

Abstract
Here, we compare the pathways by which DNA-damaging agents, NGF deprivation, and superoxide dismutase 1 (SOD1) depletion evoke apoptosis of sympathetic neurons. Previous work raised the hypothesis that cell cycle signaling plays a required role in neuronal apoptosis elicited by NGF deprivation and the DNA-damaging agent camptothecin. To test this hypothesis, we extended our investigation of DNA-damaging agents to cytosine arabinoside (AraC) and UV irradiation. As with NGF deprivation and camptothecin treatment, the cyclin-dependent kinase inhibitors flavopiridol and olomoucine protected neurons from apoptosis induced by AraC and UV treatment. These observations support the model that camptothecin, AraC, and UV treatment cause DNA damage, which leads to apoptosis by a mechanism that, as in the case of NGF deprivation, includes activation of cell cycle components. Flavopiridol and olomoucine, however, had no effect on death induced by SOD1 depletion, suggesting that CDKs do not play a role in this paradigm of neuronal death. To compare further the mechanisms of death evoked by NGF withdrawal, SOD1 depletion, and DNA-damaging agents, we investigated their responses to inhibitors of cysteine aspartases, elements of apoptotic pathways. The V-ICEinh and BAF, two peptide inhibitors of cysteine aspartases, protected neurons in all three death paradigms. In contrast, the cysteine aspartase inhibitory peptide zVAD-fmk conferred protection from NGF withdrawal and SOD1 depletion, but not DNA-damaging agents, whereas acYVAD-cmk protected only from SOD1 depletion. Taken together, these findings indicate that three different apoptotic stimuli activate separate pathways of death in the same neuron type.
Keywords: CDK, cell cycle, DNA damage, caspase, apoptosis, cytosine arabinoside
Apoptotic death of neurons, even of the same type of neuron, can be evoked by different initiating events. For instance, the death of sympathetic neurons can be caused by trophic factor deprivation (Levi-Montalcini and Angeletti, 1963), peroxynitrite-dependent oxidative stress (Troy and Shelanski, 1994;Troy et al., 1997), and exposure to DNA-damaging agents (Martin et al., 1990; Tomkins et al., 1994; Morris and Geller, 1996; Park et al., 1997).
The capacity of different initiating events to cause apoptotic death in the same type of neuron raises several issues, including (1) the extent to which the pathways leading to apoptosis in each case are distinct and (2) the degree to which they share common elements. Two specific elements of neuronal apoptotic pathways will be considered here. First, multiple lines of evidence suggest that cell cycle components are involved in neuronal death. Several agents that inhibit cell cycle progression promote survival of trophic factor-deprived sympathetic neurons and/or neuronally differentiated PC12 cells (Rydel and Greene; 1988; Rukenstein et al., 1991; Ferrari and Greene, 1994;Farinelli and Greene, 1996). Neuronal apoptosis also is accompanied by changes in cyclin-dependent kinase (CDK) activity and cyclin expression (Brooks et al., 1993; Freeman et al., 1994; Gao and Zalenka, 1995). Consistent with this, the CDK inhibitors flavopiridol and olomoucine promote survival of neuronal PC12 cells and sympathetic neurons deprived of trophic support (Park et al., 1996a).
Components of the cell cycle machinery also may play a role in neuronal death evoked by DNA-damaging agents. In support of this, flavopiridol and olomoucine, as well as several G1/S blockers, rescue sympathetic and cortical neurons from camptothecin-induced death (Park et al., 1997). The possible role of cell cycle elements in neuronal death evoked by oxidative stress, however, has not been examined.
Members of the cysteine aspartase (caspase) family of proteases represent a second element in neuronal death pathways (Ellis et al., 1991; Miura et al., 1993; Gagliardini et al., 1994). In neuronal cells, exposure to an antisense oligonucleotide that diminishes expression of the caspase Nedd-2/caspase 2 protects sympathetic neurons and PC12 cells from NGF deprivation (Troy et al., 1997). In the case of oxidative stress, studies with naive PC12 cells have established that, although death in this paradigm is blocked by the interleuken-converting enzyme-directed inhibitor acYVAD-cmk, the latter is ineffective in blocking death caused by trophic factor deprivation (Troy et al., 1996a). Moreover, the Nedd-2 antisense oligonucleotide that protects from NGF withdrawal does not protect PC12 cells from oxidative stress (Troy et al., 1997). Such findings indicate that different caspases are involved in the pathways that regulate apoptosis evoked by oxidative stress and NGF deprivation.
Caspase involvement in neuronal death caused by DNA-damaging agents has been less clear. Although the caspase inhibitor zVAD-fmk protects sympathetic neurons from NGF withdrawal and oxidative stress (Park et al., 1996b; Troy et al., 1996a), it does not protect them from the DNA-damaging agent camptothecin (Park et al., 1997). This not only indicates a divergence of death pathways but also raises the general issue of whether caspases play a required role in neuronal death evoked by DNA damage.
The aim of the present studies has been to test further and to refine our hypotheses about the pathways by which DNA-damaging agents cause neuronal apoptosis and to compare these pathways with those invoked by oxidative stress and NGF deprivation. To eliminate potential cell type-specific differences in death pathways, we focused our examination on sympathetic neurons. Here, we report that neuronal death evoked by the DNA-damaging agents cytosine arabinoside (AraC) or UV irradiation, but not oxidative stress, is inhibited by the CDK inhibitors flavopiridol and olomoucine. We also provide evidence that death evoked by DNA-damaging agents requires the actions of cysteine aspartases but that these are distinct from those involved in apoptosis caused by withdrawal of trophic support or oxidative stress.
MATERIALS AND METHODS
Materials. Human recombinant NGF was kindly provided by Genentech (South San Francisco, CA). Flavopiridol (L86-8275 [(−)cis-5,7-dihydroxy-2-(2-chlorophenyl)-8[4-(3-hydroxy-1-methyl)-piperidinyl]-4H-benzopyran-4-one]) was a generous gift from Dr. Peter J. Worland (National Cancer Institute, Bethesda, MD). Olomoucine [2-(2-hydro)-6-benzylamino-9-methylpurine] and iso-olomoucine were purchased from LC Laboratories (Woburn, MA). Aphidicolin, mouse NGF, and anti-mouse NGF antiserum were obtained from Sigma (St. Louis, MO). zVAD-fluoromethylketone (zVAD-fmk) and BAF Boc-aspartyl(OMe)-fluoromethylketone were purchased from Enzyme Systems Products (Dublin, CA). The ICEinh peptide (IQACRG; Chiron, San Diego, CA) and ICEs peptide control (ICGRQA; Chiron) and ASOD1 antisense oligonucleotide were linked to the antennapedia delivery peptide (V; Oncor, Gaithersburg, MD) as previously described (Troy et al., 1996a).
Culture and survival assay of PC12 cells. Naive PC12 cells were cultured and passaged as previously described (Greene and Tischler, 1976). Neuronally differentiated PC12 cells were generated by exposing PC12 cells to NGF in serum-free RPMI 1640 medium for 8–9 d. For survival experiments neuronally differentiated PC12 cells were plated onto collagen-coated 24-well tissue culture dishes at a density of ∼2 × 105 cells per well. Neuronally differentiated PC12 cells were cultured in serum-free RPMI 1640 medium containing NGF (100 ng/ml) throughout the course of survival experiments. At appropriate times of culture under the conditions described in the text, cells were lysed, and the numbers of viable cells were evaluated as previously described (Rukenstein et al., 1991). All experimental points are expressed as a percentage of cells plated on day 0 and are reported as mean ± SEM (n = 3).
UV irradiation. Cells were exposed to UV irradiation by using the Stratolinker UV crosslinker (Stratagene, La Jolla, CA). Sympathetic neurons were exposed to 300 J/m2, whereas neuronal PC12 cells were exposed to 650 J/m2. Each well was exposed with 200 μl of medium containing NGF with or without the appropriate drug. After irradiation 300 μl of additional medium containing NGF with or without drug was added.
Culture and survival assay of rat sympathetic neurons.Primary cultures of rat sympathetic neurons were generated from dissociated superior cervical ganglia of postnatal day 1 rats (strain, Sprague Dawley), as described previously (Lee et al., 1980). The cells were plated onto collagen-coated 24-well dishes at a density of ∼0.5 ganglia per well and maintained in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum and 60 ng/ml mouse NGF for a period of 3 d before the survival experiment. A mixture of uridine and 5-fluorodeoxyuridine (10 μm each) also was added to eliminate non-neuronal cells on day 2. No pretreatment with a survival agent, unless otherwise noted, was necessary for effective survival of neurons exposed to AraC. In the case of UV irradiation, the neurons died quite rapidly. Accordingly, neuronal cultures were pretreated overnight (18 hr) with survival-promoting agents before UV irradiation. NGF deprivation was performed by washing with NGF-free medium and the addition of anti-NGF antiserum, as previously described (Park et al., 1996a). V-ASOD1 oligonucleotides, in conjunction with the nitric oxide generator S-nitrosopenicillamine (SNAP; 100 μm), were added to the cultured sympathetic neurons as previously described (Troy et al., 1997). At appropriate times the numbers of viable phase-bright neurons were determined by strip counting, as previously described (Rydel and Greene; 1988). All experimental points are expressed relative to the original number of neurons present in each well and are reported as mean ± SEM (n = 3).
RESULTS
The CDK inhibitors flavopiridol and olomoucine promote survival of sympathetic neurons exposed to UV irradiation or treated with AraC
Our previous studies demonstrated that the camptothecin-induced apoptotic death of neurons is inhibited by G1/S-phase cell cycle blockers and CDK inhibitors (Park et al., 1997a), and it was postulated that camptothecin-induced neurotoxicity is attributable to transcriptionally mediated DNA strand break formation (Morris and Geller, 1996) and consequent cell cycle signaling components (Park et al., 1997).
To assess whether this observation could be extended to two additional potential DNA-damaging agents, AraC and UV irradiation, we first determined whether the CDK inhibitors flavopiridol and olomoucine could prevent the death of cultured sympathetic neurons exposed to UV irradiation in the presence of NGF. Flavopiridol, a flavonoid derivative, potently inhibits CDK1/2/ and 4 activities (Losiewicz et al., 1994; Filgueira de Azevedo et al., 1996) and displays poor inhibitory activities toward all other kinases examined, including cAMP-dependent kinase, epidermal growth factor receptor kinase, and protein kinase C. Olomoucine, a purine derivative, specifically blocks CDK1/2/ and 5 as well as ERK-1/MAP-kinase activities and was ineffective against >30 other kinases examined (Vesely et al., 1994). Both drugs block progression from the G1 to S- and G2 to M-phases of the cell cycle (Kaur et al., 1992; Vesely et al., 1994). As shown in Figure 1, both flavopiridol and olomoucine effectively promoted the survival of UV-treated sympathetic neurons. At 4 d after UV treatment, ∼75–80% of the neurons were alive with drug treatment, whereas only 25% were alive without the inhibitors. Maximal protection was observed with 1–3 μm flavopiridol (Fig. 1B) and 200 μm olomoucine (Fig. 1C). These are also the minimum concentrations required to fully inhibit DNA synthesis by proliferating PC12 cells (Park et al., 1996a). In addition, the doses–responses for protection against UV-induced death are nearly identical to those that rescue sympathetic neurons from both camptothecin treatment (Park et al., 1997) and NGF deprivation (Park et al., 1996a). Figure 2 shows the morphology of sympathetic neurons exposed to UV irradiation in the presence or absence of flavopiridol or olomoucine. Neurons treated with the CDK inhibitors/UV have a morphology comparable to non-UV-treated control cells (phase-bright cell bodies and intact neurites), whereas cells treated with UV alone show degenerating cell bodies and neurites. Iso-olomoucine, an analog control of olomoucine that differs in the location of one substituent methyl group and that poorly inhibits CDK activity or DNA synthesis (Park et al., 1996a), was nearly ineffective in promoting the survival of UV-treated sympathetic neurons (Fig.1D). The UV treatment experiments were extended to postmitotic, neuronally differentiated PC12 cells cultured in the presence of NGF, and similar survival-promoting effects were observed with olomoucine and flavopiridol (data no shown).
Fig. 1.

The CDK inhibitors, flavopiridol and olomoucine, inhibit the UV irradiation-induced death of rat sympathetic neurons. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before UV exposure, as indicated. Neurons were pretreated with the indicated drugs for 16 hr before UV irradiation. Each data point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each culture at the time of drug treatment. p values derived from Student’st test in comparison of the survival of UV-treated and UV/experimental agent-treated neurons at day 2 are listed.A, Effects of flavopiridol (1 μm;p < 0.025) and olomoucine (200 μm;p < 0.005) on the time course of survival of sympathetic neurons exposed to UV irradiation. B, Effects of various doses of flavopiridol on the survival of UV-treated sympathetic neurons at day 1. C, Effects of various doses of olomoucine on the survival of UV-treated sympathetic neurons at day 1. D, Effect of iso-olomoucine (200 μm; p > 0.05) on the time course of survival of sympathetic neurons exposed to UV irradiation.E, Effect of aphidicolin (10 μm) on the time course of survival of sympathetic neurons exposed to UV irradiation.
Fig. 2.

Phase-contrast micrographs of primary sympathetic neurons maintained in medium containing NGF and treated with the following: A, UV; B, no treatment;C, UV + 1 μm flavopiridol;D, UV + 200 μm olomoucine. Pictures are of cells at 1 d post-UV exposure.
The second genotoxic agent we tested was the antitumor drug AraC. Although the toxic actions of AraC on dividing cells have been postulated to be attributable to the blockade of DNA synthesis by chain termination (Ohno et al., 1988), this agent causes neuropathies and promotes the death of cultured postmitotic sympathetic neurons even in the presence of NGF (Martin et al., 1990; Tomkins et al., 1994). To account for the death-promoting actions of AraC on neurons, Martin et al. (1990) have suggested that it specifically interferes with the NGF signaling mechanism. In contrast, Tomkins et al. (1994) have proposed that AraC causes neuronal apoptosis by creating double-stranded breaks in DNA. Accordingly, we next determined whether flavopiridol and olomoucine also could inhibit the death of sympathetic neurons. As Figure 3 shows, both drugs quite effectively promoted long-term survival of AraC-treated sympathetic neurons. Approximately 80–100% of the treated neurons survived up to 5 d of AraC exposure, whereas almost all of the neurons in the control cultures were dead by this time. Beyond this time, toxicity of the CDK inhibitors was observed, even without AraC treatment (Fig.3A,C). As in the cases of UV-irradiated sympathetic neurons and neuronal PC12 cells, the maximal concentrations of flavopiridol and olomoucine required to protect sympathetic neurons from AraC correlated with the minimum concentrations required to fully inhibit DNA synthesis by proliferating PC12 cells [1 μm for flavopiridol (Fig.3B) and 200 μm for olomoucine (Fig.3D)]. The control compound iso-olomoucine was ineffective in promoting survival (Fig. 3C). Neurons treated with AraC and the CDK inhibitors showed the phase-bright morphology of viable neurite-bearing cells, whereas cells treated with AraC formed ghost-like cell bodies and degenerating processes (Fig.4). These results, along with those made previously (Park et al., 1997), demonstrate that neuronal death induced by three different forms of DNA-damaging conditions, namely UV irradiation, camptothecin, and AraC treatment, are blocked effectively by inhibitors of CDK activity.
Fig. 3.

The CDK inhibitors, flavopiridol and olomoucine, inhibit AraC-induced death of rat sympathetic neurons. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before drug treatment. Replicate cultures were treated with AraC (100 μm), as indicated. Eachdata point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each culture at the time of drug treatment.p values derived from Student’s t test in comparison of the survival of AraC-treated and AraC/experimental agent-treated neurons at day 4 are listed. A, Effect of flavopiridol (1 μm; p < 0.005) on the time course of survival of sympathetic neurons treated with AraC.B, Effect of various doses of flavopiridol on the survival of AraC-treated sympathetic neurons at day 3.C, Effects of olomoucine (200 μm;p < 0.005) and iso-olomoucine (200 μm; p > 0.05) on the time course of survival of sympathetic neurons treated with AraC. D, Effect of various doses of olomoucine on the survival of AraC-treated sympathetic neurons at day 3. E, Effect of aphidicolin (10 μm; p > 0.05) on the time course of survival of sympathetic neurons treated with AraC.
Fig. 4.

Phase-contrast micrographs of primary sympathetic neurons maintained in medium containing NGF and treated with the following: A, 100 μm AraC;B, no treatment; C, AraC + 1 μm flavopiridol; D, AraC + 200 μm olomoucine. Pictures are of cells at 3 d after AraC treatment.
The S-phase blocker aphidicolin does not promote survival of sympathetic neurons exposed to UV irradiation or AraC
Previous studies demonstrated that neurons deprived of NGF or exposed to camptothecin cannot be rescued by cell cycle blocking agents that act beyond the G1/S border (Farinelli and Greene, 1996; Park et al., 1997). For example, the S-phase blocker aphidicolin is ineffective in blocking the death of sympathetic neurons in both paradigms (Farinelli and Greene, 1996; Park et al., 1997). Consistent with this, aphidicolin also failed to block the death of neurons exposed either to UV irradiation (see Fig. 1E) or to AraC (Fig.3E). Such findings demonstrate that inhibition of DNA synthesis per se does not promote survival of neurons induced to die by UV, AraC, camptothecin, or NGF deprivation.
The CDK inhibitors flavopiridol and olomoucine do not promote survival of sympathetic neurons subjected to oxidative stress
Sympathetic neurons depleted of SOD1 by treatment with an SOD1 antisense oligonucleotide and exposed to nitric oxide-generating compounds die apoptotically, apparently because of formation of peroxynitrite (Troy et al., 1996b, 1997). We examined whether the CDK inhibitors would block the death of neurons induced by this means. As shown in Figure 5, neither flavopiridol nor olomoucine protected sympathetic neurons in this paradigm of oxidative stress. However, in the same experiments, and as anticipated from previous studies (Park et al., 1996a), both inhibitors effectively blocked the death of sympathetic neurons deprived of NGF. This suggests that cell cycle regulatory components do not control the neuronal death pathways induced by oxidative stress. The CDK inhibitors also failed to inhibit the death of neuronal PC12 cells depleted of SOD1 (data not shown).
Fig. 5.

The CDK inhibitors, flavopiridol (FL; 3 μm) and olomoucine (OL; 200 μm), do not inhibit the death of sympathetic neurons depleted of SOD1. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before drug treatment. Replicate cultures were deprived of NGF (antiNGF) or treated with ASOD1 (50 nm)/SNAP (100 μm; referred to asASOD1 in the figure), as indicated, for 3 d. Eachdata point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each culture at the time of drug treatment.p values derived from Student’s t test are p < 0.005 for anti-NGF versus anti-NGF/CDK inhibitors and p > 0.05 for ASOD1 versus ASOD1/CDK inhibitors.
Differential effects of cysteine aspartase inhibitors on sympathetic neurons deprived of NGF, depleted of SOD1, or exposed to DNA-damaging agents
We next compared the role of cysteine aspartases in our three paradigms for inducing apoptotic neuronal death. As shown in Figure6, zVAD-fmk (50 μm), an irreversible peptide inhibitor of several different cysteine aspartases (Stefanis et al., 1996; Livingston, 1997), increases the survival of sympathetic neurons from death evoked by oxidative stress (via exposure to v-ASOD1/nitric oxide generators). This inhibitor also was found to protect PC12 cells from apoptosis caused by SOD1 depletion (Troy et al., 1996a). Additionally, zVAD-fmk (50 μm) protected sympathetic neurons from NGF deprivation (Fig. 6A). In contrast to its effects on sympathetic neurons subjected to oxidative stress or NGF deprivation, 50 μm zVAD-fmk was ineffective in promoting survival of these neurons after exposure to UV irradiation (Fig. 6A) or AraC (Fig.6B). We previously reported similar findings for exposure to camptothecin (Park et al., 1997). Increasing the zVAD-fmk concentration to 100 μm had minimal effect (data not shown).
Fig. 6.
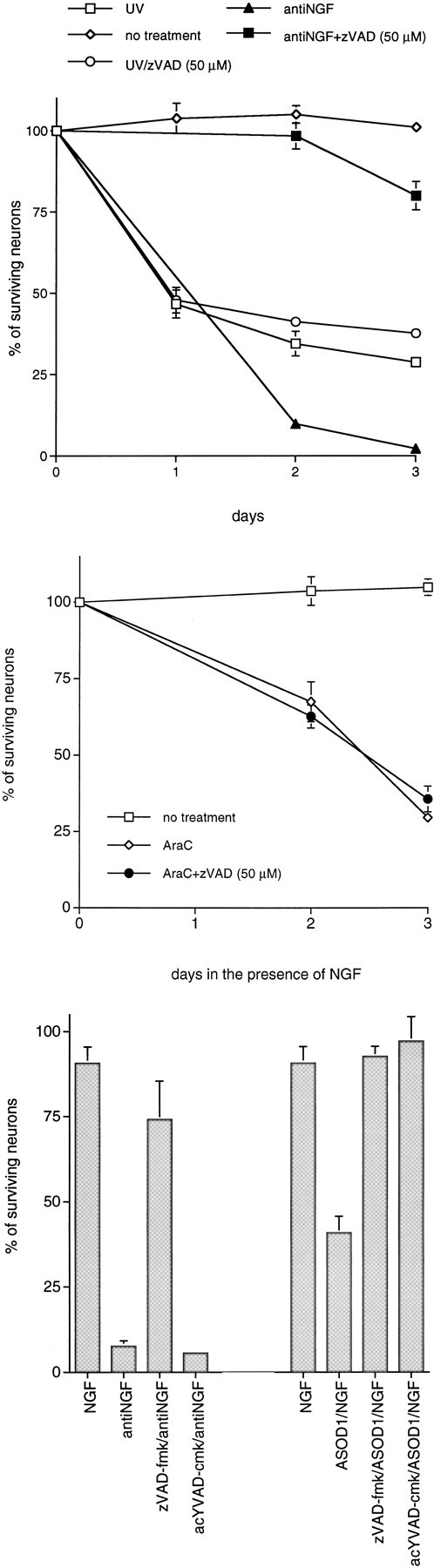
The cysteine aspartase family protease inhibitors zVAD-fmk and acYVAD-cmk differentially promote survival of sympathetic neurons exposed to DNA-damaging agents, deprived of NGF, or depleted of SOD1. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before experimentation. Cultures were deprived of NGF and treated as indicated for 3 d before assessment of neuronal survival. Neurons exposed to UV irradiation were pretreated with zVAD-fmk for 16 hr.p values derived from Student’s t test are included, as indicated. A, Effect of zVAD-fmk (50 μm) on the time course of survival of sympathetic neurons exposed to UV irradiation (p > 0.05, day 2) and deprived of NGF (p < 0.005, day 2).B, Effect of zVAD-fmk (50 μm) on the time course of survival of sympathetic neurons treated with AraC (100 μm; p > 0.05, day 3).C, Effect of acYVAD-fmk [250 μm;p > 0.05 (antiNGF);p < 0.025 (ASOD1)] and zVAD-fmk [50 μm; p < 0.005 (antiNGF); p < 0.01 (ASOD1)] on the survival of neurons deprived of NGF (antiNGF) or treated with antisense to SOD1 (ASOD1), along with SNAP (100 μm). Survival was measured 2 d after NGF deprivation or treatment with ASOD1/SNAP. Each data point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each well at the time of drug treatment.
The above observations indicate a distinction between the pathways by which DNA-damaging agents evoke death and by which oxidative stress and NGF deprivation do so. A previous study showed that sensitivity to the peptide acYVAD-cmk, a more specific inhibitor of ICE-like proteases than zVAD-fmk, blocks the death of naive PC12 cells caused by oxidative stress but has no effect in the trophic factor deprivation paradigm (Troy et al., 1996a). Figure 6Cshows that acYVAD-cmk shows similar selectivity between these two causes of death in sympathetic neurons. Thus, cysteine protease inhibitors distinguish among each of the three causes of sympathetic neuron death studied here.
Our observations raise the question as to whether any cysteine aspartase is required for death induced by DNA-damaging agents. To examine this issue, we used two additional cysteine aspartase inhibitors, the antennapedia vector peptide-linked ICEinhand the irreversible peptide inhibitor BAF. ICEinh is a peptide (IQACRG) that includes the conserved active site of ICE-family proteases (Troy et al., 1996a). Because nearly all known cysteine aspartases possess this conserved sequence, it is expected that ICEinh would compete with caspases for the binding of substrates and therefore block apoptotic death. The antennapedia vector peptide (V) crosses plasma membranes with high efficiency and functions as a vector to facilitate cellular uptake of the ICEinhpeptide (Troy et al., 1996a). The two peptides are linked by a reducible S–S bond, and V-ICEinh is cleaved within cells, releasing free ICEinh, which accumulates intracellularly (Troy et al., 1996a). A previous report demonstrated that V-ICEinh blocks the death of PC 12 cells and sympathetic neurons deprived of trophic support (Troy et al., 1996a). In addition, it has been shown to inhibit the death of PC12 cells depleted of SOD1 (Troy et al., 1996a). Figure7 shows that, under conditions in which V-ICEinh protects sympathetic neurons from NGF deprivation, it also blocks the death of sympathetic neurons exposed to UV (∼80% survival with the inhibitor vs 25% without V-ICEinhtreatment; Fig. 7A) or treated with AraC (∼90% survival with V-ICEinh vs 50% without inhibitor treatment on day 2; Fig. 7B). V-ICEinh additionally blocked the death of sympathetic neurons either exposed to camptothecin or subjected to SOD1 depletion/NO generators (data not shown). A scrambled peptide (ICGRQA) linked to antennapedia (termed V-ICEs) was used as a control for nonspecific effects and was ineffective in promoting survival of neurons in all paradigms of death (Fig. 7; Troy et al., 1996a).
Fig. 7.
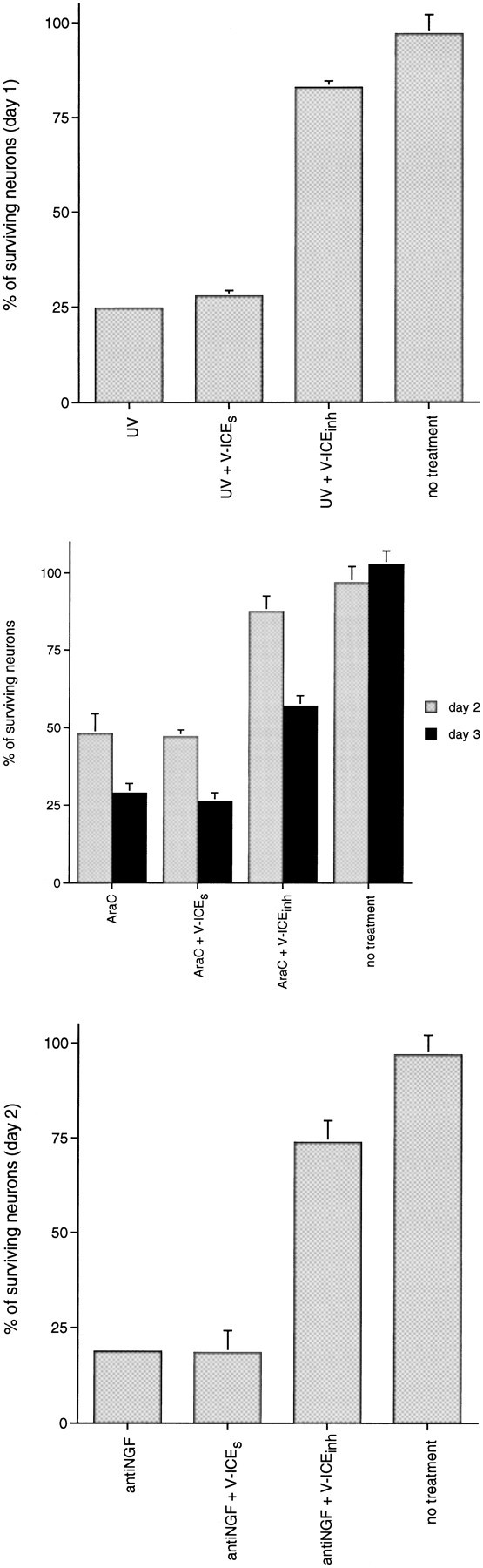
The cysteine aspartase family protease inhibitor V-ICEinh protects sympathetic neurons from NGF deprivation and UV- and AraC-induced apoptosis. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before experimentation. On the third day neurons were pretreated for 18 hr in the presence of NGF with the V-ICEinh inhibitor (200 nm) or the V-ICEs control (200 nm), as indicated. Then neurons were exposed to UV irradiation or AraC (100 μm) or were deprived of NGF. p values derived from Student’s t test are included as indicated.A, Effect of V-ICEinh inhibitor (200 nm; p < 0.005) and V-ICEs(200 nm; p > 0.05) on the survival of sympathetic neurons exposed to UV irradiation. B, Effect of V-ICEinh inhibitor (200 nm; p < 0.025, day 2) and V-ICEs (200 nm; p > 0.05, day 2) on the survival of sympathetic neurons exposed to AraC.C, Effect of V-ICEinh inhibitor (200 nm; p < 0.01) and V-ICEs(200 nm; p > 0.05) on the survival of sympathetic neurons deprived of NGF. Each data point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each well at the time of drug treatment.
BAF is a cell-permeant irreversible cysteine aspartase inhibitor that blocks the death of Fas-treated thymocytes and NGF-deprived sympathetic neurons (Deshmukh et al., 1996). Consistent with the protective effect of V-ICEinh, BAF also reduces the death of sympathetic neurons treated with all three DNA-damaging agents. Treatment with 100 μm BAF fully protected sympathetic neurons from AraC-induced death at day 3, whereas 75% death was observed in control cultures (Fig.8A). In addition, BAF protected sympathetic neurons from UV irradiation (∼80% survival with BAF treatment vs <40% without; Fig. 8B). Similar protective effects of BAF were observed with camptothecin-treated sympathetic neurons (data not shown). The IC50 of BAF for prevention of death in all three paradigms of DNA damage was ∼25 μm. As previously reported (Deshmukh et al., 1996), BAF treatment totally protected sympathetic neurons from death induced by NGF deprivation (Fig. 8C). BAF treatment alone had no toxic effect on neuronal survival (data not shown). Taken together, these data indicate that the death pathways evoked by DNA-damaging agents require the activation of one or more cysteine aspartase(s), but that these are different from those that act in the pathways triggered by NGF deprivation and SOD1 depletion.
Fig. 8.
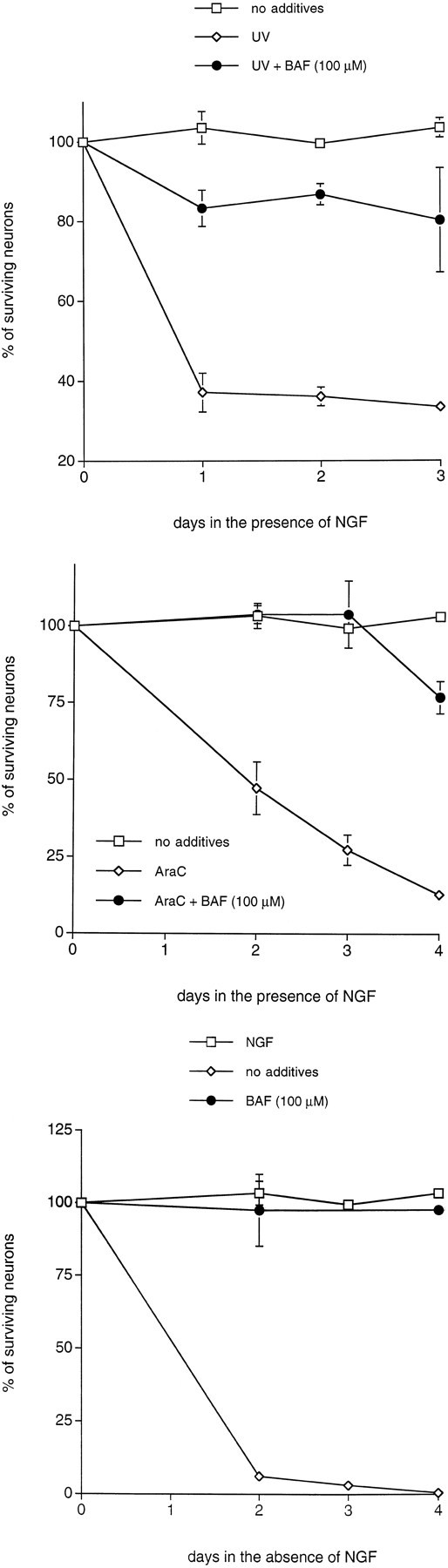
The cysteine aspartase family protease inhibitor BAF protects sympathetic neurons from NGF deprivation and UV- and AraC-induced apoptosis. Primary cultures of neonatal rat superior cervical ganglion neurons were grown in the presence of NGF for 3 d before experimentation. Cultures were deprived of NGF and treated as indicated for 3 d before assessment of neuronal survival. Neurons exposed to UV irradiation were pretreated with BAF for 16 hr.p values derived from Student’s t test are included as indicated. A, Effect of BAF (100 μm; p < 0.005, day 2) on the time course of survival of sympathetic neurons exposed to UV irradiation.B, Effect of BAF (100 μm;p < 0.025, day 3) on the time course of survival of sympathetic neurons treated with AraC (100 μm).C, Effect of BAF (100 μm;p < 0.025, day 2) on the time course of survival of neurons deprived of NGF. Each data point is the mean ± SEM (n = 3) and is expressed relative to the number of neurons present in each well at the time of drug treatment.
DISCUSSION
In the present studies we explored the signaling pathways by which DNA-damaging agents cause the death of postmitotic neurons and compared such mechanisms with those involved in neuronal apoptosis caused by the loss of trophic support and oxidative stress.
Involvement of cell cycle signals in neuronal death caused by DNA-damaging agents
Our studies considered three conditions that damage DNA and trigger apoptotic death: UV irradiation, camptothecin, and AraC treatment. UV irradiation produces direct DNA damage that is mediated primarily by the formation of pyrimidine dimers (Schreiber et al., 1995). Camptothecin binds to and inhibits DNA topoisomerase I, forming a cleavable complex consisting of topo-I covalently linked to the severed DNA strand (Hsiang et al., 1989). In postmitotic neurons it is postulated that the transcription machinery collides with this complex, resulting in a lethal DNA lesion (Morris and Geller, 1996). The chain-terminating nucleoside, AraC, also kills postmitotic neurons. However, the mechanism of AraC neurotoxicity currently remains unclear.Tomkins et al. (1994) have suggested that AraC-induced death of sympathetic neurons could be attributable to either inhibition of topo-II-mediated DNA repair or inhibition of DNA ligase, both of which would lead to the persistent formation of DNA strand breaks.
How might DNA damage lead to neuronal death? At least for proliferating cells, a growing body of evidence suggests the involvement of cell cycle-related components. For instance, the cell cycle-related tumor suppressor protein p53 appears to mediate the responses of many cell types to UV-evoked DNA damage (Sanchez and Elledge, 1995). It appears that p53 expression also is required for AraC-evoked death of neurons (Enokido et al., 1996). Furthermore, AraC treatment has been reported to increase cyclin E-associated CDK activities in HL 60 cells (Ping Dou et al., 1995), whereas camptothecin upregulates cdc2 (Shimizu et al., 1995) and cyclin D1 (Chen et al., 1995) levels in HL60 and RKO cells, respectively.
Previous evidence supports the hypothesis that components of the cell cycle machinery contribute to the death of postmitotic neurons caused by loss of trophic support (Ferrari and Greene, 1994; Farinelli and Greene, 1996; Park et al., 1996a). Here, we showed that the CDK inhibitors flavopiridol and olomoucine effectively suppress neuronal death caused by Ara C and UV treatment. Moreover, as with camptothecin (Park et al., 1997), the concentrations of CDK inhibitors required to block death correlate well with the levels required to inhibit DNA synthesis in proliferating PC12 cells. This extends our findings to two additional treatments that are quite distinct from one another and from camptothecin with respect to the initial mechanism but that also have the capacity to cause DNA damage. Such observations support the hypotheses (1) that DNA strand breaks are a common element in the pathways by which these treatments cause neuronal apoptosis and (2) that death in each instance involves components of the cell cycle machinery. For all three DNA-damaging agents, aphidicolin, which blocks S-phase by inhibiting DNA polymerase α, was ineffective in providing protection. This suggests that inhibition of active DNA synthesis per se is not sufficient to promote survival.
Interestingly, flavopiridol and olomoucine are cytotoxic to numerous oncogenic cell lines, including dividing PC12 cells (Bible and Kaufmann, 1996; Park et al., 1996a). Although the exact mechanisms by which these CDK inhibitors kill transformed cells are unknown, one interpretation is that death occurs as a result of a signaling conflict brought on by inhibition of CDK activity and constitutively present oncogenic signal(s). Presumably, in the case of neuronal PC12 cells, this conflict is avoided because of the downregulation of oncogenic/proliferative signals during differentiation. Such oncogenic/proliferative signals also would be absent in neurons, and these too are therefore resistant to the cytotoxic effects of the CDK inhibitors.
Although our findings with olomoucine and flavopiridol suggest the involvement of CDKs in the pathway by which DNA-damaging agents lead to neuronal apoptosis, alternative interpretations must be considered. In particular, we cannot rule out entirely the possibility that flavopiridol and olomoucine act on required elements in death signaling other than CDKs. We previously have eliminated effects of these drugs on JNK activity or activation (Park et al., 1996a), which appear to be required for the death of NGF-deprived neuronally differentiated PC12 cells (Xia et al., 1996). In addition, it was shown previously that attenuation of ERK activation does not affect survival of neuronal cells, suggesting that olomoucine does not act to promote survival via inhibition of ERK activity (Park et al., 1996a; Virdee and Tolkovsky, 1996). Although all our findings to date as well as those of others (Losiewicz et al., 1994; Vesely et al., 1994) have pointed to the highly selective nature of these CDK inhibitors, it remains conceivable that they affect kinases yet to be examined that play a required role in cell death.
CDK inhibitors do not protect neurons from oxidative stress
Depletion of SOD1 in the case of PC12 cells, and SOD1 depletion combined with exposure to NO generators in the case of sympathetic neurons, leads to apoptotic death by what appears to be a peroxynitrite-dependent mechanism (Troy et al., 1996b). Evidence has been presented for the role of peroxynitrite in neurodegenerative disorders, and so the mechanism by which this agent results in apoptosis is of both basic and clinical relevance (Coyle and Puttfarcken, 1993). Because peroxynitrite can lead to cellular DNA damage, we considered the possibility that, like DNA-damaging agents, it might evoke death by a mechanism dependent on cell cycle regulatory components. However, in contrast to our observations with NGF deprivation and DNA-damaging agents, death in the SOD1 depletion paradigm was insensitive to flavopiridol and olomoucine. This suggests that CDKs do not regulate the death of sympathetic neurons in all paradigms of neuronal death.
Different cysteine aspartases mediate the death of neurons deprived of NGF, depleted of SOD1, or exposed to DNA-damaging conditions
Although our findings suggest that CDK activity mediates some part of the apoptotic signaling response in the case of trophic factor deprivation and DNA damage, it is not clear to what extent the signaling pathways activated by these two apoptotic stimuli are identical and to what extent they may differ. In addition, a notable feature of our experiments was that AraC, UV irradiation, and camptothecin each evoked apoptosis in the presence of NGF. Accordingly, an alternative interpretation, as has been suggested for AraC (Martin et al., 1990), is that all or some of the apoptosis-evoking conditions we used work by blocking the signaling pathway by which NGF promotes survival. To address these issues, we examined whether the death of sympathetic neurons caused by different stimuli can be blocked by inhibitors of cysteine aspartases. The cysteine aspartase inhibitor zVAD-fmk blocked death evoked by NGF deprivation and SOD1 depletion, but not by AraC or UV irradiation. We previously reported similar findings for camptothecin (Park et al., 1997a). In addition, we show here that apoptosis in response to NGF deprivation and oxidative stress in sympathetic neurons, as in PC12 cells (Troy et al., 1996a), also differ in their response to caspase inhibitors. acYVAD-cmk, an inhibitor more specific to the ICE-like caspases, blocks death evoked by SOD1 depletion, but not death caused by NGF deprivation. These observations indicate that the death pathways triggered by NGF deprivation, SOD1 depletion, and DNA-damaging agents are distinct from one another. They also rule out the action of DNA-damaging agents, including AraC, as inhibitors of proximal elements of the NGF signaling mechanism.
Our observations suggest that death elicited by NGF withdrawal and DNA-damaging agents might involve different members of the cysteine protease family, because death caused by DNA-damaging agents is insensitive to zVAD-fmk. It must be noted that the exact nature of the inhibitory efficacies and potencies of zVAD-fmk on various known members of the cysteine aspartase family has yet to be reported. Although we have shown that zVAD-fmk can block the processing of IL-Iβ by ICE (L. Stefanis, unpublished results) and cleavage of a fluorogenic DEVD substrate by a CPP32-like activity in cell extracts taken from trophic factor-deprived PC12 cells (Stefanis et al., 1996), others have reported that zVAD-fmk is not a potent inhibitor of PARP-cleaving protease activity in THP.1 cells (Slee et al., 1996). This suggests that zVAD-fmk is not a universal inhibitor of cysteine aspartases. To address whether different caspases might mediate the death of sympathetic neurons exposed to differential apoptotic stimuli, we tested other known caspase inhibitors. BAF has been shown previously to inhibit ICE and CPP32 in vitro, but not calpain I or II, indicating that it acts as an inhibitor of cysteine aspartases, but not other cysteine proteases (Deshmukh et al., 1996). In addition, it was reported to block the death of Fas-treated thymocytes and NGF-deprived sympathetic neurons (Deshmukh et al., 1996). Similarly, V-ICEinh mimics the active site of almost all known cysteine aspartase family members and should function as a general inhibitor of these enzymes (Troy et al., 1996a). We show here that both BAF and V-ICEinh inhibit the death of sympathetic neurons evoked by all three DNA-damaging agents and NGF deprivation, implicating the action of one or more caspases. Taken together, these data indicate that apoptosis caused by DNA-damaging agents is dependent on one or more cysteine aspartase family members and that the identity of this/these is different from the family member(s) that mediate apoptosis caused by NGF withdrawal and oxidative stress.
The identity of the cysteine aspartase(s) required for neuronal death caused by DNA-damaging agents presently is unknown. Previous work compared the cysteine aspartases that mediate the death of PC12 cells and sympathetic neurons caused by either NGF deprivation or downregulation of SOD1 (Troy et al., 1996a). Although death in both cases was inhibited by V-ICEinh and zVAD-fmk, antisense oligonucleotides to Nedd-2 rescued PC12 cells and sympathetic neurons from withdrawal of NGF, but not from SOD1 downregulation (Troy et al., 1996a, 1997). Conversely, acYVAD-cmk, as well as blocking antibodies to mature IL-1β and an IL-1 receptor antagonist, protected PC12 cells from SOD1 depletion, but not NGF withdrawal (Troy et al., 1996a). Our experiments extend the observations with acYVAD-cmk to sympathetic neurons and thus lend further support to the model that distinct caspase-dependent pathways mediate neuronal apoptosis evoked by NGF withdrawal (Nedd-2 dependent) and SOD1 depletion (ICE-like dependent).
In summary, we report that NGF deprivation, DNA-damaging agents, and oxidative stress activate different apoptotic pathways in sympathetic neurons. The properties of these pathways are summarized in Figure9. As shown in this figure, past studies have placed cell cycle signals upstream of caspase activation (Stefanis et al., 1996). For oxidative stress, formation of peroxynitrite appears to be the initiating cause of death, and therefore this also lies upstream of caspase activation (Troy et al., 1996b). In the cases of NGF deprivation and DNA damage-induced neuronal death, there is evidence for the involvement of CDKs, whereas this is not the case for the oxidative stress model. In addition, each of the three pathways of death uses distinct cysteine aspartases. Thus, even in thesame neuron type, multiple apoptotic pathways can be activated, depending on the initiating stimulus of death.
Fig. 9.

Model for the differential death pathways induced by trophic factor deprivation, DNA damage, and superoxide dismutase 1 reduction in sympathetic neurons.
Footnotes
This work was supported in part by Grants from National Institutes of Health (NS33689), March of Dimes, Blanchette Rockefeller Foundation, Amyotrophic Lateral Sclerosis Foundation, and the Aaron Diamond Foundation (to L.A.G.); by Grants NS36443 and ES06897 from National Institutes of Health and grants from the Cancer Institute of New Jersey (to H.M.G.) and from Muscular Dystrophy Association (to C.M.T.). DSP is an Aaron Diamond Foundation Fellow. We thank Dr. Peter J. Worland for kindly donating flavopiridol and for his helpful discussions.
Correspondence should be addressed to Dr. David S. Park, Department of Pathology, Taub Center for Alzheimer’s Disease Research, Columbia University College of Physicians and Surgeons, 630 West 168th Street, New York, NY 10032.
REFERENCES
- 1.Bible KC, Kaufmann SH. Flavopiridol: a cytotoxic flavone that induces cell death in noncycling A549 human lung carcinoma cells. Cancer Res. 1996;56:4856–4861. [PubMed] [Google Scholar]
- 2.Brooks SF, Gibson LA, Rubin LL. Apoptosis induced by NGF withdrawal from differentiated PC12 cells involves activation of P34 cdc2 kinase. Soc Neurosci Abstr. 1993;19:885. [Google Scholar]
- 3.Chen X, Bargonetti J, Prives C. p53 through p21 (waf cip1) induced cyclin d1 synthesis. Cancer Res. 1995;55:4257–4263. [PubMed] [Google Scholar]
- 4.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 5.Deshmukh M, Vasilakos J, Deckworth TL, Lampe P, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 7.Enokido Y, Araki T, Aizawa S, Hatanaka H. p53 involves cytosine arabinoside-induced apoptosis in cultured cerebellar granule neurons. Neurosci Lett. 1996;203:1–4. doi: 10.1016/0304-3940(95)12247-8. [DOI] [PubMed] [Google Scholar]
- 8.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari G, Greene LA. Proliferative inhibition by dominant-negative Ras rescues naive and neuronally differentiated PC12 cells from apoptotic death. EMBO J. 1994;13:5922–5928. doi: 10.1002/j.1460-2075.1994.tb06937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filgueira de Azevedo W, Mueller-Dieckmann H-J, Schulze-Gahmen U, Worland PJ, Sausville E, Kim S-H. Structural basis for specificity and potency of a flavonoid inhibitor of human CDK2, a cell cycle kinase. Proc Natl Acad Sci USA. 1996;93:2735–2740. doi: 10.1073/pnas.93.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman RF, Estus S, Johnson EM., Jr Analysis of cell-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 12.Gagliardini V, Fernandez P-A, Lee RKK, Drexler HCA, Rotello RJ, Fishman MC, Yuan J. Prevention of vertebrate neuronal death by the crmA gene. Science. 1994;263:826–828. doi: 10.1126/science.8303301. [DOI] [PubMed] [Google Scholar]
- 13.Gao CY, Zalenka PS. Induction of cyclin B and H1 kinase activity in apoptotic PC12 cells. Exp Cell Res. 1995;219:612–618. doi: 10.1006/excr.1995.1271. [DOI] [PubMed] [Google Scholar]
- 14.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which responds to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 16.Kaur G, Stetler-Stevenson M, Sebers S, Worland P, Sedlacek H, Myers C, Czech J, Naik R, Sausville E. Growth inhibition with reversible cell cycle arrest of carcinoma cells by flavone L86-8275. J Natl Cancer Inst. 1992;22:1736–1740. doi: 10.1093/jnci/84.22.1736. [DOI] [PubMed] [Google Scholar]
- 17.Lee VM, Shelanski ML, Greene LA. Characterization of antisera raised against cultured rat sympathetic neurons. Neuroscience. 1980;5:2239–2245. doi: 10.1016/0306-4522(80)90140-2. [DOI] [PubMed] [Google Scholar]
- 18.Levi-Montalcini R, Angeletti PU. Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol. 1963;7:653–657. doi: 10.1016/0012-1606(63)90149-0. [DOI] [PubMed] [Google Scholar]
- 19.Livingston DJ. In vitro and in vivo studies of ICE inhibitors. J Cell Biochem. 1997;64:19–26. [PubMed] [Google Scholar]
- 20.Losiewicz MD, Carlson BA, Kaur G, Sausville EA, Worland PJ. Potent inhibition of CDC2 kinase activity by the flavonoid L86-8275. Biochem Biophys Res Commun. 1994;201:589–595. doi: 10.1006/bbrc.1994.1742. [DOI] [PubMed] [Google Scholar]
- 21.Martin DP, Wallace TL, Johnson EM., Jr Cytosine arabinoside kills postmitotic neurons in a fashion resembling trophic factor deprivation: evidence that a deoxycytidine-dependent process may be required for nerve growth factor signal transduction. J Cell Biol. 1990;10:184–193. doi: 10.1523/JNEUROSCI.10-01-00184.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miura M, Zhu H, Rotello R, Hartweig EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1 beta-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 23.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell-cycle independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohno Y, Spriggs D, Matsukage A, Ohno T, Kufe D. Effects of 1-β-d-arabinofuranosylcytosine incorporation on elongation of specific DNA sequences by DNA polymerase beta. Cancer Res. 1988;48:1494–1498. [PubMed] [Google Scholar]
- 25.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of postmitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996a;271:8161–8170. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 26.Park DS, Stefanis L, Farinelli SE, Yan CYI, Greene LA. Ordering the cell death pathway: differential effects of BCL2, an ICE-family protease inhibitor, and other survival agents on JNK activation in serum/NGF-deprived PC12 cells. J Biol Chem. 1996b;271:21898–21906. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 27.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ping Dou Q, An B, Yu C. Activation of cyclin E-dependent kinase by DNA-damage signals during apoptosis. Biochem Biophys Res Commun. 1995;214:771–780. doi: 10.1006/bbrc.1995.2353. [DOI] [PubMed] [Google Scholar]
- 29.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez Y, Elledge SJ. Stopped for repairs. BioEssays. 1995;17:545–548. doi: 10.1002/bies.950170611. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber M, Baumann B, Cotten M, Angel P, Wagner EF. Fos is an essential component of the mammalian UV response. EMBO J. 1995;14:5338–5349. doi: 10.1002/j.1460-2075.1995.tb00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu T, O’Connor PM, Kohn KW, Pommier Y. Unscheduled activation of cyclin B1/Cdc2 kinase in human promyelocytic leukemia cell line HL60 cells undergoing apoptosis induced by DNA damage. Cancer Res. 1995;55:228–231. [PubMed] [Google Scholar]
- 34.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. Benzyloxycarbonyl-val-ala-asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of cpp32. Biochem J. 1996;315:21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stefanis L, Park DS, Yan CYI, Farinelli SE, Troy CM, Shelanski ML, Greene LA. Induction of CPP-32-like activity in PC12 cells by withdrawal of trophic support: dissociation from apoptosis. J Biol Chem. 1996;271:30663–30671. doi: 10.1074/jbc.271.48.30663. [DOI] [PubMed] [Google Scholar]
- 36.Tomkins CE, Edwards SN, Tolkovsky AM. Apoptosis is induced in post-mitotic rat sympathetic neurons by arabinosides and topoisomerase II inhibitors in the presence of NGF. J Cell Sci. 1994;107:1499–1507. doi: 10.1242/jcs.107.6.1499. [DOI] [PubMed] [Google Scholar]
- 37.Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase causes apoptotic death in PC12 neuronal cells. Proc Natl Acad Sci USA. 1994;91:6384–6387. doi: 10.1073/pnas.91.14.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family protease and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA. 1996a;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Troy CM, Derossi D, Prochiantz A, Greene LA, Shelanski ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide–peroxynitrite pathway. J Neurosci. 1996b;16:253–261. doi: 10.1523/JNEUROSCI.16-01-00253.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Troy CM, Stefanis L, Green LA, Shelanski ML. Nedd2 is required for apoptosis after trophic support withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vesely J, Havlicek L, Strnad M, Blow JJ, Donella-Deanna A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S, Mieijer L. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- 42.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 43.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1996;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]