

Abstract
Metabotropic glutamate receptors (mGluRs) couple to heterotrimeric G-proteins and regulate cell excitability and synaptic transmission in the CNS. Considerable effort has been focused on understanding the cellular and biochemical mechanisms that underlie regulation of signaling by G-proteins and their linked receptors, including the mGluRs. Recent findings demonstrate that regulators of G-protein signaling (RGS) proteins act as effector antagonists and GTPase-activating proteins for Gα subunits to inhibit cellular responses by G-protein-coupled receptors. RGS4 blocks Gq activation of phospholipase Cβ and is expressed broadly in rat brain. The group I mGluRs (mGluRs 1 and 5) couple to Gq pathways to regulate several effectors in the CNS. We examined the capacity of RGS4 to regulate group I mGluR responses. InXenopus oocytes, purified RGS4 virtually abolishes the mGluR1a- and mGluR5a-mediated but not the inositol trisphospate-mediated activation of a calcium-dependent chloride current. Additionally, RGS4 markedly attenuates the mGluR5-mediated inhibition of potassium currents in hippocampal CA1 neurons. This inhibition is dose-dependent and occurs at concentrations that are virtually identical to those required for inhibition of phospholipase C activity in NG108–15 membranes and reconstituted systems using purified proteins. These findings demonstrate that RGS4 can modulate mGluR responses in neurons, and they highlight a previously unknown mechanism for regulation of G-protein-coupled receptor signaling in the CNS.
Keywords: RGS4, RGS proteins, metabotropic glutamate receptor, hippocampus, Gα-proteins, synaptic regulation, Xenopus
Glutamate is the principal excitatory amino acid neurotransmitter in the CNS, and its actions are mediated by specific interactions with two distinct families of receptors: the ionotropic and metabotropic glutamate receptors (mGluRs) (Hollmann and Heinemann, 1994). The mGluRs couple to heterotrimeric GTP-binding proteins (G-proteins) and regulate cell excitability and synaptic transmission at glutamatergic synapses throughout the brain (Saugstad et al., 1995a; Conn and Pin, 1997). The mGluR family consists of at least eight distinct receptors, many with alternatively spliced isoforms, that are classified into three major subgroups (Conn and Pin, 1997). Members of the group I mGluRs (mGluRs 1 and 5) couple to the Gq signaling pathway, whereas members of the group II (mGluRs 2 and 3) and III (mGluRs 4, 6, 7, and 8) mGluRs couple to Gi/Go signaling pathways in heterologous expression systems.
Net transmission through glutamatergic circuits can be impacted dramatically by the mGluRs present at a given synapse, and signaling by mGluRs is dynamically regulated under various physiological and pathological conditions (Akiyama et al., 1987; Seren et al., 1989;Yamada et al., 1989; Aronica et al., 1991; Chen et al., 1992; Mayat et al., 1994). Delineation of the mechanisms involved in regulating mGluR function will be critical for development of a complete understanding of regulation of transmission at glutamatergic synapses.
Considerable effort has been focused on understanding the cellular and biochemical mechanisms that underlie regulation of signaling by G-proteins and their linked receptors, including the mGluRs. Recent findings demonstrate that G-proteins interact directly with a newly identified family of regulatory proteins, regulators of G-protein signaling (RGS) proteins, that block G-protein functions (Dohlman and Thorner, 1997; Koelle, 1997). Although RGS proteins were first identified by genetic analysis of lower eukaryotic organisms, more than 20 unique mammalian RGS isoforms have been identified by molecular cloning, with predicted molecular mass ranging from 140 to 17 kDa (Druey et al., 1996; Koelle and Horvitz, 1996). Biochemical evidence indicates that RGS proteins serve as GTPase-activating proteins (GAPs) to markedly accelerate Gα-catalyzed GTP hydrolysis (Berman et al., 1996; Hunt et al., 1996; Watson et al., 1996) or as effector antagonists (Hepler et al., 1997) to negatively regulate G-protein signaling by members of the Gq or Gifamilies, but not Gs or G12.
We reported recently that RGS4, a protein found exclusively in brain (Druey et al., 1996), blocks Gq activation of phospholipase Cβ in vitro (Hepler et al., 1997) and receptor and Gq-directed inositol lipid signaling in intact cells (Huang et al., 1997; Yan et al., 1997). To determine whether RGS4 could play an important role in regulation of mGluR signaling, we examined the effects of RGS4 on characteristic cellular responses stimulated by group I mGluRs in Xenopus oocytes or neurons. We report that RGS4 blocks the mGluR1a- and mGluR5a-mediated activation of the calcium-dependent chloride current (ICl(Ca)) in Xenopus oocytes and the mGluR5-mediated inhibition of calcium-activated potassium currents (IAHP) in hippocampal CA1 neurons. This is the first demonstration that RGS proteins can modulate mGluR responses, and it reveals a previously unknown mechanism for the regulation of G-protein-coupled receptor (GPCR) responses in the CNS.
MATERIALS AND METHODS
Reagents. 3,5-Dihydroxyphenylglycine (DHPG) was purchased from Tocris Cookson (Bristol, UK). Glutamate, inositol 1,4,5-trisphosphate [Ins(1,4,5)P3], and isopropylthio-β-galactoside (IPTG) were obtained from Sigma (St. Louis, MO). RNA transcription kits were obtained from Stratagene (La Jolla, CA), 32P-dCTP was purchased from New England Nuclear, and Xenopus laevis were purchased from Nasco, Ft. Atkinson, WI. Purified recombinant PLCβ1 was a generous gift of Drs. R. Ball and P. Sternweis (University of Texas Southwestern Medical Center). Recombinant hexahistidine-tagged Gqα was prepared and purified as described (Hepler et al., 1996). The polyclonal antiserum that specifically recognizes RGS4 (R374) was kindly provided by Dr. A. G. Gilman (University of Texas Southwestern Medical Center) and was made in rabbits against the synthetic peptide YKGAKSSADCTSLVPQ that corresponds to the C-terminal residues 189–203 of RGS4. The cDNA clones were generously shared by Dr. A. Gilman (University of Texas Southwestern Medical Center; RGS4), Dr. E. Mulvihill (Darwin; mGluR1a), Dr. J. P. Pin (Centre CNRS–INSERM de Pharmacologie–Endocrinologie; mGluR5a), Dr. H. Lester (California Institute of Technology; Kir3.4), and Dr. J. Adelman (Vollum Institute; Kir3.1).
Preparation of RGS protein. Bacterial cultures (JM109) harboring the hexahistidine-tagged RGS4 cDNA in a bacterial expression vector (His6-RGS4/pQE60) were grown to OD6000.5, and protein production was induced by the addition of 0.1 mmIPTG. Cells were grown for 12 hr at 30°C with constant shaking (150 rpm) and harvested by centrifugation. Cell lysates were prepared, and the supernatant containing RGS4 was recovered in Buffer A [50 mm HEPES, pH 8.0, 20 mm β-mercaptoethanol, 100 mm NaCl, 0.1 mm phenylmethylsulfonyl fluoride (PMSF)]. This material was applied to a column containing 5 ml of Ni-NTA affinity resin (Qiagen, Hilden, Germany), and the column was washed with 100 ml of Buffer A containing 500 mm NaCl and 15 mm imidazole. RGS4 was eluted with 20 ml of Buffer A containing 150 mm imidazole. Eluted protein was concentrated to 1 ml and further enriched by size exclusion chromatography. Sample was applied to a column containing 50 ml Sephacryl S-200 media (Pharmacia, Piscataway, NJ) equilibrated with 50 mm HEPES, pH 7.4, 100 mm KCl, and 1 mm DTT, and fractions enriched with RGS4 were pooled, concentrated, and stored at −80°C.
Reconstitution of RGS4 with NG108–15 membranes. NG108–15 membranes were prepared exactly as described previously (Hepler et al., 1997). Confluent NG108–15 cells were grown at 37°C in DMEM supplemented with 10% fetal bovine serum, 0.1 mmhypoxanthine, 0.4 μm aminopterine, and 16 μm thymidine, and harvested in buffer containing 50 mm Na-HEPES, pH 8.0, 1 mm EDTA, 150 mm NaCl, and 0.1 mm PMSF. Homogenates were prepared by nitrogen cavitation and centrifugation at 500 ×g to remove nuclei and unbroken cells. Supernatants were centrifuged at 100,000 × g, and membranes were suspended in Buffer B (50 mm Na-HEPES, pH 7.2, 1 mm EDTA, 3 mm EGTA, 5 mmMgCl2, 150 mm NaCl, 2 mmdithiothreitol, and 0.1 mm PMSF) and then stored at −80°C. Reactions were performed as described previously (Hepler et al., 1997) with minor modifications. NG108–15 membranes (6.5 μg/assay) in 10 μl of Buffer A were mixed with an equal volume of RGS4 in Buffer C (50 mm Na-HEPES, pH 7.2, 3 mmEGTA, 100 mm NaCl2, 2 mmdithiothreitol, and 80 mm KCl). Membranes and RGS4 were incubated at 4°C for 30 min and then mixed with 30 μl of sonicated phospholipid vesicles containing [3H]phosphatidyl inositol 4,5-bisphosphate and phosphatidylethanolamine (Hepler et al., 1993) and 100 μm GTPγS in Buffer C. Reactions were initiated by the addition of 10 μl of 9 mmCaCl2 in Buffer C, and assays were performed for 30 min at 30°C. Reactions were stopped, and the samples were processed as described (Hepler et al., 1993).
Reconstitution of RGS4 with Gqα and phospholipase Cβ1. Reactions were performed essentially as described (Hepler et al., 1997) with minor modifications. Assays were performed in a final volume of 60 μl. Purified recombinant Gqα was activated with 1 mm GTPγS for 1 hr at 30°C in Buffer A. Activated Gqα (10 μl; 6 nm) was mixed with RGS4 in 10 μl of Buffer C. The Gqα/RGS sample was incubated for 30 min at 4°C and then mixed with 10 μl of 9 mm CaCl2 in Buffer B. The reactions were started by the addition of 30 μl of sonicated phospholipid vesicles containing [3H]phosphatidyl inositol 4,5-bisphosphate and phosphatidylethanolamine (Hepler et al., 1993) and purified recombinant PLCβ1 (1 ng) in Buffer B. Assays were performed for 20 min at 30°C. Reactions were stopped, and the samples were processed as described (Hepler et al., 1993).
RNA isolation and analysis. Total RNA was isolated from adult rat brain tissue using RNA-Stat 60 (Tel-Test, Friendswood, TX). The RNA was separated on agarose/formaldehyde gels and transferred by capillary action to membrane overnight. The resulting RNA blots were stained with methylene blue to visualize the ribosomal RNA bands and then hybridized with RGS4 cDNA probe labeled with 32P-dCTP at 42°C for 12–16 hr. The blots were washed to a final stringency of 65°C in 0.2× SSC (1× = 150 mm NaCl, 15 mmsodium citrate), 1.0% SDS to remove unbound probe, and the RNA was visualized by autoradiography using Kodak BiomaxMS film.
Oocyte electrophysiology. Oocytes were harvested from anesthetized Xenopus laevis and enzymatically defolliculated as described previously (Saugstad et al., 1995b). Stage V–VI oocytes were injected with 50–100 ng of a cRNA consisting of mGluR1a or mGluR5a in combination with Kirs 3.1 and 3.4. The oocytes were maintained at 16°C in 1× Barth’s culture solution (90 mm NaCl, 1 mm KCl, 10 mm HEPES, 4 mm NaOH) supplemented with 100 μg/ml gentamicin and 50 μg/ml penicillin/streptomycin. Electrophysiological recordings were performed 72–96 hr after injection of the cRNA mixtures. For the RGS4 experiments, 50 nl of a 48 μm stock of purified RGS4 protein or heat-inactivated RGS4 protein (30 min/100°C) was injected into each oocyte, and recordings were performed 30 min to 1 hr after injection. Patch pipettes with tip diameters of 1–2 μm were used as electrodes and filled with 3 m KCl. Oocytes were voltage-clamped at −60 mV in a two-electrode voltage-clamp mode (Oocyte Clamp OC-725C, Warner Instrument Corporation). Currents were acquired at 5 kHz with Axotape 2.0.2. The oocytes were placed in a 1 ml chamber and bathed continuously in 1× Barth’s culture containing 1.5 mm CaCl2 and 1.5 mmMgCl2 at ∼2 ml/min. Solutions were changed using a solenoid valve controller (Valvelink 8, Automate Scientific) with an exchange time of 30–45 sec. Glutamate diluted to a final concentration of 100 μm into 1× Barth’s bathing solution was used to elicit ICl(Ca). ForIGirk, oocytes were initially bathed in 1× Barth’s and then changed to a high potassium (hK) solution (2 mm NaCl, 96 mm KCl, 10 mm HEPES, 1.5 mm CaCl2, 1.5 mmMgCl2). Glutamate diluted to a final concentration of 100 μm into hK solution was applied when the basal high potassium current reached equilibrium. The hK current was subtracted from the total current to obtain the agonist-induced current. Current amplitudes were measured off line, and statistical analysis was performed using SigmaPlot 4.0 (SPSS). A value of <0.05 was considered significant.
Whole-cell electrophysiology. For patch-clamp studies, thick (400 μm) transverse hippocampal slices were prepared and maintained as described previously (Gereau and Conn, 1994a,b). Slices were maintained fully submerged on the stage of a brain slice chamber and perfused continuously with artificial cerebrospinal fluid (ACSF; 1 ml/min). ACSF contained 124 mm NaCl, 2.5 mmKCl, 1.3 mm MgSO4, 2.0 mmCaCl2, 1.0 mmNaH2PO4, 26 mmNaHCO3, and 10 mm glucose, equilibrated with 95%O2/5%CO2. DHPG was applied through the perfusion system to give a final concentration of 10 μm. Whole-cell recording was performed as described (Gereau and Conn, 1994b,c). Briefly, patch electrodes (3–7 MΩ) were pulled from thin-walled borosilicate glass and filled with 150 mm potassium gluconate, 5 mm HEPES, 2 mm ATP, 0.1 mm GTP, 0.1 mm DTT, pH adjusted to 7.4 with 0.5 m KOH. In preliminary experiments with RGS4 protein we found that the inclusion of EGTA/calcium buffer in the patch solution caused rapid clogging of the electrode, possibly by promoting aggregation of the protein. The patch solution used here allows for recording of IAHP for >45 min, with no discernible rundown; however, the IAHPexhibits faster kinetics than is usually reported because of the absence of calcium buffer in our patch solution (Engisch et al., 1996). Recordings from CA1 pyramidal cells were established using the “blind” patch/slice technique described in detail (Blanton et al., 1989) using a Warner PC-501A patch-clamp amplifier.IAHP was measured by recording tail currents after a 100 msec step to 0 mV from a holding potential of −50 mV. We found that this protocol and filling solution produced a robust and stable IAHP that was modulated by DHPG. For protein dialysis, the tips of patch electrodes were filled with normal patch solution and then filled with active RGS4 (0.25–2.5 μm in patch pipette) or heat-inactivated RGS4 (2.5 μm) in normal patch solution. Series resistance was monitored throughout the experiment and often increased as protein dialyzed into the cell. Cells were not included if series resistance increased above 20 MΩ or increased during drug application.
RESULTS
Purified RGS4 blocks Gq-directed activation of phospholipase Cβ1 in cell membranes and reconstituted systems
Members of the Gq family of G-proteins link a wide variety of neurotransmitter receptors, including the group I mGluRs 1 and 5, to activation of the β isoforms of phospholipase C (PLC) (Hepler and Gilman, 1992). We have shown previously that RGS4 is a GAP for Gqα and that it blocks receptor and Gqfunctions in reconstituted systems (Hepler et al., 1997) or when overexpressed in intact cells (Huang et al., 1997; Yan et al., 1997). To examine possible roles for RGS4 as a regulator of mGluR signaling, we first prepared purified recombinant RGS4 and examined its capacity to block Gq functions (Fig.1). RGS4 synthesized in Escherichia coli was purified to near homogeneity and is recognized by a specific anti-RGS4 sera (Fig. 1A). NG108–15 neuroblastoma X glioma cells express Gqα and G11α (Gutowski et al., 1991), and activation of endogenous Gq/11 in NG108–15 cell membranes with GTPγS, a poorly hydrolyzable analog of GTP, stimulates activation of endogenous PLCβ isoforms (Gutowski et al., 1991; Hepler et al., 1997). Although RGS4 is an activator of Gqα- and Giα-catalyzed GTP hydrolysis, it also can inhibit Gqα functions by binding directly to the GTPγS-activated form of Gqα to occlude interactions with PLCβ (Hepler et al., 1997). Reconstitution of purified RGS4 with NG108–15 membranes completely blocked the capacity of GTPγS-Gq/11 to stimulate PLCβ activity (Fig.1B). This observation implies that RGS4 directly blocks Gqα interactions with PLCβ1, and this idea was confirmed in complementary experiments demonstrating that RGS4 blocks GTPγS-Gqα activation of PLCβ1 when reconstituted as purified proteins with phospholipid vesicles (Fig. 1C). These findings are consistent with our previous observations (Hepler et al., 1997) and demonstrate that the recombinant RGS4 can block Gq functions in vitro. This material was used for all subsequent experiments.
Fig. 1.

Purified RGS4 blocks G-protein-directed activation of phospholipase C in NG108–15 membranes and Gqα-directed activation of purified phospholipase Cβ1. A, Purified RGS4 (0.5 μg) was resolved by SDS-PAGE and visualized by staining with Coomassie blue (left, stain) or transferred to nitrocellulose and visualized by immunostaining with specific anti-RGS4 sera R374 (right, blot). B, NG108–15 membranes (6.5 μg) were mixed with [3H]phosphatidyl inositol 4,5-bisphosphate-containing phospholipid vesicles and incubated for 30 min at 30°C with 100 μm GTPγS (open circles) or GTPγS and various concentrations of RGS4 (closed circles). Synthesis of [3H] inositol 1,4,5-trisphosphate ([3H]InsP3) was measured, and [3H]InsP3 accumulation in the absence of NG108–15 membranes (blank = 118 pmol) was subtracted from each value. The data presented are duplicate values and representative of three independent experiments, each with similar results. C, Purified recombinant Gqα was activated with 1 mm GTPγS for 1 hr at 30°C. Activated Gqα (1 nm final) was mixed with purified recombinant PLCβ1 (1 ng) and in the absence (open circles) or presence of various concentrations of RGS4 (closed circles). Synthesis of [3H]InsP3 was measured, and [3H]InsP3 accumulation in the absence of PLCβ1 (blank = 170 pmol) was subtracted from each value. The data presented are duplicate values and representative of three independent experiments, each with similar results.
Distribution of RGS4 messenger RNA in adult rat brain
To determine the relative distribution and levels of expression of RGS4 mRNA within the CNS, we isolated total RNA from adult rat whole brain and eight well defined brain subregions and subjected these samples to RNA blot analysis using a 32P-labeled RGS4 cDNA as a probe (Fig. 2). RGS4 mRNA is detected in all brain regions examined, with the highest levels of RNA expression in cerebral cortex, brainstem, and thalamus. The relative levels of 28S ribosomal RNA are also shown to confirm equal loading of the RNA.
Fig. 2.

Distribution of RGS4 RNA in rat brain. RNA blot analysis of adult rat brain hybridized with a 32P-labeled RGS4 cDNA probe and visualized by autoradiography. RGS4 mRNA was detected as one size class of ∼3.5 kb in all brain regions, with the highest levels seen in cortex, brainstem, and thalamus.
The distribution of group I mGluR mRNA and protein in various adult rat brain regions has been described (Masu et al., 1991; Abe et al., 1992;Martin et al., 1992; Baude et al., 1993; Romano et al., 1995). Both mGluR1 and mGluR5 are expressed broadly within rat brain, and mGluR1 is particularly abundant in cerebellum, thalamus, hippocampus, and olfactory bulb, whereas mGluR5 is enriched in cortex, hippocampus, and olfactory bulb. These distribution patterns indicate that group I mGluRs and RGS4 coexist in some of the same brain regions, supporting our hypothesis that RGS4 could serve as a modulator of mGluR signaling in neurons.
RGS4 blocks mGluR1a-mediated activation of ion currents inXenopus oocytes
We next examined the capacity of purified RGS4 to regulate mGluR1a-mediated ion currents in Xenopus oocytes. The cRNA encoding mGluR1a was coexpressed in oocytes with the cRNAs encoding Kirs 3.1 and 3.4, the two protein subunits that form the functional G-protein-coupled inward-rectifying potassium channel Girk (Dascal et al., 1993; Kubo et al., 1993; Krapivinsky et al., 1995). Two-electrode voltage-clamp recording was performed 3–4 d after injection. In these oocytes, the application of glutamate (100 μm) elicits two large inward currents; the ICl(Ca) that is mediated by activation of PLC and the subsequent Ins(1,4,5)P3-induced release of calcium, andIGirk, the potassium current carried by activation of the exogenously expressed Kir channels (Fig.3A, left). TheICl(Ca) is only slightly reduced by pertussis toxin (PTX) treatment (Houamed et al., 1991; Masu et al., 1991), suggesting that the current is mediated through the Gqsignaling pathway. In contrast, the IGirkcurrent is inhibited by PTX treatment (Saugstad et al., 1996) and is most likely mediated by Gβγ (Reuveny et al., 1994;Wickman et al., 1994) released from Gi/Go. Microinjection of purified RGS4 protein (50 nl of 48 μm) markedly reduced the peakICl(Ca) amplitude (82.4% of control) (Fig.3A, middle). Microinjection of RGS4 also reduced, to a lesser extent, the observed peak current amplitude for theIGirk (59.3% of control), consistent with previous reports that RGS4 acts as a GAP for Gi proteins (Berman et al., 1996; Watson et al., 1996). These effects were caused by active RGS4 protein, because microinjection of heat-inactivated RGS4 failed to elicit a statistically significant reduction of either the mGluR1a-mediated ICl(Ca) orIGirk (Fig. 3A, right). These data show that RGS4 blocks the mGluR1a-mediated Gq-linked activation of ICl(Ca)(Fig. 3B), and to a lesser extent the mGluR1a-mediated Gi/o-linked activation of IGirk(Fig. 3C).
Fig. 3.

Differential effects of RGS4 on mGluR1a-induced currents in Xenopus oocytes.A, Oocytes coinjected with mGluR1a and Kirs 3.1 and 3.4 were recorded under two-electrode voltage clamp at −60 mV. Application of 100 μm glutamate in 1× Barth’s solution to a control oocyte (left) resulted in the activation of theICl(Ca). Subsequently, the bathing solution was changed to a high potassium solution (hK), and the application of 100 μm glutamate in hK resulted in a large agonist-induced IGirk. In a separate oocyte injected with 50 nl of purified RGS4 (48 μm) at least 30 min before recording (middle), glutamate application did not result in the activation ofICl(Ca). The hK solution evoked an inward current attributable to basally activated potassium channels, whereas the subsequent addition of glutamate in hK resulted in activation ofIGirk. In an oocyte injected with 50 nl of heat-inactivated RGS4 (right), theICl(Ca) was induced by application of glutamate, as was the IGirk. Eachtrace is one representative oocyte. B, Bar graph depicting the effect of RGS4 on the mGluR1a-induced activation of ICl(Ca) in control, RGS4-injected, or heat-inactivated RGS4-injected oocytes. RGS4 resulted in an 82.4% reduction in theICl(Ca), whereas the heat-inactivated RGS4 produced currents that were not significantly different from the control, as determined by unpaired Student’s t test. The numbers for each value are in parentheses, and * indicates a p value of <0.05, as determined by unpaired Student’s t test. C, Bar graph depicting the effect of RGS4 on the mGluR1a-induced activation of IGirk in control, RGS4-injected, or heat-inactivated RGS4-injected oocytes. Injection of RGS4 resulted in a 59.3% reduction in theIGirk, and the boiled RGS4 also resulted a small reduction in current compared with control. The numbers for each value are in parentheses, and * indicates a p value of <0.05, as determined by unpaired Student’s t test.
RGS4 blocks mGluR5a-mediated activation ofICl(Ca) in Xenopus oocytes
To extend the observation that RGS4 blocks Gq-linked, mGluR-mediated responses in oocytes (Fig. 3), we examined the effect of RGS4 on cellular responses mediated by mGluR5a, another group I mGluR linked to Gq signaling (Abe et al., 1992). In oocytes expressing mGluR5a, the application of 100 μm glutamate elicited a robust ICl(Ca) (Fig.4A, left). Microinjection of purified active RGS4 protein (50 nl of 48 μm) nearly abolished the ICl(Ca)(93.7% of control) (Fig. 4A, middle), whereas heat-inactivated RGS4 was without effect (Fig.4A, right). The summary data (Fig.4B) indicate that RGS4 blocked the mGluR5a-mediated activation of ICl(Ca). These results demonstrate that RGS4 blocks signaling by both of the Gq-coupled group I mGluRs in Xenopus oocytes, consistent with the idea that RGS4 acts directly on Gqα.
Fig. 4.
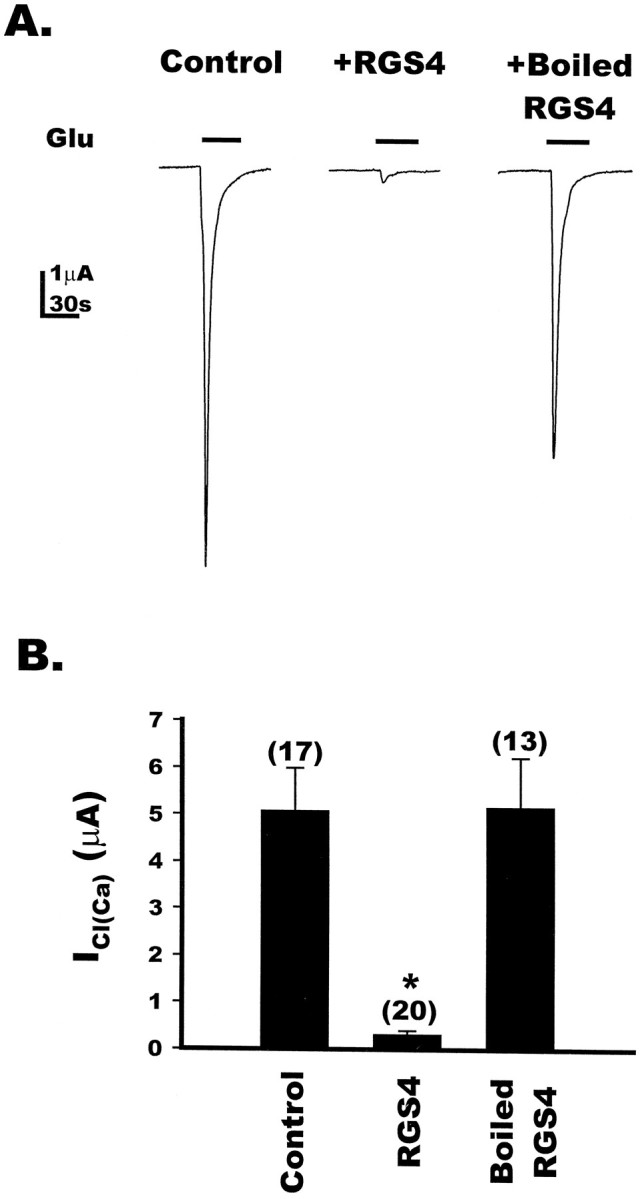
RGS4 blocks mGluR5a-induced calcium-activated chloride currents in Xenopus oocytes. A, Oocytes expressing mGluR5a were recorded under two-electrode voltage clamp at −60 mV. Application of 100 μm glutamate in 1× Barth’s solution to a control oocyte (left) resulted in the activation of theICl(Ca). In a separate oocyte injected with 50 nl of purified RGS4 (48 μm) 30 min to 1 hr before recording (middle), glutamate application did not result in the activation ofICl(Ca); however, sister oocytes injected with 50 nl of heat-inactivated RGS4 (right) were able to evoke currents similar to those in control oocytes. Eachtrace is one representative oocyte. B, Bar graph depicting the effect of RGS4 on the mGluR5a-induced activation of ICl(Ca) in control, RGS4-injected, or heat-inactivated RGS4-injected oocytes. RGS4 resulted in a 93.7% reduction in the ICl(Ca), whereas the current amplitudes evoked in oocytes injected with heat-inactivated RGS4 were not significantly different from the control, as determined by unpaired Student’s t test. The numbers for each value are in parentheses, and * indicates a p value of <0.05, as determined by unpaired Student’s t test.
RGS4 does not act directly on the calcium-dependent chloride channel in Xenopus oocytes
Figure 1 demonstrates that RGS4 directly blocks Gqαfunctions. These findings, together with those of the mGluR1a- and mGluR5a-mediated ICl(Ca) in oocytes, suggest strongly that RGS4 blocks the Gqα-directed formation of Ins(1,4,5)P3 and calcium release rather than directly blocking the endogenous oocyte chloride channel. To strengthen this argument, we examined the capacity of RGS4 to block the Ins(1,4,5)P3-mediated activation of the calcium-dependent chloride channel in Xenopus oocytes. Ins(1,4,5)P3 was microinjected directly into control oocytes or oocytes that had been microinjected with active RGS4 or heat-inactivated RGS4. The injection of 10–15 nl of 1 mmIns(1,4,5)P3 into control oocytes evoked a small inward current attributable to the local release of calcium from Ins(1,4,5)P3-sensitive intracellular stores and activation of ICa(Cl) (Fig.5A, left). In oocytes injected with either active RGS4 (50 nl, 48 μm) or heat-inactivated RGS4, subsequent microinjection of Ins(1,4,5)P3 also elicited an inward current equal to or greater than that observed in the absence of RGS4 (Fig. 5A,middle and left). The summary data (Fig.5B) show that RGS4 does not directly block chloride channel functions in oocytes, and they support the idea that RGS4 acts upstream of Ins(1,4,5)P3 formation, most likely by blocking Gq function directly.
Fig. 5.
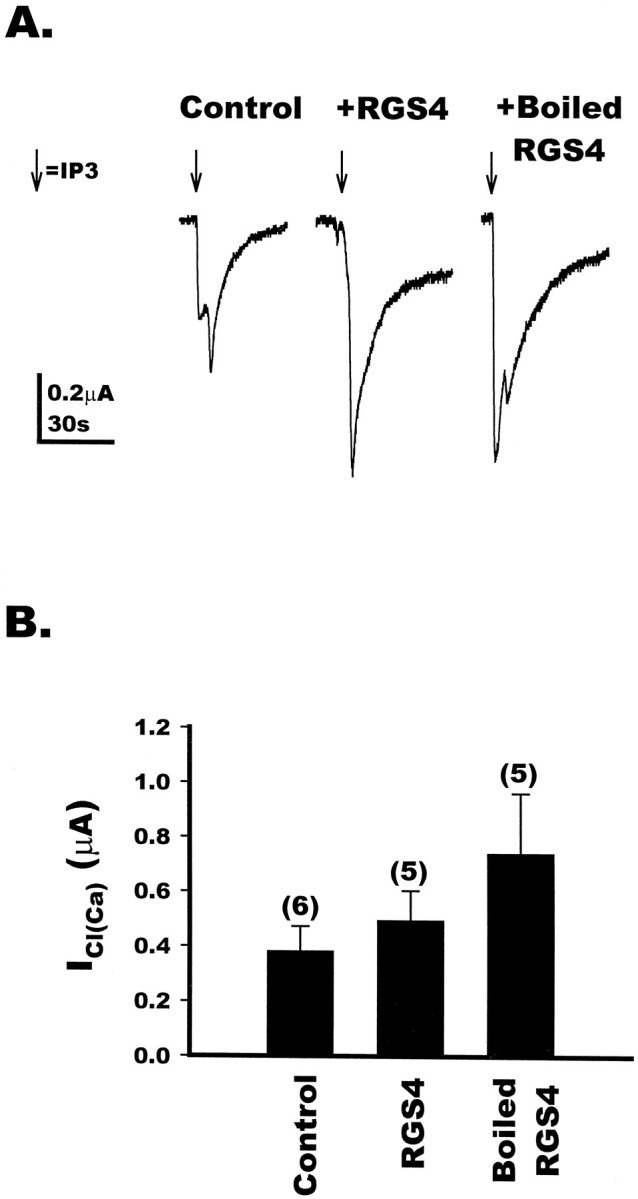
Direct activation of the calcium-activated chloride channel by inositol 1,4,5-trisphosphate is not blocked by RGS4 in Xenopus oocytes. A, Uninjected oocytes (control, left) were two-electrode voltage-clamped at −60 mV. Ins(1,4,5)P3 (1 mm) was injected directly into the oocytes, and the resulting calcium-activated chloride currents were measured. Oocytes that had been injected with 50 nl of purified RGS4 (48 μm) for 30 min to 1 hr before recording (middle) also resulted in the activation ofICl(Ca) in response to the direct injection of Ins(1,4,5)P3. In addition, oocytes injected with 50 nl of heat-inactivated RGS4 (right) resulted in the activation of ICl(Ca) by Ins(1,4,5)P3. Each trace represents one individual oocyte. B, Bar graph depicting the direct activation of ICl(Ca) in control, RGS4-injected, or heat-inactivated RGS4-injected oocytes. The numbers for each value are in parentheses, and each set of conditions was analyzed by unpaired Student’s t test and shown not to be statistically different.
RGS4 blocks mGluR5-mediated inhibition ofIAHP in hippocampal neurons
Results in oocytes demonstrate that RGS4 can block signaling by group I mGluRs in an experimental model system. As shown in Figure 2, RGS4 is expressed in various regions of rat brain where it could regulate mGluR signaling. This includes expression of RGS4 mRNA in hippocampus, an area that is particularly enriched in mGluR5 (Abe et al., 1992). In the hippocampus, a calcium-activated potassium current (IAHP) that underlies a slow afterhyperpolarization is known to be inhibited by activation of mGluRs (Charpak et al., 1990; Desai and Conn, 1991). This modulation is mediated by the phosphoinositide-coupled mGluRs and requires intracellular calcium release but not protein kinase C (PKC) or protein kinase A (PKA) (Abdul-Ghani et al., 1996). Pharmacological studies suggest that inhibition of IAHP in hippocampal CA1 pyramidal cells is mediated by a group I mGluR (Gereau and Conn, 1995; Manzoni and Bockaert, 1995). In addition, immunocytochemistry experiments reveal that mGluR5 is expressed in CA1 pyramidal cells (Romano et al., 1995; Lujan et al., 1996), whereas mGluR1a is not expressed in these cells (Martin et al., 1992). To determine whether RGS4 can regulate signaling by group I mGluRs in a native system, we tested the hypothesis that RGS4 could block the mGluR5-mediated modulation of IAHP by the group I-selective mGluR agonist DHPG (Ito et al., 1992). IAHP was recorded immediately after a 100 msec depolarization to 0 mV from a holding potential of −50 mV. This protocol produces a robust and stable IAHP, presumably by promoting calcium influx and subsequent activation of calcium-activated potassium channels. Application of 10 μm DHPG reduced theIAHP (Fig.6A,B) by ∼45% of control, on average. Introduction of active RGS4 by whole-cell perfusion (0.25, 1.0, or 2.5 μm in patch pipette) attenuated the mGluR5-mediated reduction in IAHPin a dose-dependent manner (Fig. 6C, filled circles). DHPG (10 μm) applied to cells perfused with 0.25, 1.0, or 2.5 μm RGS4 produced on average 46, 27, and 12% inhibition of IAHP, respectively. This indicates that 2.5 and 1 μm RGS4 inhibited the mGluR5-mediated inhibition of IAHPby 74% and 41%, respectively, whereas 0.25 μm RGS4 was without significant effect. Heat-inactivated RGS4 was also without effect (Fig. 6C, filled triangle), indicating that the blockade of mGluR5-mediated IAHPmodulation was not caused by nonspecific effects of cell loading. The concentration-dependent inhibition of the DHPG-induced inhibition ofIAHP by RGS4 occurs at protein amounts that are virtually identical to those required for RGS4 inhibition of phospholipase C activation in in vitro reconstitution studies (Fig. 1B,C). These results extend the observation that RGS4 blocks signaling by group I mGluRs in oocytes to show that RGS4 also inhibits mGluR5-mediated actions in a concentration-dependent manner in hippocampal CA1 neurons, suggesting a role for RGS proteins in the regulation of GPCR responses in the CNS.
Fig. 6.

RGS4 blocks group I-mediatedIAHP inhibition in CA1 pyramidal cells.A, AHP currents were recorded from CA1 pyramidal cells by whole-cell voltage clamp. Currents were elicited by a 100 msec step from −50 mV to 0 mV. The truncated currents elicited by the step have been eliminated for clarity. After 30 min of cell dialysis, application of 10 μm DHPG produced ∼50% reduction inIAHP (left). Addition of 2.5 μm RGS4 to the patch pipette markedly reduced the effect of DHPG (middle), whereas addition of 2.5 μm heat-inactivated RGS4 had no effect. Eachpanel is an average of three traces. B, Time course from representative cells of the effect of 2.5 μm RGS4 on DHPG-induced reduction of AHP currents. Control = filled circles; RGS4 = open circles. C, Dose–response curve depicting the mean data for the effect of RGS4 on DHPG-induced inhibition ofIAHP. Filled circles indicate the average percent inhibition of IAHPinduced by 10 μm DHPG in control cells or cells dialyzed with purified RGS4 (250 nm, 1 μm, or 2.5 μm), whereas the filled trianglerepresents cells dialyzed with 2.5 μm RGS4 that has been heat-inactivated. Data are represented as mean ± SEM;n = 4 for each set (* = p < 0.05 relative to control; unpaired Student’s ttest).
DISCUSSION
mGluRs and other G-protein-coupled receptors have been shown to play a critical role in the regulation of neuronal excitability, synaptic transmission, and various other functions of neurons and glia. Because of the important roles of these receptors in regulation of cell function, their activity is tightly regulated by mechanisms such as desensitization, receptor expression levels, and the specific localization of receptors in the cell. We now report that mGluR signaling through Gq-mediated pathways can be regulated by a new family of proteins that regulate G-protein signaling, the RGS proteins. One family member, RGS4, is widely expressed in the rat brain and is shown to inhibit phospholipase C activation in vitroin a dose-dependent manner. When microinjected into oocytes expressing either of the Gq-coupled mGluRs, mGluR1a or mGluR5a, RGS4 completely blocks activation of ICl(Ca) by mGluRs. This inhibition is attributable to a block of the Gq signaling, because RGS4 did not block activation ofICl(Ca) by direct injection of Ins(1,4,5)P3 into oocytes. On the basis of the calculated volume of an oocyte, we estimate the final concentra-tion of RGS4 inside the oocyte to be ∼1–5 μm. At this concentration of RGS4, the extent of RGS4 inhibition of theICl(Ca) in oocytes (80–90%) correlates very well with the extent of RGS4 blockade of Gqα-activation of PLCβ observed in NG108–15 membranes (Fig. 1B). It should be noted that RGS4 was synthesized in E. coli, and although the protein is clearly active, we cannot rule out the possibility that potential post-translational modifications on RGS4 could increase its affinity for the plasma membrane and/or target Gα subunits, thereby decreasing its effective concentration in a native system. To date, however, RGS4 has not been isolated from native brain tissue for comparative study.
In addition to blocking mGluR effects on the Gqα-regulated ICl(Ca), we found that RGS4 also attenuates effects of group I mGluRs on the Gβγ-regulated IGirk. RGS4 is a GAP for both Gqα (Hepler et al., 1997; Huang et al., 1997; Yan et al., 1997) and members of the Gi family of Gα subunits (Berman et al., 1996), and RGS4 blocks receptor and Gi-directed inhibition of adenylyl cyclase activity in cell membranes and intact cells (Hepler et al., 1997; Huang et al., 1997; Yan et al., 1997). We propose that the inhibitory effect of RGS4 on IGirk is attributable to deactivation of Xenopus Gi/oα, which in turn serves as a sink for free Gβγ and promotes formation of inactive G-protein heterotrimer. Evidence to support this idea can be found in yeast, where the prototypical RGS protein Sst2 clearly acts on Gα to block the signaling functions of Gβγ (Dohlman et al., 1996).
In addition to the oocyte expression system, RGS4 was also shown to modulate mGluR responses in hippocampal neurons. Activation of mGluRs has a number of physiological effects in the hippocampus (Pin and Duvoisin, 1995; Saugstad et al., 1995a; Conn and Pin, 1997) that are likely to play a critical role in regulating transmission through the hippocampal circuit and to be important for both normal hippocampal function and in certain pathological states involving the hippocampus, such as temporal lobe epilepsy. In addition to their inhibition ofIAHP, evidence suggests that group I mGluRs mediate the other direct excitatory effects of mGluR activation on hippocampal pyramidal cells (Charpak et al., 1990, Davies et al., 1995; Gereau and Conn, 1995; Manzoni and Bockaert, 1995; Fitzjohn et al., 1996). Although we focused on modulation ofIAHP in the current study, it is likely that RGS4 would block other group I mGluR-mediated effects in CA1 pyramidal cells, including inhibition of a leak potassium current, inhibition of the M-current, potentiation of NMDA receptor currents, and activation of a nonselective cation current.
Although the present findings clearly demonstrate that RGS4 is capable of inhibiting signaling by group I mGluRs, the exact role of endogenous RGS4 in regulation of mGluR-mediated responses and the conditions under which endogenous RGS proteins regulate mGluR signaling are not yet clear. If the cellular levels of RGS4 are elevated in resting neurons or glia, then RGS4 likely serves an important role in tonic regulation of signaling events by mGluRs and other GPCRs. In this case, mGluR-mediated responses could be fine-tuned by regulating levels of RGS4 or other RGS proteins under different physiological and pathological conditions. For instance, signaling by group I mGluRs is known to be tightly regulated during postnatal development so that mGluR-mediated increases in phosphoinositide hydrolysis are much greater in neonatal than in adult animals. Furthermore, a number of manipulations can lead to long-term increases in mGluR-mediated phosphoinositide hydrolysis in adult that are not correlated with changes in the levels of group I mGluRs (Iversen et al., 1994; Rosdahl et al., 1994; Akbar et al., 1996). The present findings suggest that mGluR signaling could be regulated by altering levels of RGS4 or another RGS protein that regulates Gq-mediated responses. Additional studies will be needed to determine whether developmental regulation of resting cellular levels of RGS4 participates in developmental regulation of mGluR-mediated responses.
Another possible role of RGS proteins is in receptor-mediated desensitization of mGluR responses. If RGS4 levels are low in resting neurons, activation of mGluRs and other Gq-linked neurotransmitter receptors may increase the cellular levels of RGS4 or other RGS proteins and thereby inhibit subsequent Gq-mediated signaling events. For instance, Desai et al. (1996) reported recently that incubation of cells expressing mGluR1a with an mGluR agonist results in a long-term desensitization of mGluR1a-mediated increases in phosphoinositide hydrolysis. Ongoing protein synthesis was required for this desensitization, and the loss of response did not appear to be caused by downregulation of cell-surface mGluR1a receptors. In another recent report Balazs et al. (1997) showed that treatment of cortical astrocytes with mGluR agonists results in a biphasic pattern of desensitization of mGluR5, which includes a rapid desensitization phase that arises within 1–3 hr, and a delayed phase, which peaks at 24 hr. It is tempting to speculate that these delayed forms of desensitization of group I-mediated responses are mediated by mGluR-induced increases in levels of RGS proteins. It is possible that Gq-linked receptors act to regulate the cellular levels of RGS4 and other RGS proteins to serve a role in heterologous desensitization of neurotransmitter signaling. What RGS4 and other RGS proteins contribute to desensitization events relative to other well studied mechanisms (e.g., phosphorylation, internalization) remains unexplored, and clarification of these issues in future studies will prove to be of great interest.
In the present study we chose to focus on RGS4 because it is one of only two RGS proteins to date that have been reported to inhibit Gq-regulated pathways. However, it is important to note that RGS proteins comprise a large and diverse family of regulatory proteins. All mammalian family members share a 120 amino acid core domain that, at least in the case of RGS4, forms a bundle of nine α helices. Specific residues within this helical domain contact Gα subunits and act to accelerate Gα-catalyzed GTP hydrolysis and block Gα-effector interactions (Popov et al., 1997; Tesmer et al., 1997). Beyond this shared feature, RGS proteins differ in most other regards. They range in size from 140 to 17 kDa, share little overall amino acid sequence identity outside the core RGS domain, and are distributed very differently across tissues and cells (Dohlman and Thorner, 1997; Koelle, 1997). These striking differences strongly suggest diversity of function and may enable RGS proteins to interact with target proteins and to incorporate post-translational modifications that confer important (and as yet unappreciated) cellular functions independent of their established roles as regulators of G-protein signaling at the plasma membrane.
All RGS proteins characterized to date, including RGS4, serve as inhibitors of members of the Gi but not the Gsor G12 family of proteins. RGS4 was the first family member identified that also regulates Gq functions (Hepler et al., 1997). Although poorly understood, roles for RGS4 in desensitization necessarily are complex because it regulates both Gq- and Gi-directed signaling pathways. We have recently found that RGS2 is also a selective and potent inhibitor of Gqfunctions (Heximer et al., 1997). Thus, of the RGS proteins characterized to date, RGS4 and RGS2 are the most likely to be involved in regulation of signaling by receptors coupled to Gqpathways, such as the group I mGluRs. Interestingly, RGS4 is expressed exclusively in brain (Druey et al., 1996), whereas RGS2 is found in many tissues including brain (Chen et al., 1997). It is likely that this tissue-specific expression, as well as many other factors, contribute to RGS regulation of mGluR and GPCR signaling within an individual neuron or glial cell. Little is known about cellular localization of RGS proteins and how this process may be regulated. Whether RGS4 and other RGS proteins are found in the cytosol or plasma membrane, and whether they are localized to the soma, dendrites, or axons of neurons is unknown. How individual neurons and glia regulate the cellular levels and localization of RGS proteins in time and space will necessarily determine specific roles for these regulatory proteins in CNS signaling and which GPCR-mediated responses they impact.
Footnotes
This work was supported by in part by National Institutes of Health Grants NS28405, NS31373, and NS34876 (P.J.C.), and the American Heart Association, Georgia Affiliate, the Pharmaceutical Researchers and Manufacturers of America Foundation, and the American Cancer Society/Winship Cancer Center (J.R.H.). We thank Dr. Dieter Jaeger for helpful discussions and Hazar Awad for help with the RGS4 protein purification.
Correspondence should be addressed to Dr. P. J. Conn, Department of Pharmacology, Rollins Research Building, Atlanta, GA 30322.
REFERENCES
- 1.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- 2.Abdul-Ghani MA, Valiante TA, Carlen PL, Pennefather PS. Tyrosine kinase inhibitors enhance a Ca2+-activated K+ current (IAHP) and reduce IAHP suppression by a metabotropic glutamate receptor agonist in rat dentate granule neurones. J Physiol (Lond) 1996;496:139–144. doi: 10.1113/jphysiol.1996.sp021671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akbar MT, Rattray M, Powell JF, Meldrum BS. Altered expression of group I metabotropic glutamate receptors in the hippocampus of amygdala-kindled rats. Brain Res. 1996;43:105–116. doi: 10.1016/s0169-328x(96)00162-3. [DOI] [PubMed] [Google Scholar]
- 4.Akiyama K, Yamada N, Sato M. Increase in ibotenate-stimulated phosphatidylinositol hydrolysis in slices of the amygdala/pyriform cortex and hippocampus of rat by amygdala kindling. Exp Neurol. 1987;98:499–508. doi: 10.1016/0014-4886(87)90259-7. [DOI] [PubMed] [Google Scholar]
- 5.Aronica E, Frey U, Wagner M, Schroeder H, Krug M, Ruthrich H, Catania MV, Nicoletti F, Reymann KG. Enhanced sensitivity of metabotropic glutamate receptors after induction of long-term potentiation in rat hippocampus. J Neurochem. 1991;57:376–383. doi: 10.1111/j.1471-4159.1991.tb03763.x. [DOI] [PubMed] [Google Scholar]
- 6.Balazs R, Miller S, Romano C, de Vries A, Chun Y, Cotman CW. Metabotropic glutamate receptor mGluR5 in astrocytes: pharmacological properties and agonist regulation. J Neurochem. 1997;69:151–163. doi: 10.1046/j.1471-4159.1997.69010151.x. [DOI] [PubMed] [Google Scholar]
- 7.Baude A, Nusser Z, Roberts JDB, Mulvihill E, McIlhinney RAJ, Somogyi P. The metabotropic glutamate receptor (mGluR1a) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- 8.Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 9.Blanton MG, Turco JJL, Kriegstein AR. Whole cell recording from neurons in slices of mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 10.Charpak S, Gahwiler BH, Do KQ, Knopfel T. Potassium conductances in hippocampal neurons blocked by excitatory amino-acid transmitters. Nature. 1990;347:765–767. doi: 10.1038/347765a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen C, Zheng B, Han J, Lin S-C. Characterization of a novel mammalian RGS protein that binds to Gα proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997;272:8679–8685. doi: 10.1074/jbc.272.13.8679. [DOI] [PubMed] [Google Scholar]
- 12.Chen SJ, Winder DG, Desai MA, Klann E, Sweatt JD, Conn PJ. Amygdala kindling alters protein kinase C activity and increases levels of a 17 kDa protein kinase C substrate in the dentate gyrus. J Neurochem. 1992;59:1761–1769. doi: 10.1111/j.1471-4159.1992.tb11008.x. [DOI] [PubMed] [Google Scholar]
- 13.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 14.Dascal N, Schreibmayer W, Lim NF, Wang W, Chavkin C, DiMagno L, Labarca C, Kieffer BL, Gaveriaux-Ruff C, Trollinger D, Lester HA, Davidson N. Atrial G-protein-activated K+ channel: expression cloning and molecular properties. Proc Natl Acad Sci USA. 1993;90:10235–10239. doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies CH, Clarke VRJ, Jane DE, Collingridge GL. Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurons. Br J Pharmacol. 1995;116:1859–1869. doi: 10.1111/j.1476-5381.1995.tb16674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai MA, Conn PJ. Excitatory effects of ACPD receptor activation in the hippocampus are mediated by direct effects on pyramidal cells and blockade of synaptic inhibition. J Neurophysiol. 1991;66:40–52. doi: 10.1152/jn.1991.66.1.40. [DOI] [PubMed] [Google Scholar]
- 17.Desai MA, Burnett JP, Mayne NG, Schoepp DD. Pharmacological characterization of desensitization in a human mGlu1a-expressing non-neuronal cell line cotransfected with a glutamate transporter. Br J Pharmacol. 1996;118:1558–1564. doi: 10.1111/j.1476-5381.1996.tb15574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dohlman HG, Thorner J. RGS proteins and signaling by heterotrimeric G proteins. J Biol Chem. 1997;272:3871–3874. doi: 10.1074/jbc.272.7.3871. [DOI] [PubMed] [Google Scholar]
- 19.Dohlman HG, Song J, Ma D, Courshesne WJ, Thorner J. Sst2: a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression localization genetic interaction and physical association with Gpα1 (G protein α subunit) Mol Cell Biol. 1996;16:5194–5209. doi: 10.1128/mcb.16.9.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Druey KM, Blumer KJ, Kang VH, Kehrl JJ. Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature. 1996;379:742–746. doi: 10.1038/379742a0. [DOI] [PubMed] [Google Scholar]
- 21.Engisch KM, Wagner JJ, Alger BE. Whole-cell voltage-clamp investigation of the role of PKC in muscarinic inhibition of IAHP in rat CA1 hippocampal neurons. Hippocampus. 1996;6:183–191. doi: 10.1002/(SICI)1098-1063(1996)6:2<183::AID-HIPO8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 22.Fitzjohn SM, Irving AJ, Palmer MJ, Harvey J, Lodge D, Collingridge GL. Activation of group I mGluRs potentiates NMDA responses in rat hippocampal slices. Neurosci Lett. 1996;203:211–213. doi: 10.1016/0304-3940(96)12301-6. [DOI] [PubMed] [Google Scholar]
- 23.Gereau RW, Conn PJ. A cyclic AMP-dependent form of associative synaptic plasticity induced by coactivation of α-adrenergic receptors and metabotropic glutamate receptors in rat hippocampus. J Neurosci. 1994a;14:3310–3318. doi: 10.1523/JNEUROSCI.14-05-03310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gereau RW, Conn PJ. Presynaptic enhancement of excitatory synaptic transmission by α-adrenergic receptor activation. J Neurophysiol. 1994b;72:1438–1442. doi: 10.1152/jn.1994.72.3.1438. [DOI] [PubMed] [Google Scholar]
- 25.Gereau RW, Conn PJ. Potentiation of cAMP responses by metabotropic glutamate receptors depresses excitatory synaptic transmission by a kinase-independent mechanism. Neuron. 1994c;12:1121–1129. doi: 10.1016/0896-6273(94)90319-0. [DOI] [PubMed] [Google Scholar]
- 26.Gereau RW, Conn PJ. Roles of specific metabotropic glutamate receptor subtypes in hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 27.Gutowski S, Smrcka AV, Nowak L, Wu D, Simon MI, Sternweis PC. Antibodies to the alpha q subfamily of guanine nucleotide-binding regulatory protein alpha subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis by hormones. J Biol Chem. 1991;266:20519–20524. [PubMed] [Google Scholar]
- 28.Hepler JR, Gilman AG. G proteins. Trends Biochem Sci. 1992;17:383–387. doi: 10.1016/0968-0004(92)90005-t. [DOI] [PubMed] [Google Scholar]
- 29.Hepler JR, Kozasa T, Smrcka AV, Simon MI, Rhee SG, Sternweis PC, Gilman AG. Purification from Sf9 cells and characterization of recombinant Gqαand G11α. J Biol Chem. 1993;268:14367–14375. [PubMed] [Google Scholar]
- 30.Hepler JR, Biddlecome GH, Kleuss C, Camp LA, Hofmann SL, Ross EM, Gilman AG. Functional importance of the amino terminus of Gqα. J Biol Chem. 1996;271:496–504. doi: 10.1074/jbc.271.1.496. [DOI] [PubMed] [Google Scholar]
- 31.Hepler JR, Berman DM, Gilman AG, Kozasa T. RGS4 and GAIP are GTPase-activating proteins for Gqα and block activation of phospholipase Cβ by γ-thio-GTP-Gqα. Proc Natl Acad Sci USA. 1997;94:428–432. doi: 10.1073/pnas.94.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/GOS8 is a selective inhibitor of Gqα function. Proc Natl Acad Sci USA. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 34.Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O’Hara PJ, Mulvihill ER, Almers W, Hagen FS. Cloning expression gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991;252:1318–1321. doi: 10.1126/science.1656524. [DOI] [PubMed] [Google Scholar]
- 35.Huang CF, Hepler JR, Gilman AG, Mumby SM. Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc Natl Acad Sci USA. 1997;94:6159–6163. doi: 10.1073/pnas.94.12.6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of Gαi GTPase activity. Nature. 1996;383:175–177. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- 37.Ito I, Kohda A, Tanabe S, Hirose E, Hayashi E, Mitsunaga S, Sugiyama H. 3,5-dihydroxyphenylglycine: a potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- 38.Iversen L, Mulvihill E, Haldeman B, Diemer NH, Kaiser F, Sheardown M, Kristensen P. Changes in metabotropic glutamate receptor mRNA levels following global ischemia: increase of a putative presynaptic subtype (mGluR4) in highly vulnerable rat brain areas. J Neurochem. 1994;63:625–633. doi: 10.1046/j.1471-4159.1994.63020625.x. [DOI] [PubMed] [Google Scholar]
- 39.Koelle MR. A new family of G-protein regulators: the RGS proteins. Curr Opin Cell Biol. 1997;9:143–147. doi: 10.1016/s0955-0674(97)80055-5. [DOI] [PubMed] [Google Scholar]
- 40.Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:115–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 41.Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+-channel IkAch is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 42.Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY. Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- 43.Lujan R, Nusser Z, Roberts JDB, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 44.Manzoni O, Bockaert J. Metabotropic glutamate receptors inhibiting excitatory synapses in the CA1 area of rat hippocampus. Eur J Neurosci. 1995;7:2518–2523. doi: 10.1111/j.1460-9568.1995.tb01051.x. [DOI] [PubMed] [Google Scholar]
- 45.Martin LJ, Blackstone GD, Huganir RL, Price DL. Cellular localization of a metabotropic glutamate receptor in rat brain. Neuron. 1992;9:259–270. doi: 10.1016/0896-6273(92)90165-a. [DOI] [PubMed] [Google Scholar]
- 46.Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- 47.Mayat E, Lerner-Natoli M, Rondouin G, Lebrun F, Sassetti I, Récasens M. Kainate-induced status epilepticus leads to a delayed increase in various specific glutamate metabotropic receptor responses in the hippocampus. Brain Res. 1994;645:186–200. doi: 10.1016/0006-8993(94)91652-7. [DOI] [PubMed] [Google Scholar]
- 48.Pin J-P, Duvoisin R. Neurotransmitter receptors I: the metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 49.Popov S, Yu K, Kozasa T, Wilkie TM. The regulation of G protein signaling (RGS) domains of RGS4, RGS10 and GAIP retain GTPase activating protein activity in vitro. Proc Natl Acad Sci USA. 1997;94:7216–7220. doi: 10.1073/pnas.94.14.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhl JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- 51.Romano C, Sesma MA, McDonald CT, O’Malley K, van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355:455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 52.Rosdahl D, Seitzberg DA, Christensen T, Balchen T, Henrik Diemer N. Changes in mRNA for metabotropic glutamate receptors after transient cerebral ischemia. NeuroReport. 1994;5:593–596. doi: 10.1097/00001756-199401000-00015. [DOI] [PubMed] [Google Scholar]
- 53.Saugstad JA, Segerson TP, Westbrook GL. Modulation of ion channels and synaptic transmission by metabotropic glutamate receptors. In: Wheal H, Thompson A, editors. Excitatory amino acids and synaptic transmission, Ed 2. Academic; New York: 1995a. pp. 77–88. [Google Scholar]
- 54.Saugstad JA, Segerson TP, Westbrook GL. l-2-amino-3-phosphonopropionic acid competitively antagonizes metabotropic glutamate receptors 1a and 5 in Xenopus oocytes. Eur J Pharmacol Mol Pharmacol. 1995b;289:395–397. doi: 10.1016/0922-4106(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 55.Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seren MS, Aldinio C, Zanoni R, Leon A, Nicoletti F. Stimulation of inositol phospholipid hydrolysis by excitatory amino acids is enhanced in brain slices from vulnerable regions after transient global ischemia. J Neurochem. 1989;53:1700–1705. doi: 10.1111/j.1471-4159.1989.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 57.Tesmer JJG, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4−-activated Giα1: stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 58.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature. 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 59.Wickman KD, Iniguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;378:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 60.Yamada N, Akiyama K, Otsuki S. Hippocampal kindling enhances excitatory amino acid receptor-mediated polyphosphoinositide hydrolysis in the hippocampus and amygdala/pyriform cortex. Brain Res. 1989;490:126–132. doi: 10.1016/0006-8993(89)90437-x. [DOI] [PubMed] [Google Scholar]
- 61.Yan YB, Chi PP, Bourne HR. RGS4 inhibits Gq-mediated activation of mitogen-activated protein kinase and phosphoinositide synthesis. J Biol Chem. 1997;272:11924–11927. doi: 10.1074/jbc.272.18.11924. [DOI] [PubMed] [Google Scholar]