (NT-3) can improve the life-span and decrease the loss of myelinated axons in the phrenic nerve; CNTF also increases the survival of motoneurons (Sendtner et al., 1992; Sagot et al., 1995a;Haase et al., 1997). In contrast, glial-derived neurotrophic factor (GDNF), a molecule that has been shown to be very potent in preventing motoneuron cell death after axotomy in the neonate (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995), was found to act uniquely on preserving the motoneuron cell soma without affecting myelinated axons or life-span in the pmn (Sagot et al., 1996). This discrepancy between neuronal survival and preservation of axonal integrity was also observed in pmn mice that overexpressed the human Bcl-2 protein (Sagot et al., 1995b). These experiments suggested the existence of two intracellular pathways, one that is necessary for the survival of the cell body and a second that is required for axonal maintenance.
Impairment of axonal maintenance is a common parameter in motor degenerative diseases. For example, abnormalities in axonal transport/uptake have been reported in several models of motoneuron degeneration, such as the wobbler mouse, and in mice overexpressing the heavy chain of neurofilament protein (NF-H) and SOD-I (Bird et al., 1971; Mitsumoto and Gambetti, 1986; Collard et al., 1995; Tu et al., 1996). Interestingly, in aged rats, basal forebrain cholinergic neurons also show a diminution in their ability to retrogradely transport nerve growth factor (NGF) or Fluorogold (Cooper et al., 1994; De Lacalle et al., 1996). Finally, in human neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), impairment of axonal transport and axonal abnormalities are commonly observed (Sasaki and Iwata, 1996; for review, see Hirano, 1991).
Therefore it was of interest to study the retrograde axonal transport combined with neurotrophic factor administration in pmn mice. For this we have examined the rate of motoneuron labeling using fluorescent dyes injected into the gastrocnemius muscle or via direct application onto the nerve stump in the presence or absence of neurotrophic factors. These factors were selected because they belong to three different families of neurotrophic factors: the neurotrophins (for review, seeBarbacid, 1994), the neurocytokine CNTF (for review, see Ip and Yancopoulos, 1996), and GDNF, a member of the TGF-β superfamily (for review, see Lindsay and Yancopoulos, 1996). All of the receptors for these molecules, apart from trkA, have been shown to be present on motoneurons (Yan et al., 1988; DiStefano et al., 1992; Ehlers et al., 1995; Pachnis et al., 1993; Colucci-D’Amato et al., 1996).
The present results confirmed that molecules that promote cell survival do not necessarily compensate for axonal deficiency; in fact, there is a strong correlation between molecules that can act on axonal properties and those that have been shown to beneficially affect the life-span of pmn mice. This study showed that when the first clinical signs of the disease appear, there was no loss of motoneuron cell bodies, whereas the axonal retrograde transport was already impaired. These results substantiate the existence of a peripheral location for the first pathological changes in the pmn disease as have been observed in other motor degenerative disorders.
MATERIALS AND METHODS
Injections of tracers
Intramuscular injection of Fast blue. Mice (16 or 29 d old) were anesthetized with 250 μg of tribromoethanol/gm body weight in PBS (Aldrich, Milwaukee, WI). A small incision was made in the left calf skin to expose the gastrocnemius muscle. A total volume of 5 μl of 0.5% Fast blue (Sigma, St. Louis, MO) in PBS was injected in three different parts of the muscle (median, proximal, and distal) using a Hamilton syringe with a G31 gauge needle. In some experiments the 5 μl injection also contained neurotrophic factors (1 μg/μl) in PBS. The skin was thereafter sutured with 6-0 polyamide thread (Supramid), and the mice were kept at 35°C until they recovered from the narcosis. They were then returned to their cage where all animals received food and water ad libitum. At different times after surgery (see Results), mice were perfused and processed for histological analysis.
Biological activity of the studied neurotrophic factors has been verified in vitro on ventral spinal cord cultures orin vivo on models of axotomy-induced cell death (Sagot et al., 1995a, 1996; Vejsada et al., 1998).
Fluorogold labeling of motoneurons. The left sciatic nerve of anesthetized mice (16, 29, and 37 d old) was sectioned in the mid-thigh. A small polyethylene tube containing 2.5% Fluorogold was applied to the central nerve stump (Vejsada et al., 1995). In some experiments, the Fluorogold solution was mixed with neurotrophic factors (1 μg/μl) in PBS. The incision was sutured, and the animals were returned to their cages after they had recovered from the narcosis. After a 1, 4, or 10 d survival period (see Results), the mice were perfused and processed for histological analysis.
Histological analysis and motoneuron counting
At different times after surgery, mice deeply anesthetized with pentobarbital were perfused with PBS followed by 4% paraformaldehyde (PAF, Sigma) in PBS. Spinal cords were removed and processed for cryosectioning as described previously (Sagot et al., 1995b). Cryostat serial sections (30 μm) were viewed under a Reichert-Jung fluorescent microscope using a wide-band UV filter. Fluorogold or Fast blue-labeled cells, identifiable as motoneurons by their size, shape, and location in the left ventral horn, were counted in every section. Figure1 shows sections of the spinal cord after either Fast blue labeling after injections into the muscle or Fluorogold labeling after application onto the cut nerve. No contralateral motoneurons were labeled (Fig. 1) using the intramuscular injection or the nerve-capping techniques. In some experiments the fluorescent sections were scanned with a high sensitivity Photonic Science Coolview color camera (Carl Zeiss, Oberkochen, Germany) connected to a 486DX2/66 Intel PC. The captured images were analyzed for the mean cross-sectional area of motoneurons using the software package KS 400 (release 2.0, Kontron Electronik, Eching, Germany). Because no difference in the mean cross-sectional area of labeled cells was observed between control and pmn mice (485 ± 12 μm2, n = 338 for control vs 500 ± 12μm2, n = 268 for pmn), we did not correct the neuronal counts according to the Abercrombie formula. Some experiments were subsequently evaluated using cresyl violet staining of the sections. To exclude motoneurons that do not project into the sciatic nerve, only the motoneuron pools that contained Fluorogold were counted. Using this auxiliary criteria, large cells in the ventral horn with abundant cytoplasm and a prominent nucleolus were counted on both sides, without making allowance for split nucleoli. The total nerve length was determined after exposure of the whole nerve starting at the spinal exit of L4 ventral root to the entry into the gastrocnemius muscle. In some cases the gastrocnemius muscles were removed after perfusion and weighed. All data were submitted to ANOVA and an unpaired two-tailed Student’s ttest.
Fig. 1.
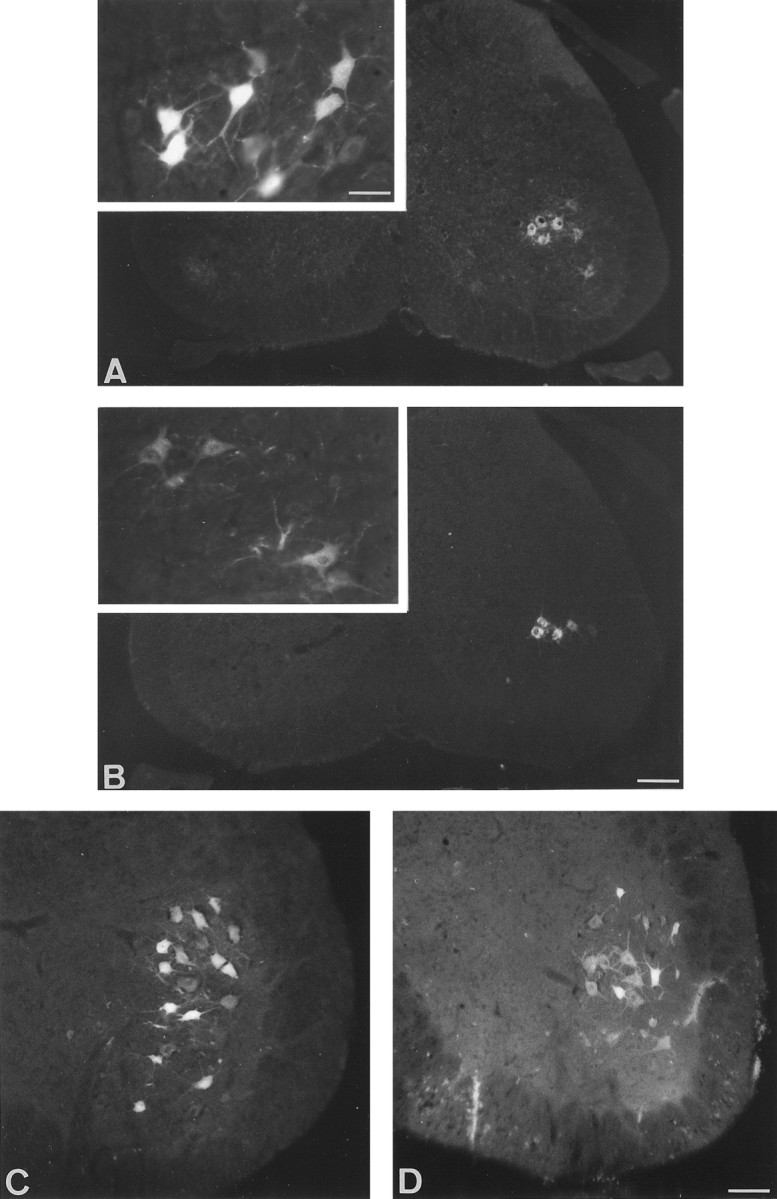
Transverse section of lumbar spinal cord from control (A, C) and pmn (B, D) mice. The labeling in pmn mice is generally weaker than in controls, particularly for short survival times (inset). A, B, Fast blue labeling of motoneurons 6 d after injection into the gastrocnemius muscle of 29-d-old control and pmn mice. Note the absence of contralateral labeling. Inset, High magnification of Fast blue-labeled motoneurons 24 hr after dye injection showing the distribution of fluorescent particles. C, D, Fluorogold labeling of motoneurons 4 d after section of the sciatic nerve in 38-d-old control and pmn mice. Scale bars: A, B, 170 μm; C, D, 70 μm; inset, 35 μm.
Production of pmn homozygote mice overexpressing the human Bcl-2
pmn/pmn bcl-2/+ were generated and selected by PCR analysis as described previously (Sagot et al., 1995b) using transgenic mice that overexpress Bcl-2 (line 71) (Martinou et al., 1994).
RESULTS
Motoneurons degenerate during the pmn disease progression
The pmn disease begins in the hind limbs during the third week after birth, leading rapidly to an atactic gait and eventually to severely impaired locomotion. To determine whether these clinical signs were accompanied by motoneuron cell loss in the lumbar spinal cord, we compared the counts of Fluorogold retrogradely labeled motoneurons projecting into the sciatic nerve in normal mice and in pmn homozygotes of a corresponding age. At day 20, 4 d after the first clinical signs appeared, there was no difference between pmn mice and age-matched control animals in the number of motoneurons labeled with Fluorogold (Fig. 2). However, as the disease progressed, the cell counts in pmn mice decreased by as much as 36% as compared with age-matched controls (p < 0.01) and by 31% as compared with 20-d-old pmn mice (p < 0.01) (for raw numbers see Table1).
Fig. 2.

A, Quantification of motoneuron cell loss during pmn/pmn disease progression using the Fluorogold retrolabeling technique. Each point represents three animals. As early as day 33, the number of retrogradely labeled motoneurons is significantly lower in pmn as compared with controls (*p < 0.05). At 41 d of age the number of motoneurons is significantly lower than in controls and in 20-d-old pmn animals (**p < 0.01). B, The same sections were counterstained with cresyl violet and recounted. At day 41, the number of motoneurons is smaller than in controls or 20-d-old pmn mice (****p < 0.0001). Note the similarity in the pattern of motoneuron cell loss with the two techniques.
Table 1.
Motoneuron cell loss in pmn mice
| Mouse strain | Motoneuron number Fluorogold | Motoneuron number cresyl violet ipsilateral | Motoneuron number cresyl violet contralateral |
|---|---|---|---|
| pmn 20 d | 1799 ± 133 | 2044 ± 50 | 2067 ± 39 |
| pmn 33 d | 1379 ± 84* | n.d. | n.d. |
| pmn 41 d | 1242 ± 105** | 1348 ± 60*** | 1226 ± 16*** |
| Control 20 d | 1834 ± 119 | 2073 ± 137 | 2041 ± 150 |
| Control 33 d | 1961 ± 96 | 2005 ± 64 | 1996 ± 116 |
Raw data from cell counting. Column 1: Fluorogold labeling of motoneurons projecting into the sciatic nerve, 4 d after axotomy. Columns 2 and 3: Cell counts of counterstained sections in column 1. Column 2: Ipsilateral to Fluorogold-labeled cells. Column 3: Contralateral to Fluorogold-labeled column. Number of animals in each group was 3; n.d., not determined.
*p < 0.05; **p < 0.01; ***p < 0.001.
To exclude the possibility that some motor fibers were in the process of degenerating and thus could not take up the Fluorogold after the lesion, we counterstained the sections with cresyl violet and counted motoneurons on both sides of the spinal cord. In accord with the above experiments using retrograde labeling, a significant decrease in motoneuron numbers was observed at later stages of the disease (Fig. 2, Table 1). In addition, this reduction in motoneuron numbers could not be attributed to the nerve lesion itself because the number of motoneurons was identical on the operated and unoperated sides (Table1).
In accordance with the results obtained in the facial nucleus (Sagot et al., 1995a,b; 1996), the loss of spinal lumbar motoneurons was hardly discernible statistically at the onset of the clinical symptoms, but with disease progression it increased rapidly. Assuming that the motoneuron loss was approximately linear with time, there was a loss of 32 (Fluorogold) to 36 (cresyl violet) sciatic motoneurons per day between 20 and 41 d of age. This value is similar to what we observed previously in the facial nucleus of this mouse mutant (2863 ± 109, n = 4 and 2160 ± 51,n = 7 respectively, for 18- and 43-d-old pmn mice, representing a loss of 28 motoneurons per day) (Sagot et al., 1996). Therefore, the rate of loss would be ∼1.6–2.0% lumbar motoneurons per day in pmn mice, a value twofold higher than what has been found in the G1H line of SOD transgenic mice (Chiu et al., 1995). The shorter life-span in pmn mouse mutants (42 d) as compared with the G1H–SOD transgenics (150 d) may be directly correlated with the increased rate of motoneuron loss.
Motoneurons in pmn mice are not hypersusceptible to axotomy
To determine whether axotomy could exacerbate the “natural” degeneration of motoneurons in these mutant mice, we increased the postlesion survival time up to 10 d after surgery in 29-d-old animals. In these conditions, the number of Fluorogold-labeled motoneurons was determined and compared with that obtained with a short survival time, namely 4 d. Both in wild-type and in pmn mice, there was a significant reduction of labeled cells at 10 d as compared with 4 d; in wild-type animals there was a 21% decrease (1549 ± 89, n = 4 vs 1961 ± 96,n = 3; p < 0.05), and in pmn there was an 18% diminution (1126 ± 88, n = 4 vs 1379 ± 84, n = 3; p < 0.01). Taking into account that in pmn the “natural” loss between 33 and 41 d of age is ∼10%, the decrease in motoneuron number attributable to axotomy is only 9–10% (i.e., a lower value than in wild-type controls).
Retrograde labeling of motoneurons is significantly delayed in pmn mice
The pmn mouse model is considered to be a retrograde neurodegenerative disease (Schmalbruch et al., 1991). To document this initial description, we investigated the rate of motoneuron retrograde labeling in 29-d-old control and pmn mice (Fig.3A,B) by injecting Fast blue, a fluorescent tracer, into the gastrocnemius muscle.
Fig. 3.

A, Chronological study of retrolabeling of motoneurons projecting into the gastrocnemius muscle. Injection of Fast blue was performed in 29-d-old animals. Each point represents at least three animals (typically four). During the entire time course, the number of motoneurons labeled during a given time was larger in controls (p < 0.0001). Note the steady labeling from 6 to 15 d in controls. B, Enlargement of the first hours shown in A. Fifteen hours after injection (arrow), 12.5% of control motoneurons are labeled; none were observed in pmn mice. Half-maximal values (dashed lines) are attained at 24 hr in controls; 48 hr was required for pmn mice (p < 0.0001).
A plateau was reached 6 d after Fast blue injection for both pmn and non-pmn animals; however, the maximum number of motoneurons labeled in pmn was ∼46% lower than in control mice. To exclude the possibility that the quantity of Fast blue that was injected might be a limiting factor in these experiments and would thus explain the difference between pmn and controls, injections into the gastrocnemius muscle were performed with a 1% Fast blue solution rather than 0.5%. Six days after injection, the number of motoneurons labeled with the higher dose of Fast blue was identical to that obtained with a 0.5% solution in pmn mice (Table 2).
Table 2.
Axonal transport deficit in pmn mice
| Strain of mouse | pmn (16 d) | Control (16 d) | pmn (29 d) | Control (29 d) |
|---|---|---|---|---|
| Nerve length (mm) | 23.9 ± 0.3 | 24.3 ± 0.2 | 27.9 ± 0.2* | 32.1 ± 0.2 |
| (n = 4) | (n = 4) | (n = 5) | (n = 6) | |
| Rate of labeling (mm/day) | n.d. | n.d. | 37.2 | 64 |
| Gastrocnemius weight (mg) | 18.62 ± 1.4** | 30.8 ± 1.3.5 | 19.02 ± 1.2* | 51.7 ± 5.7 |
| (n = 5) | (n = 6) | (n = 7) | (n = 6) | |
| Maximum of motoneurons at 6 d, 0.5% Fast blue | 291 ± 25 | 509 ± 24 | 262 ± 11 | 483 ± 34 |
| (n = 4) | (n = 3) | (n = 4) | (n = 4) | |
| Maximum of motoneurons at 6 d, 1% Fast blue | n.d. | n.d. | 280 ± 26 | n.d. |
| (n = 3) | ||||
| % of maximum at 48 hr | 51.2 ± 15* | 89.0 ± 7.3 | 47.9 ± 2.7* | 73.7 ± 12 |
| % of maximum at 96 hr | n.d. | n.d. | 90.4 ± 14.8 | 93.6 ± 10.5 |
pmn mice show deficits in axonal retrotransport as soon as the first clinical symptoms appear. The difference in motoneuron retrolabeling between controls and pmn mice was compared with nerve length and muscle weight. At day 16, there was no significant difference in the length of the sciatic nerve; however, the percentage of motoneurons labeled at 48 hr was significantly lower in pmn than in controls. At day 29, despite the shorter sciatic nerve in pmn, the percentage of labeled motoneurons was lower than in controls. The rate of labeling has been calculated by dividing the distance (nerve length) by the time necessary to obtain the first motoneurons labeled (12 hr for controls, 18 hr for pmn mice). n, Number of animals in each group; n.d., not determined.
*p < 0.001; **p < 0.01.
Second, the time necessary to obtain 50% of the maximum (t50%) was reached 2 d after injection in pmn mice; meanwhile, it took only 1 d in control animals. Because in 29-d-old pmn mice the sciatic nerve was 10% shorter than in age-matched controls (Table 2), the delay observed in pmn mice was actually even larger than what was measured.
Third, no motoneurons were labeled 15 hr after Fast blue dye injection in pmn. In contrast, after this short survival time in wild-type animals, there were 61 ± 12 motoneurons already labeled, which represents 12.5% of the maximum value. In control animals the first motoneurons labeled appeared 12 hr after Fast blue injection, whereas in pmn mice the first motoneurons were detectable only 18 hr after injection. When expressed as a function of sciatic nerve length, the rate of transport for the first motoneurons to be labeled was ∼37 mm/d for pmn and 64 mm/d for control animals (Table 2).
Finally, we studied the retrolabeling in younger animals (i.e., 16 d old) just at the onset of the first clinical signs to make allowance for the difference in the nerve length and to a lesser extent the muscle size between control and pmn mice. The rate of retrograde labeling of motoneurons was found to be lower in 16-d-old pmn animals than in controls, although the nerve lengths were identical in both groups at this age. These results also indicate that the impairment of uptake/retrograde axonal transport coincides with the first symptoms of the disease.
To eliminate the potential problem of dye diffusion into the muscles, we performed backlabeling from the cut nerve by applying a Fluorogold solution directly onto the central nerve stump. Twenty-four hours later, labeled cells were counted, and the results were compared with the maximal values obtained with longer survival times (i.e., 4 d) (Fig. 4). Under these conditions, the number of labeled motoneurons in pmn represented only 30% of the maximum. In contrast, in control mice, 65% of motoneurons were already labeled 24 hr after Fluorogold application. Therefore, even in the absence of possible problems related to dye diffusion, we could detect severe impairment of retrograde labeling in pmn mice.
Fig. 4.

Total number of Fluorogold-positive cells at 24 or 96 hr (also see Fig. 2) after application of the Fluorogold tracer onto the central nerve stump of the cut sciatic nerve of 29-d-old control and pmn mice. In controls (n = 4) the total number of labeled cells at 24 hr represents 65% of the maximum obtained at 96 hr; in pmn (n = 4) only 30% of the cells are labeled (****p < 0.0001 for both). These results and those presented in Figure 3A,B suggest an impairment in uptake/retrotransport properties in pmn.
Influence of neurotrophic factors on retrolabeling of motoneurons
Previous studies have shown that certain neurotrophic factors such as CNTF or NT-3 could slow down the disease progression in pmn mice (Sendtner et al., 1992; Sagot et al., 1995a; Haase et al., 1997). We therefore tested the effects of various neurotrophic factors on motoneuron backfilling by coinjecting them with Fast blue.
As compared with injections with Fast blue alone, coinjections with CNTF, BDNF, or NT-3 increased the number of labeled motoneurons at 24 hr in pmn mice by as much as 46% (Fig.5A). In contrast, coinjection of NGF or GDNF did not alter motoneuron retrolabeling (Fig.5B). It is noteworthy that 4 d later, when >90% of the maximal value was attained (Table 2), there was no longer a difference in motoneuron counts between animals coinjected or simply treated with Fast blue (Fig. 5C). Because GDNF has been reported to be a very potent molecule for motoneuron survival (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995), different concentrations of GDNF were examined. No increase in retrolabeling rate was observed at any of the tested concentrations (0.1 μg/μl: 113% ± 8.5; 1 μg/μl: 105.6% ± 9.5; 6 μg/μl: 91.6% ± 4.9; n = 4 for each). Interestingly, we did not observe any alteration in the rate of backfilling in control animals with any of the neurotrophic factors tested at 1 μg/μl (Fig. 5A,B).
Fig. 5.
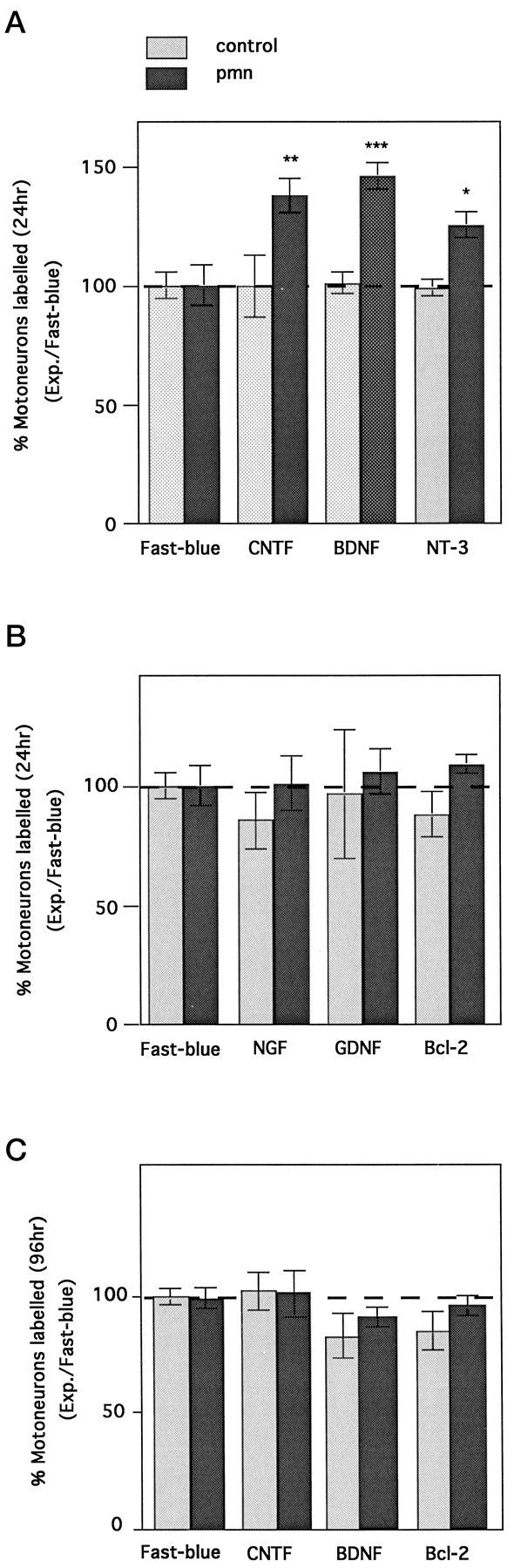
Influence of neurotrophic factors on retrolabeling of pmn motoneurons. Neurotrophic factors (1 μg/μl) mixed with Fast blue were coinjected into the gastrocnemius muscle of 29-d-old pmn or control mice. A, Twenty-four hours later, the number of labeled motoneurons was determined and expressed as the percentage of the values obtained with Fast blue alone (dashed line). BDNF, NT-3, and CNTF displayed a significant increase in the number of labeled motoneurons (*p < 0.01; **p < 0.005; ***p < 0.001). B, In contrast NGF, GDNF, and Bcl-2 did not compensate for the axonal impairment observed in pmn. None of the factors tested induced any changes in the control animals. C, Four days after coinjection, the initial increase disappeared; the size of the motoneuron pool was unchanged. Each group represents the average value obtained from at least three animals (typically four).
Because a decay in rate of retrolabeling was also observed when Fluorogold was applied onto the nerve stump of pmn mice, we also tested the effect of neurotrophic factors in these same conditions (Figs.6, 7). Results were even more striking as compared with those obtained with Fast blue. Figure 6 shows photographs of spinal cord sections 24 hr after Fluorogold application onto the end of the sciatic nerve stump. In control animals (Fig. 6A), there is an intense labeling of the motoneurons in the ventral part of the spinal cord, whereas in pmn mice (Fig. 6B) the labeling is considerably weaker. The number of motoneurons and the intensity of their labeling (not quantified) were augmented by treatment with BDNF (Fig. 6C) or CNTF (Fig. 6E). In contrast, NGF (Fig. 6D) and GDNF (Fig. 6F) did not visibly alter the number of motoneurons that were detectable. Indeed, as shown in Figure 7, the BDNF or CNTF application increased the number of motoneurons labeled at 24 hr by as much as 100% (809 ± 81 for CNTF and 898 ± 35 for BDNF vs 442 ± 23 for Fluorogold alone); meanwhile, GDNF and NGF had no effect (488 ± 58 for GDNF and 478 ± 35 for NGF). Therefore, in pmn treated with CNTF or BDNF, the rate of labeling becomes equivalent to that of control animals. To exclude the possibility that the increase in motoneurons counted was caused by a modification in cell size, we measured the cross-sectional area of fluorescent cells in untreated pmn or in pmn treated with BDNF or CNTF. The size of the motoneurons remained unchanged, independent of the treatment (504 ± 21 μm2, n = 99 for FG alone; 482 ± 15 μm2, n = 89 for BDNF; 514 ± 25 μm2, n=87 for CNTF).
Fig. 6.

Transverse sections of lumbar spinal cord from 29-d-old control (A) and pmn (B–F) mice showing the effects of neurotrophic factors on retrograde labeling of motoneurons 24 hr after application onto the sciatic nerve stump. Compared with untreated pmn mice (B), the number and the intensity of Fluorogold labeled cells were significantly increased after BDNF (C) or CNTF (E) application. In contrast, NGF (D) or GDNF (F) had no effect on retrolabeling rate. Occasionally strongly labeled cells are visible in NGF- and GDNF-treated or untreated pmn mice (F). Note also the punctate labeling in the cell bodies, which may indicate the trapping of Fluorogold in lysosomes and endosomes (Wessendorf, 1991). Scale bars: A–D, 90 μm; E, F, 60 μm.
Fig. 7.

Restoration of motoneuron retrolabeling rate by BDNF and CNTF but not NGF or GDNF. After section of the sciatic nerve of 29-d-old pmn or control mice, neurotrophic factors (1 μg/μl) mixed with Fluorogold have been applied onto the proximal nerve stump. Twenty-four hours later, the number of labeled motoneurons was determined and expressed as a percentage of the values obtained with Fluorogold alone. The values obtained with CNTF and BDNF were significantly different from those of other groups (one-way ANOVA and Student’s t test; ***p < 0.005; ****p < 0.001). As in Figure 5, none of the factors tested induced any changes in the control animals.
Bcl-2 overexpression does not stimulate the uptake or axonal transport
Because Bcl-2 as well as GDNF have been shown to slow down motoneuron cell death without acting on axonal maintenance (Sagot et al., 1995b; 1996), it was of interest to study the effect of Bcl-2 overexpression on retrograde labeling in pmn. Pmn mice overexpressing the human Bcl-2 transgene were generated by crossing pmn/+ breeders with Bcl-2 transgenic mice (Sagot et al., 1995b). Despite its strong expression in motoneurons (Sagot et al., 1995b), Bcl-2 did not influence the rate of labeling either in pmn or in control animals (Fig. 5B,C).
DISCUSSION
pmn is a motoneuronopathy and not an axonopathy
Using three different techniques of assessment of motoneuron cell numbers, we have demonstrated in the present report that there was indeed a significant loss of spinal cord motoneurons in pmn mice. First, we examined the motoneurons in the sciatic nerve pool by means of retrolabeling of Fluorogold applied onto the cut nerve. Second, the same sections of lumbar spinal cord were counterstained with cresyl violet, and the motoneuron pools that had been previously labeled with Fluorogold were recounted. In this way, it was possible to confirm that the loss observed using Fluorogold backlabeling from the cut sciatic nerve was not simply caused by the presence of degenerating axons that were unable to take up the dye. Furthermore, the loss of motoneurons was not induced by the peripheral nerve lesion, because motoneuron counts were identical on the unlesioned contralateral side. Third, a smaller pool of motoneurons in the lumbar spinal cord were retrolabeled by means of injections of a fluorescent tracer (Fast blue) into the gastrocnemius muscle (see below); 46% fewer motoneurons were labeled as compared with control animals injected at 29 d of age.
Taken together, these results strongly suggest that pmn is a motoneuronopathy, with a significant loss of motoneuron cell bodies, and not simply a disease restricted to the axon. However, the issue of whether spinal cord motoneurons degenerate in pmn mice has been questioned because of the initial report by Schmalbruch et al. (1991), who claimed that the number of ventral roots in pmn was unaffected. It is possible that the ventral root counts are not an correct estimation of the motoneuron pool; as reported by Schmalbruch et al. (1991), the number of myelinated fibers of a given root may vary considerably. Therefore, the difference in the techniques for motoneuron assessment may be the source of the differences in results observed between the study by Schmalbruch et al. (1991) and this report.
Impairment of retrotransport in pmn mice
Axonal impairment was examined by studying the capacity of the sciatic nerve to retrogradely transport a fluorescent tracer (Fast blue) injected into the gastrocnemius muscle. The maximum number obtained in control animals is in accordance with results obtained byMcHanwell and Biscoe (1981), who used horseradish peroxidase tracing. As expected, there were fewer motoneurons labeled in the mutant mice as compared with the controls; however, this loss of motoneurons (i.e., 46%) surpassed that observed with either Fluorogold or cresyl violet staining in the lumbar spinal cord (∼23% at 33 d of age). This difference supports the hypothesis of a “dying-back” neuronopathy, whereby motor fibers degenerate from the muscle before death of the cell body ensues.
This method of examining retrolabeling of motoneurons using a fluorescent tracer lends itself to studying not only the total number of motoneurons but also to analyzing the rate of motoneuron backfilling. The rate of 64 mm/d for control animals agrees with other reports that examined the rate of horseradish peroxidase retrotransport (Kristensson, 1975; Bisby, 1980). By means of a chronological study of motoneuron labeling from 12 to 400 hr after dye injection, it was possible to show that there was a decreased rate of motoneuron labeling in pmn as compared with controls. This impairment of uptake/retrograde transport properties demonstrated with Fast blue injections into the gastrocnemius muscle was also observed when another tracer (i.e., Fluorogold) was applied onto the proximal stump of the cut nerve.
The first motoneurons to be labeled with Fast blue appeared several hours earlier in controls than in pmn mice. This latter result suggests that even motoneurons in the pmn, which maintain a contact with the muscle, suffer from an impairment in uptake or axonal transport. Finally, this impairment in pmn mice was detectable as soon as the first clinical signs appeared (i.e., 16 d of age), and it preceded motoneuron cell death.
BDNF, NT-3, and CNTF can increase the rate of retrolabeling
Previous studies have demonstrated the beneficial effect of neurotrophic factors on the pmn disease (Sendtner et al., 1992; Sagot et al., 1995a; Haase et al., 1997). Therefore, it was of interest to examine the effects of an anti-apoptotic molecule and different neurotrophic factors for their ability to alter the properties of retrograde transport. The neurocytokine CNTF and both neurotrophins, BDNF and NT-3, were found to increase the rate of retrolabeling by ∼40%, whereas GDNF and NGF had no effect. This difference between CNTF/BDNF and GDNF/NGF was even more pronounced when the retrolabeling assay was performed on the cut sciatic nerve. This differential response cannot be related to the inability of the axons to transport the neurotrophic factors. In fact, all of these factors, even NGF, whose cognate receptor trkA is absent on motoneurons (Yan et al., 1988;Ehlers et al., 1995), have been shown to be retrogradely transported to motoneuron cell bodies (DiStefano et al., 1992; Curtis et al., 1993;Yan et al., 1995). It seems improbable that the effects of CNTF, BDNF, or NT-3 are mediated by an action on motoneuron survival. Although <2% of motoneurons are lost per day, there is already an increase in the number of labeled neurons that exceeds 40% for Fast blue and >100% for Fluorogold after 24 hr in the presence of neurotrophic factors. Furthermore, we can exclude the possibility that other motoneurons (non-gastrocnemius) are labeled, because the maximum number of labeled motoneurons remained the same at 4 d.
It is interesting that whatever the experimental paradigm, neurotrophic factors do not affect the speed of retrolabeling in normal, control animals. Possibly the rate of transport in these animals is already at an optimal level and cannot be influenced by an exogenous source of factors. A similar phenomenon has been observed in experiments designed to examine retrograde transport of neurotrophic factors in unlesioned and lesioned motor nerves (Curtis et al., 1993, 1994, 1995). Indeed, in undamaged motor nerves there was almost no transport of CNTF, leukemia inhibitory factor, or NT-4, whereas nerve lesion produced a significant increase in the retrograde axonal flow of these molecules. Taken together, these results support the idea that normal or undamaged axons would not be capable of upregulating their retrograde transport mechanism in response to exogenous ligands.
We can only speculate about the actual mechanism(s) by which BDNF, NT-3, or CNTF increase the rate of backfilling. It is generally considered that after binding to their cognate receptors, neurotrophic factors become internalized into vesicles and are then retrogradely transported to the cell body (Curtis and DiStefano, 1994). It seems improbable, however, that such a mechanism would increase the rate of retrolabeling with Fast blue or Fluorogold in pmn mice, because in control animals none of the neurotrophic factors examined were capable of increasing the rate of labeling. Furthermore, in pmn mice we have shown that peripherally applied GDNF or CNTF could slow down the loss of motoneurons (Sagot et al., 1995a; 1996), suggesting therefore that both of them are retrogradely transported to the cell body.
It seems remarkable that these factors can increase the number of retrogradely labeled cells at 24 hr. If their action is mediated via the cell soma, it is difficult to imagine how they can act so rapidly. The action of neurotrophic factors in pmn could result from various local intracellular events, including uptake and transport. Indeed, peripheral action of neurotrophic factors, independent of the soma, have already been reported in vitro (Stoop and Poo, 1995). It is known that neurotrophic factors such as NGF induce changes in membrane structures (Connolly et al., 1979) and increase the number and velocity of transported particles on cultured neuronal cells (Yuki et al., 1996). Because receptors to CNTF and neurotrophins are known to activate common pathways for NGF (for review, see Ip and Yancopoulos, 1996; Segal and Greenberg, 1996), it is tempting to propose that what may contribute to the stimulation of retrolabeling is a direct action on uptake at the motor terminals as well as an increase in the rate of axonal retrograde transport.
Molecules that activate the retrolabeling also increase the life-span of pmn mice
The difference between the two classes of molecules, namely BDNF, CNTF, and NT-3 versus GDNF, NGF, and Bcl-2, with respect to retrograde transport, may have a biological relevance. Indeed two of the three molecules, namely CNTF and NT-3, which stimulate the transport, significantly increase the pmn life-span (Sendtner et al., 1992; Sagot et al., 1995a; Haase et al., 1997). In contrast, those that act only on cell body survival, such as GDNF and Bcl-2, do not act on the disease progression (Sagot et al., 1995b, 1996). Interestingly, such a difference has also been found in other animal models of ALS. For example, CNTF and BDNF almost arrested the disease progression in wobbler mice (Mitsumoto et al., 1994), whereas Bcl-2 had no effect on the disease (Aït-Ikhlef et al., 1995; Coulpier et al., 1996).
Impairment of uptake/axonal transport is not restricted to pmn mice but appears to be a key feature in many diseases as well as in aging (for review, see Lee and Cleveland, 1996). It remains to be determined whether defective axonal transport is the primary event in pmn and other motoneuron diseases or rather a consequence of some as yet uncharacterized pathology. At least for the pmn disease, if the impairment of uptake/axonal transport is not the primary cause, it belongs to the earliest manifestations of the disease.
Footnotes
This work was supported by the Association Française contre les Myopathies (France) and the Swiss National Science Foundation. We thank Dr. E. Johnson for reading this manuscript, B. King for her excellent technical assistance, F. Pillonel for assistance with the photographs, and M. L. Bochaton-Piallat and K. Grandchamp for their help with image analysis. We thank Genentech (San Francisco) for the gift of GDNF, and Regeneron (Tarrytown) for providing us with CNTF, BDNF, NGF, and NT-3.
Correspondence should be addressed to Dr. Y. Sagot, Department of Pharmacology, Centre Médical Universitaire, 1 avenue Champel, 1211 Geneva 4, Switzerland.
REFERENCES
- 1.Aït-Ikhlef A, Murawska M, Blondet B, Hantaz-Ambroise D, Martinou JC, Rieger F. The motoneuron degeneration in the wobbler mouse is independent of the overexpression of a Bcl2 transgene in neurons. Neurosci Lett. 1995;199:163–166. doi: 10.1016/0304-3940(95)12061-8. [DOI] [PubMed] [Google Scholar]
- 2.Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 3.Bird MT, Shuttleworth E, Jr, Koestner A, Reinglass J. The wobbler mouse mutant: an animal model of hereditary motor system disease. Acta Neuropathol. 1971;19:39–50. doi: 10.1007/BF00690953. [DOI] [PubMed] [Google Scholar]
- 4.Bisby MA (1980) Retrograde axonal transport. Adv Cell Neurobiol 69–117.
- 5.Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- 6.Collard JF, Côté F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 7.Colucci-D’Amato GL, D’Alessio A, Filliatreau G, Florio T, Di Giamberardino L, Chiappetta G, Vecchio G, Fusco A, Santoro M, de Franciscis V. Presence of physiologically stimulated RET in adult rat brain: induction of RET expression during nerve regeneration. Cell Growth Differ. 1996;7:1081–1086. [PubMed] [Google Scholar]
- 8.Connolly JL, Greene LA, Viscarello RR, Riley WD. Rapid, sequential changes in surface morphology of PC12 pheochromocytoma cells in response to nerve growth factor. J Cell Biol. 1979;82:820–827. doi: 10.1083/jcb.82.3.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper JD, Lindholm D, Sofroniew MV. Reduced transport of (125I)NGF by cholinergic neurons and down-regulated TrkA expression in the medial septum of aged rats. Neuroscience. 1994;62:625–629. doi: 10.1016/0306-4522(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 10.Coulpier M, Junier MP, Peschanski M, Dreyfus PA. Bcl-2 sensitivity differentiates two pathways for motoneuronal death in the wobbler mutant mouse. J Neurosci. 1996;16:5897–5904. doi: 10.1523/JNEUROSCI.16-19-05897.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis R, DiStefano PS. Neurotrophic factors, retrograde axonal transport and cell signalling. Trends Cell Biol. 1994;4:383–386. doi: 10.1016/0962-8924(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 12.Curtis R, Adryan KM, Zhu Y, Harkness PJ, Lindsay RM, DiStefano PS. Retrograde axonal transport of ciliary neurotrophic factor is increased by peripheral nerve injury. Nature. 1993;365:253–255. doi: 10.1038/365253a0. [DOI] [PubMed] [Google Scholar]
- 13.Curtis R, Scherer SS, Somogyi R, Adryan KM, Ip NY, Zhu Y, Lindsay RM, DiStefano PS. Retrograde axonal transport of LIF is increased by peripheral nerve injury: correlation with increased LIF expression in distal nerve. Neuron. 1994;12:191–204. doi: 10.1016/0896-6273(94)90163-5. [DOI] [PubMed] [Google Scholar]
- 14.Curtis R, Adryan KM, Stark JL, Park JS, Compton DL, Weskamp G, Huber LJ, Chao MV, Jaenisch R, Lee KF, Lindsay RM, DiStefano PS. Differential role of the low affinity neurotrophin receptor (p75) in retrograde axonal transport of the neurotrophins. Neuron. 1995;14:1201–1211. doi: 10.1016/0896-6273(95)90267-8. [DOI] [PubMed] [Google Scholar]
- 15.De Lacalle S, Cooper JD, Svendsen CN, Dunnett SB, Sofroniew MV. Reduced retrograde labelling with fluorescent tracer accompanies neuronal atrophy of basal forebrain cholinergic neurons in aged rats. Neuroscience. 1996;75:19–27. doi: 10.1016/0306-4522(96)00239-4. [DOI] [PubMed] [Google Scholar]
- 16.DiStefano PS, Friedman B, Radziejewski C, Alexander C, Boland P, Schick CM, Lindsay RM, Wiegand SJ. The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal transport in peripheral and central neurons. Neuron. 1992;8:983–993. doi: 10.1016/0896-6273(92)90213-w. [DOI] [PubMed] [Google Scholar]
- 17.Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haase G, Kennel P, Pettmann B, Vigne, Akli S, Revah F, Schmalbruch H, Kahn A. Gene therapy of murine motor neuron disease using adenoviral vectors for neurotrophic factors. Nat Med. 1997;3:429–436. doi: 10.1038/nm0497-429. [DOI] [PubMed] [Google Scholar]
- 19.Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- 20.Hirano A. Cytopathology of amyotrophic lateral sclerosis. In: Rowland LP, editor. Advances in neurology, Vol 56. Amyotrophic lateral sclerosis and other motor neuron diseases. Raven; New York: 1991. pp. 91–101. [PubMed] [Google Scholar]
- 21.Ip NY, Yancopoulos GD. The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Annu Rev Neurosci. 1996;19:491–515. doi: 10.1146/annurev.ne.19.030196.002423. [DOI] [PubMed] [Google Scholar]
- 22.Kristensson K. Retrograde axonal transport of protein tracers. In: Cowan WM, Cuénod M, editors. The use of axonal transport for studies of neuronal connectivity. Elsevier; New York: 1975. pp. 70–82. [Google Scholar]
- 23.Lindsay RM, Yancopoulos GD. GDNF in a bind with known orphan: accessory implicated in new twist. Neuron. 1996;17:571–574. doi: 10.1016/s0896-6273(00)80189-0. [DOI] [PubMed] [Google Scholar]
- 24.Lee MK, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci. 1996;19:187–217. doi: 10.1146/annurev.ne.19.030196.001155. [DOI] [PubMed] [Google Scholar]
- 25.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1–20. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 26.McHanwell S, Biscoe TJ. The localisation of motoneurons supplying the hindlimb muscles of the mouse. Philos Trans R Soc Lond [Biol] 1981;293:477–508. doi: 10.1098/rstb.1981.0082. [DOI] [PubMed] [Google Scholar]
- 27.Mitsumoto H, Gambetti P. Impaired slow axonal transport in wobbler mouse motor neuron disease. Ann Neurol. 1986;19:36–43. doi: 10.1002/ana.410190108. [DOI] [PubMed] [Google Scholar]
- 28.Mitsumoto H, Ikeda K, Klinkosz B, Cedarbaum JM, Wong V, Lindsay RM. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science. 1994;265:1107–1110. doi: 10.1126/science.8066451. [DOI] [PubMed] [Google Scholar]
- 29.Oppenheim RW, Houenou LJ, Johnson JE, Lin LFH, Li L, Lo AC, Newsome AL, Prevette DM, Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 30.Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–1017. doi: 10.1242/dev.119.4.1005. [DOI] [PubMed] [Google Scholar]
- 31.Sagot Y, Tan SA, Baetge E, Schmalbruch H, Kato AC, Aebischer P. Polymer encapsulated cell lines genetically engineered to release ciliary neurotrophic factor can slow down progressive motor neuronopathy in the mouse. Eur J Neurosci. 1995a;7:1313–1322. doi: 10.1111/j.1460-9568.1995.tb01122.x. [DOI] [PubMed] [Google Scholar]
- 32.Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995b;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sagot Y, Tan SA, Hammang JP, Aebischer P, Kato AC. GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmm/pmm mice. J Neurosci. 1996;16:2335–2341. doi: 10.1523/JNEUROSCI.16-07-02335.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagot Y, Vejsada R, Kato AC. Clinical and molecular aspects of motoneuron diseases: animal models, neurotrophic factors and Bcl-2 oncoprotein. Trends Pharmacol. 1997;18:330–337. doi: 10.1016/s0165-6147(97)01094-8. [DOI] [PubMed] [Google Scholar]
- 35.Sasaki S, Iwata M. Impairment of fast axonal transport in the proximal axons of anterior horn neurons in amyotrophic lateral sclerosis. Neurology. 1996;47:535–540. doi: 10.1212/wnl.47.2.535. [DOI] [PubMed] [Google Scholar]
- 36.Schmalbruch H, Jensen HJS, Bjaerg M, Kamieniecka Z, Kurland L. A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol. 1991;50:192–204. doi: 10.1097/00005072-199105000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 38.Sendtner M, Schmalbruch H, Stöckli KA, Carroll P, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature. 1992;358:502–504. doi: 10.1038/358502a0. [DOI] [PubMed] [Google Scholar]
- 39.Stoop R, Poo MM. Potentiation of transmitter release by ciliary neurotrophic factor requires somatic signaling. Science. 1995;267:695–699. doi: 10.1126/science.7839148. [DOI] [PubMed] [Google Scholar]
- 40.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VMY. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vejsada R, Sagot Y, Kato AC. Quantitative comparison of the transient rescue effects of neurotrophic factors on axotomized motoneurons in vivo. Eur J Neurosci. 1995;7:108–115. doi: 10.1111/j.1460-9568.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- 42.Vejsada R, Tseng JL, Lindsay RM, Acheson A, Aebischer P, Kato AC (1998) Synergistic but transient rescue effects of BDNF and GDNF on axotomized neonatal motoneurons. Neuroscience, in press. [DOI] [PubMed]
- 43.Wessendorf MW. Fluoro-Gold: composition, and mechanism of uptake. Brain Res. 1991;553:135–148. doi: 10.1016/0006-8993(91)90241-m. [DOI] [PubMed] [Google Scholar]
- 44.Yan Q, Snider WD, Pinzone JJ, Johnson EM. Retrograde transport of nerve growth factor (NGF) in motoneurons of developing rats: assessment of potential neurotrophic effects. Neuron. 1988;1:335–343. doi: 10.1016/0896-6273(88)90082-7. [DOI] [PubMed] [Google Scholar]
- 45.Yan Q, Matheson C, Lopez OT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 46.Yuki S, Takenaka T, Kawakami T, Sakai I, Saito KI. Control of axonal transport by recombinant human NGF in cultured mouse dorsal root ganglion cells. Soc Neurosci Abstr. 1996;22:746. [Google Scholar]