

Abstract
Vesicles and/or other membranous structures that form after axolemmal damage have recently been shown to repair (seal) the axolemma of various nerve axons. To determine the origin of such membranous structures, (1) we internally dialyzed isolated intact squid giant axons (GAs) and showed that elevation of intracellular Ca2+ >100 μm produced membranous structures similar to those in axons transected in Ca2+-containing physiological saline; (2) we exposed GA axoplasm to Ca2+-containing salines and observed that membranous structures did not form after removing the axolemma and glial sheath but did form in severed GAs after >99% of their axoplasm was removed by internal perfusion; (3) we examined transected GAs and crayfish medial giant axons (MGAs) with time-lapse confocal fluorescence microscopy and showed that many injury-induced vesicles formed by endocytosis of the axolemma; (4) we examined the cut ends of GAs and MGAs with electron microscopy and showed that most membranous structures were single-walled at short (5–15 min) post-transection times, whereas more were double- and multi-walled and of probable glial origin after longer (30–150 min) post-transection times; and (5) we examined differential interference contrast and confocal images and showed that large and small lesions evoked similar injury responses in which barriers to dye diffusion formed amid an accumulation of vesicles and other membranous structures. These and other data suggest that Ca2+ inflow at large or small axolemmal lesions induces various membranous structures (including endocytotic vesicles) of glial or axonal origin to form, accumulate, and interact with each other, preformed vesicles, and/or the axolemma to repair the axolemmal damage.
Keywords: axotomy, calcium, endocytosis, plasmalemmal repair, vesicles, axolemmal lesions
Vesicles and other membranous structures have well documented roles in many normal functions of eukaryotic cells (Lodish et al., 1995). However, although vesicles and other membranous structures have been reported to occur after injury in many cell types (Délèze, 1970; Gingell, 1970; DeMello, 1973; Jeon and Jeon, 1975; Nishiye, 1977; Foissner, 1988; Severs et al., 1990), only recently have such structures been proposed to repair plasmalemmal damage. That is, vesicles or other membranous structures (e.g., myelin delaminations) have been reported to form, migrate, and accumulate at cut ends of unmyelinated (Fishman et al., 1990) and myelinated axons (Ballinger et al., 1997) where ion- and dye-diffusion barriers are subsequently established by interactions of such membranous structures with each other, preformed vesicles, and/or the axolemma (Krause et al., 1994; Ballinger et al., 1997;Eddleman et al., 1997; Godell et al., 1997). Repair of plasmalemmal injury has been reported to be mediated by exocytosis of preformed vesicles in endothelia (Miyake and McNeil, 1995) and oocytes (Steinhardt et al., 1994; Bi et al., 1995; Terasaki et al., 1997). Considering these and other data, we (Eddleman et al., 1997) have hypothesized that eukaryotic cells repair plasmalemmal damage by whatever membranous sources are available.
The origin(s) and mechanism(s) of formation of membranous structures that repair axolemmal damage are not yet known, nor is whether similar membranous structures seal both complete axonal transections and small axolemmal holes. To obtain such data, we examined the formation of injury-induced membranous structures in two unmyelinated invertebrate axons, the crayfish medial giant axon (MGA) and the squid giant axon (GA).
Our data from GAs and/or MGAs show that increases in intracellular Ca2+ rather than injury per se induce endocytotic vesiculation of the plasmalemma and the formation of other membranous structures (e.g., invaginations). These vesicles often fuse with each other while moving toward the cut end where a barrier (seal) to dye diffusion has been reported to form in MGAs transected in physiological saline (Eddleman et al., 1997) or GAs transected in physiological saline containing exogenous calpain (Godell et al., 1997). We report that most vesicles and other membranous structures are single-walled and appear to have an axolemmal origin, although many double- and multi-walled structures, some of apparent glial origin, are also observed. Finally, we show that a diffusion barrier to hydrophilic dyes at small axolemmal lesions (1- to 40-μm-diameter micropunctures) forms amid an accumulation of Ca2+-induced vesicles and other membranous structures similar to that previously reported for complete axonal transections (Eddleman et al., 1997). From these and other data, we suggest that repair of the axolemma in unmyelinated axons at completely transected axonal ends and at small holes involves vesicles and other membranous structures that primarily arise from the axolemma by endocytosis with some contribution from membranous structures that arise from glia.
MATERIALS AND METHODS
Squid GAs. Squid were obtained at the Marine Biological Laboratory (Woods Hole, MA; Loligo pealei) or the Marine Biomedical Institute (Galveston, TX; Sepioteuthis lessoniana). No significant differences in morphology or physiology of the GAs were noted between the two species for the experiments reported in this study. Axon lengths of 2–4 cm in the hindmost stellate nerve were isolated by tying the ends with cotton thread, removed from the animal, and fine-cleaned in control external saline, as described previously (Gilbert et al., 1990; Krause et al., 1994).
Damaged GAs having injury-induced vesicles or opaque regions were discarded. Transections were made with microscissors (Vannas RS-5610; Roboz Surgical Instruments), and small holes (up to 40 μm in diameter) were made by axonal impalement with glass micropipettes 1 μm in diameter.
Control external saline consisted of (in mm): 430 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, and 5 Tris-Cl, pH 7.4. Divalent cation-free external saline was the same as control external saline with osmolal replacement of CaCl2and MgCl2 with TMA-Cl and addition of 1 mmEDTA. Control internal saline consisted of (in mm): 440 K glutamate, 2.5 EGTA-Tris, 280 glycine, and 5 Tris-Cl. Fluorescent dyes were injected intracellularly in a buffer consisting of (in mm): 440 KAc, 0.54 EGTA, 5 Tris-Cl, and 150 sucrose. The axoplasm of GAs was replaced by internal perfusion with a saline consisting of (in mm): 500 KI and 5 Tris-Cl. The percentage of axoplasm removed by the perfusion was calculated from the expression (VT −VL)/VT × 100, where VT is the total volume of axoplasm before perfusion, assuming a cylinder of 500 μm diameter and 1 cm length, and VL is the displaced volume after perfusion, assuming a 1-cm-long cylinder of 480 μm in diameter [corresponding to a residual cortical layer of axoplasm 10 μm thick (R. A. Sheller and G. D. Bittner, unpublished observation)]. The osmolality of all internal salines, measured with an osmometer (5100; Wescor, Logan, UT), was adjusted with sucrose to 975 mOsm.
Crayfish MGAs. Crayfish (Procambarus clarkii), ∼2–3 inches in body length (Atchafalaya Biological Supply, Raceland, LA), were anesthetized on ice for 5–10 min. The ventral nerve cord containing the paired MGAs was removed from the animal, as described previously by Eddleman et al. (1997), and bathed in control external saline. MGAs were isolated and fine-cleaned by carefully teasing away connective and other surrounding tissue with minutien pins.
Damaged MGAs having injury-induced vesicles or opaque regions were discarded. Undamaged MGAs were transected with microscissors, and small holes (up to 20 μm in diameter) were made by axonal impalement with glass micropipettes having a tip diameter of 1–3 μm.
Crayfish control external saline consisted of (in mm): 205 NaCl, 5.4 KCl, 2.6 MgCl2, 13.5 CaCl2, and 10.0 HEPES, pH 7.4. Divalent cation-free external saline consisted of (in mm): 205 NaCl, 5.4 KCl, 10.0 HEPES, 24.15 TMA-Cl, 1.0 EGTA, and 1.0 EDTA, pH 7.4. Fluorescent dyes were injected intracellularly in a buffer consisting of (in mm): 109 KF, 37 KAc, 15 NaCl, 96 mannitol, and 5 HEPES. The average osmolality of the hemolymph of three crayfish was 383 mOsm. The osmolality of crayfish physiological saline was ∼424 mOsm. All other crayfish salines were adjusted to this value.
Internal microdialysis of axons. Internal microdialysis of GAs (Mullins and Brinley, 1990) was combined with differential interference contrast (DIC) microscopy to compare the extent of vesiculation induced by the systematic addition of various ions to the axoplasm (Fishman and Metuzals, 1993). This technique required cannulation with two glass capillaries, one inserted at each end of 1 cm lengths of finely dissected GAs (∼500 μm in diameter). A 160-μm-diameter dialysis tube (cellulose acetate) with porous length of 1.5 cm delimited by black marks on the tube was inserted into one of the glass capillaries and advanced through the axon until the nonporous end of the dialysis tube protruded out of the farthest end of the other glass capillary. After insertion, the porous section of the dialysis tube was located midway between the ends of the axon and was not surrounded by a glass capillary. Flow of dialysate through the 160-μm-diameter tube was maintained at 12 drops/min. The amount of calcium buffer EGTA required to obtain a specific free calcium concentration in dialysis solutions was calculated with a computer program (Fabiato, 1988).
Confocal microscopy and fluorescent dyes. GAs and MGAs were viewed in vitro with the laser-scanning confocal microscopes LSM 410 (Zeiss) or TCS-4D (Leica, Nussloch, Germany) equipped with DIC optics, Zeiss Achroplan objective lenses [model 440090; 40× water immersion (numerical aperture (NA), 0.75; working distance (WD), 1.9 mm) and 63× water immersion (model 440067; NA, 0.9; WD, 1.5 mm)], an Ar/Kr laser (488, 568, and 648 nm), and FITC, tetramethylrhodamine isothiocyanate, and/or Cy5 filter sets. Axons were placed on a 0.1-mm-thick glass coverslip that replaced part of the base of a plastic Petri dish (∼3 ml). Fluorescent dye in the tip of a glass micropipette was pressure-injected (PLI-100; Medical Systems Corp., Greenvale, NY) into an axon after impalement. To facilitate impalement, an axon was supported along one side and parallel to its long axis by applying a continuous border of silicone grease (Dow Corning) with a syringe through an 18-gauge Luer stub adapter. Fluorescent dyes were injected at a concentration and volume to approximate a final concentration in the axon of 0.01%.
The external membranes (axolemma and glialemma) of intact axons were pulse-labeled with styryl dye (25 μm of FM 1-43 or FM 4-64; Molecular Probes, Eugene, OR) by briefly (5–10 min) placing axons in control external saline containing FM dye and subsequently removing FM dye from the bath by washing the axons several times with control external saline without dye. The results obtained with the two different FM dyes were very similar: FM styryl dyes partition readily into membranes, do not cross a membrane, and fluoresce substantially only when incorporated into a membrane (Betz et al., 1992). This brief (pulse) exposure of axons to FM dye in the bath saline was sufficient to pulse label the external membranes in MGAs and GAs and enabled us to observe plasmalemmal fluorescence for >1 hr after the FM dye was removed from the bath. Membrane-impermeant fluorescent hydrophilic dyes (0.01% w/v Texas Red-dextran, FITC-dextran, and Cy5-dextran; molecular weight, 3000) were added to the bath by complete replacement of the control external saline with saline-containing dye. The fluorescent hydrophilic dyes did not cross intact axons when left in the external bath or in the axoplasm for up to 3 hr. Membrane-permeable hydrophilic dye (1 μm calcein AM) that fluoresced once the acetoxymethyl ester group was removed was also used to label gliaplasm and/or axoplasm (Eddleman et al., 1997). All membrane-impermeant hydrophilic dyes were obtained from Molecular Probes. Cy5 was obtained from Amersham (Arlington Heights, IL). To enhance signal-to-noise ratio, each line of a confocal image was usually built up from the average of eight line scans. Time-lapse video images were acquired at regular intervals of ∼6 sec, unless otherwise stated.
Electron microscopy. GAs (intact, n > 15; severed, n > 30) and MGAs (intact, n> 15; severed, n > 30) were fixed for 2 hr in 1.25–4% glutaraldehyde in 0.1 m sodium phosphate or cacodylate buffer, pH 7.4. These axons were taken from preparations not used for confocal studies to avoid additional trauma to the axon before fixation. Most axons were post-fixed for 1.5 hr in 1–2% OsO4 in the same buffer used for the primary fixation. All fixed axons were rinsed in three changes of buffer for 1 hr, dehydrated in a graded ethanol series, and embedded in Spurrs plastic (Ballinger and Bittner, 1980; Krause et al., 1994). Thick (0.5 μm) sections were collected on glass slides, stained with Richardson’s stain, and photographed through a Zeiss CM 45 inverted microscope. Thin sections were collected on Formvar-coated, carbon-stabilized, single-slot grids, poststained with lead citrate, and viewed on a Siemens 1A transmission electron microscope at 60 kV. GAs and MGAs were mounted, trimmed, and sectioned to examine midsagittal (longitudinal) sections of their cut ends.
RESULTS
Structure of control squid GAs and crayfish MGAs
Control (intact) squid GAs (200–600 μm in diameter) are surrounded by a 6- to 10-μm-thick nonmyelinated glial sheath consisting of a single layer of adaxonal glial cells, a basal lamina, and alternating layers of collagen and fibrocytes (Villegas and Villegas, 1984; Brown et al., 1991; Brown and Abbott, 1993; Krause et al., 1994; Godell et al., 1997). Compared with the axoplasmic core (Villegas and Villegas, 1984), the cortical (subaxolemmal) region of axoplasm in an intact GA contains a greater density of neurofilaments, microtubules, smooth endoplasmic reticulum (SER), and some single-walled vesicles but hardly any double-walled structures other than an occasional mitochondrion. A 1- to 3-μm-thick single adaxonal layer of glia runs parallel to and between the axolemma and a collagenous basal lamina (Fig.1A). The most prominent cytoplasmic layer of the glial sheath is almost always the adaxonal layer, containing glial nuclei and cytoplasm with rough endoplasmic reticulum (RER), SER, intermediate filaments, microtubules, and a transverse tubular lattice that consists of hexagonal arrays of membranes that span the adaxonal glial cell layer (Brown et al., 1991;Brown and Abbott, 1993). The axolemma and glialemma occasionally form invaginations or evaginations 0.1–0.5 μm long.
Fig. 1.
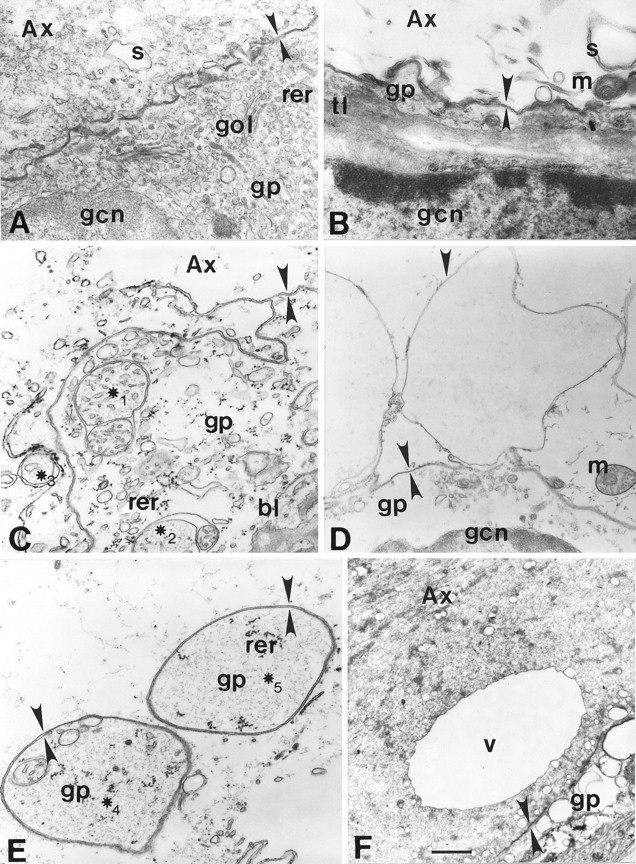
Electron micrographs of GAs (A,C, E, F) and MGAs (B, D). A, Normal squid (Sepioteuthis) axonal–glial interface.Ax, Axoplasm; bl, basal lamina;gp, gliaplasm; gcn, glial cell nucleus;gol, Golgi body; m, mitochondrion;rer, rough endoplasmic reticulum; s, smooth endoplasmic reticulum; tl, transverse tubular lattice. Apposing arrowheads indicate double-layered membranous structures identified as the axolemma and glialemma, except for E. ∗1–∗5, Double- or multi-walled membranous structures. B, Normal crayfish MGA glial interface. C, Discontinuous axolemma and intermingling of axonal and glial elements ∼20 μm from the cut end of a transected GA fixed at 5 min after transection. D, Invaginating axolemma (Figure legend continued) (downward arrowhead not facing an upward arrowhead) within 50 μm of the cut end of a crayfish MGA fixed at 20 min after transection. Glialemma (apposing arrowheads) does not evaginate at this site of axolemmal invagination. E, Double-walled vesicles containing gliaplasm ∼20 μm from the cut end of a transected GA fixed at 5 min after transection. F, Large (10 μm) single-walled vesicle (V) in axoplasm and highly vesiculated gliaplasm ∼150 μm from the cut end of a GA fixed at 30 min after transection. Scale bars: A–C, E, 0.45 μm;D, 1.2 μm; F, 2.0 μm.
Control (intact) crayfish MGAs (80–150 μm in diameter) are surrounded by a 3- to 10-μm-thick nonmyelinated glial sheath in which layers of glial processes alternate with extracellular matrix containing collagen (Ballinger and Bittner, 1980; Viancour et al., 1987; Eddleman et al., 1997). The axoplasm, axolemma, and sheath of the MGA (Fig. 1B) are similar to control GAs (as described above) with the following exceptions. The MGA axoplasm lacks ultrastructural evidence of neurofilaments, the core axoplasm is almost completely devoid of any cytoplasmic organelles, and the axolemmal invaginations and glialemmal evaginations are larger and more frequent. The glial sheath of the MGA does not exhibit a well defined basal lamina and contains a greater number of alternating layers of glial cytoplasm (the innermost being the adaxonal glial layer) and collagenous extracellular matrix.
Effectiveness of Ca2+ and other ions to induce vesicles or other membranous structures
Vesicles or other membranous structures are formed when GAs (Fishman et al., 1990; Krause et al., 1994; Gallant et al., 1995;Godell et al., 1997) and MGAs (Tanner et al., 1995; Eddleman et al., 1997) are severed in bath salines that contain Ca2+. GAs or MGAs severed in Ca2+-free salines do not produce large numbers of vesicles or form other injury-induced membranous structures (Fishman et al., 1990; Krause et al., 1994;Eddleman et al., 1997; Godell et al., 1997). These results suggest that external Ca2+ that enters the axon could induce the formation of membranous structures.
Intact GAs (n = 6) bathed in and dialyzed with control internal saline [containing (in mm): 0 Ca2+, 440 glutamate, 440 K+, and 1 EGTA; see Materials and Methods] for >60 min showed no significant formation of membranous structures in cortical axoplasm (Fig.2A) or in any other regions of axoplasm in the vicinity of the dialyzed portion of the axon. Approximately 45 min after the glutamate in the control internal saline dialysate was replaced by an equal amount of chloride, small (<0.5 μm in greatest diameter) vesicles or other membranous structures were discernible in cortical axoplasm in the vicinity of the dialyzed portion of the axon (Fig. 2B). Approximately 30 min after most (430 mm) of the K+ in the dialysate was replaced by Na+, some slightly larger vesicles (∼1 μm) appeared in cortical axoplasm (Fig.2C). Finally, ∼15 min after Ca2+buffered to 1 mm was added to the Na+dialysate, many large (>2 μm) vesicles or other membranous structures formed in the cortical axoplasm of the dialyzed portion of the axon (Fig. 2D). These data suggested that K+, the predominant cation in axoplasm, did not induce formation of membranous structures and that the order of effectiveness of other ions that induced formation of membranous structures was Ca2+ ≫ Na+ > Cl. All the data reported above also suggested that an increase in axoplasmic Ca2+ rather than plasmalemmal damage per se induced the formation of large numbers of vesicles or other membranous structures in the intact segment of these dialyzed axons.
Fig. 2.
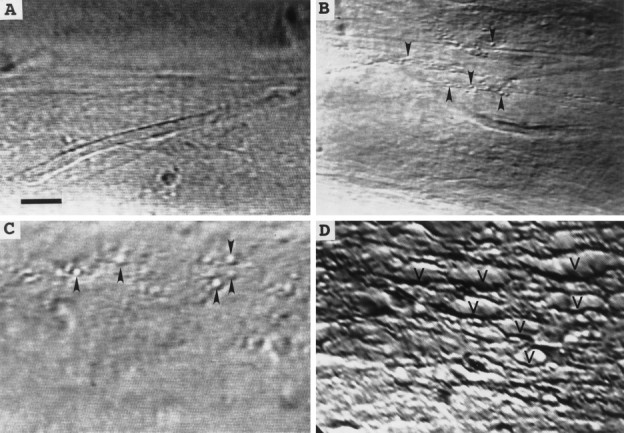
DIC images showing extent of invagination and vesiculation in the dialyzed region of a squid GA bathed in control internal saline after changes in the ionic composition of the solution used to internally microdialyze the axoplasm. A, No invagination or vesiculation after dialysis with control internal saline (buffered K glutamate). B, Small amount of invagination or vesiculation (arrowheads) after dialysis with the internal saline in A modified by Cl− replacement of glutamate. C, Moderate amount of invagination or vesiculation after dialysis with the internal saline in B modified by Na+replacement of K+. D, Extensive invagination or vesiculation (V) after dialysis with the internal saline used in C modified to ensure that the free concentration of Ca2+ equaled 1 mm. All images were acquired 30 min after introducing a new microdialysate solution to ensure equilibration of the axoplasmic ionic concentrations with those of the dialysate. Scale bar (inA), 25 μm.
To describe the relationship between the intracellular unbound Ca2+ concentration and the formation of vesicles or other membranous structures, we bathed GAs (n = 16) in Ca2+-free control internal saline and dialyzed them with Ca2+-free internal salines (see Materials and Methods) buffered with EGTA and calcium to obtain various internal Ca2+ concentrations ranging from 100 nmto 1 mm (Fabiato, 1988). [Equilibration of the axoplasmic Ca2+ concentration to >90% of the free concentration in the dialysis saline takes 0.5 hr from the time the dialysis saline is changed from the control level (100 nm) to a test solution with a higher level of free Ca2+(Mullins and Brinley, 1990).] Membranous structures of >2 μm diameter did not form in axoplasm for up to 2 hr (maximum observation time) after the Ca2+ concentration in the dialysis solution was increased from 100 nm to <100 μm (n = 6). Membranous structures of 2 μm diameter were often observed 90–120 min after the dialysis solution was changed from 100 nm to 100 μmfree Ca2+ (n = 3). Further increases in Ca2+ concentration of the test solution produced larger structures even more rapidly. For example, membranous structures were observed 15–20 min (n = 3) after changing to a test solution containing 1 mm free Ca2+, i.e., well before the axoplasmic concentration had equilibrated with the dialysate concentration. These and other observations suggested that the formation of vesicles or other membranous structures was not a continuous monotonic function of intracellular free Ca2+, but rather that a threshold level of ∼100 μm Ca2+ was necessary to induce such structures. Increases in axoplasmic free Ca2+ >100 μm produced more membranous structures of larger diameter in shorter times.
Ca2+-induced formation of membranous structures occurs in cortical axoplasm and requires the axolemma and/or glial sheath
We used DIC microscopy to compare Ca2+-induced formation of membranous structures in severed GAs versus desheathed GA axoplasm lacking an axolemma and the glial sheath. When an intact GA was severed in control external saline containing 10 mmCa2+, large (5–20 μm) membranous structures formed in cortical regions of axoplasm within 10 min (Fig.3A). These membranous structures were not present before transection (Fig. 3A,inset). When GAs placed in control internal saline (a Ca2+-free saline; see Materials and Methods) containing 1 mm EGTA were desheathed by removing the axolemma and glial sheath to expose GA axoplasm for a length of several millimeters, membranous structures did not appear in axoplasm within 30 min after addition of 10 mm Ca2+ to the control internal saline (Fig. 3B). These data suggested that Ca2+-induced and/or injury-induced membranous structures occurred only if cytoplasm were surrounded by a plasmalemma (axolemma or glial sheath), and that such membranous structures do not arise from the fusion of small preformed membranous structures (e.g., transport vesicles or smooth endoplasmic reticulum).
Fig. 3.

DIC images of squid GAs showing Ca2+- or injury-induced invagination and vesiculation. A, Extensive invagination and vesiculation (arrows) ∼400 μm from the cut end in GA axoplasm 30 min after transecting the GA in control external saline containing 10 mm Ca2+. Inset, No invagination or vesiculation of the intact GA in control external saline before its transection. B, No invagination or vesiculation 30 min after adding 1 mmCa2+ to the control internal saline that bathed the isolated desheathed axoplasm (see text). C, No invagination or vesiculation in a GA after microdialysis for 1 hr with buffered KI that removed >99% of the axoplasm. D, Invagination and vesiculation (arrows) ∼350 μm from the cut end induced 15 min after severance in control external saline (containing 10 mm Ca2+) of the same axon shown in C. This result suggests that vesiculation or invagination requires a plasmalemma but does not require the presence of axoplasm. Scale bar (in D): A–D, 25 μm; A, inset, 100 μm.
To determine whether axoplasm was necessary for the formation of Ca2+-induced membranous structures, we internally perfused GAs (n = 4) with 0.5 m KI (see Materials and Methods) for 1 hr to remove almost all (>99%) of the axoplasm but leaving the axolemma and glia intact (Baumgold et al., 1981). To prevent axonal collapse after removal of axoplasm, each of these perfused GAs was ligated proximal to the exit hole from which the perfusate flowed. This proximal ligation stopped the flow of perfusate through the GA and sustained the inflation pressure so that the GA could also be ligated at the perfusate entry point without collapse. Then the intact, doubly ligated portion of the GA was excised and placed in control external saline. No large axoplasmic vesicles or other membranous structures were seen when these GAs lacking axoplasm were bathed in control external saline (10 mmCa2+) for 20 min (Fig. 3C). Large spherical vesicles (diameters ranging from 2 to 12 μm) or other membranous structures did form just beneath the axolemma within 15 min after these GAs were transected in control external saline (Fig.3D). These observations suggested that Ca2+-induced vesicles or other membranous structures arise primarily from the axolemma and/or glia. These data from desheathed and internally perfused GAs did not eliminate the possibility that membranous structures of axoplasmic origin (e.g., smooth endoplasmic reticulum, transport vesicles, and mitochondria) may fuse with membranous structures of axolemmal or glial origin (see Discussion).
Origin and mode of formation of vesicles and other membranous structures
The Ca2+-induced membranous structures described above that appear in axoplasm could arise from any combination of single-walled structures formed by axolemmal invaginations, double-walled structures formed by axolemmal invaginations apposed by glialemmal evaginations, single-walled vesicles formed by endocytosis of axolemma or glialemma, or double- or multi-walled vesicles formed by axolemmal phagocytosis of glialemma or other mechanisms that may not involve a plasmalemma.
To obtain data on the origin and mode of formation of membranous structures, we first injected intact MGAs with FITC-dextran so that the final axoplasmic concentration was ∼0.01%. FITC-dextran did not induce vesiculation. The MGA was then transected and maintained in control external saline. At 10 min after transection, a midsection confocal image of the FITC fluorescence from the MGA axoplasm showed black holes (circular areas lacking FITC fluorescence in a confocal image) near the axolemma (Fig.4A,B), suggesting the presence of membranous structures whose contents were isolated from the axoplasm that was labeled with FITC fluorescence. When viewed with DIC (data not shown), these black holes were clearly membranous structures (invaginations or vesicles). When Texas Red-dextran (0.01%) was added to the bath, and the same optical section was imaged for Texas Red fluorescence, many black holes such as those shown in Figure 4A did not take up the Texas Red from the extracellular medium (Fig. 4C). The absence of Texas Red uptake was consistent with the hypothesis that many of these black holes were Ca2+-induced axoplasmic vesicles isolated from the extracellular space. Other black holes (Fig.4B) in the same MGAs did take up dye (Fig.4D), consistent with the hypothesis that those black holes were membranous invaginations that retained a connection with the bath saline.
Fig. 4.
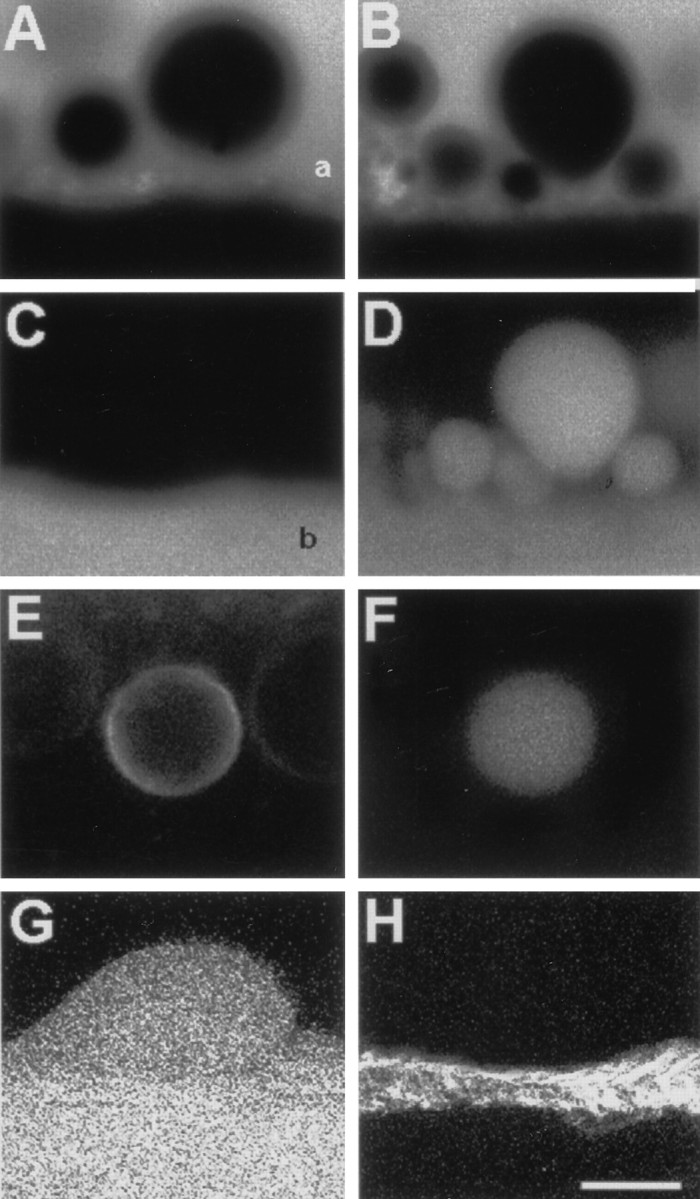
Confocal images taken 50 μm from the cut ends of lesioned MGAs exposed to fluorescent dyes.A, B, Membranous structures identified in DIC (data not shown) in the axoplasm of an MGA injected with 0.01% FITC-dextran before transection and viewed 10 min after transection. Absence of fluorescence (black holes) indicates membranous structures, the contents of which do not include FITC-dextran. Fluorescent area (white region) at the top ofA and B delineates the axoplasm (a) just interior to the axolemma; theblack region at the bottom is the extracellular space. Black holes were not observed in the axoplasm before transecting the MGA. C, Same axoplasmic region shown in A but imaged for fluorescence of Texas Red-dextran placed in the bath after black holes had formed. Many of the black holes in A remained unlabeled, indicating that they are axoplasmic vesicles. Fluorescent area delineates the extracellular bath (b). D, Same axoplasmic region shown in B but imaged for fluorescence of Texas Red-dextran placed in the bath after black holes had formed. The black holes in B are dye-filled in D, indicating that they are invaginations that are connected to the extracellular space. E, Ring-shaped fluorescence of injury-induced membranous structure in axoplasm 10 min after transection of an MGA that was pulse-labeled with FM 4-64 (25 μm for 10 min) before transection. F, Same axoplasmic region shown in E but imaged for fluorescence of FITC-dextran placed in the bath 2 min after transection and washed off at 10 min after transection. After dye washout from the bath, the membranous structure retained the dye, indicating its isolation from the bath saline; i.e., this structure was an axoplasmic vesicle. The images in E and F are consistent with a vesicle that formed from FM-labeled plasmalemmal membrane after an axolemmal invagination which subsequently budded off to become an axoplasmic vesicle. G, Image of a membranous structure invaginating into the axoplasm. The contents of this structure filled with Texas Red-dextran that was added to the bath saline 10 min after transection. H, Same optical section as inG, but imaged for calcein (glial cytosolic marker) (Eddleman et al., 1995) showing no evagination of the glial cell associated with the axoplasmic invagination in G. The images in G and H are consistent with an axolemmal invagination not associated with a glialemmal evagination. Scale bar (in H): A–D, 5 μm;E, F, 1 μm; G, H, 15 μm.
To determine the origin of the membranes of many Ca2+-induced structures, we FM pulse-labeled all external plasmalemmal membranes of an intact MGA. We observed many ring-shaped FM-fluorescing structures near the axolemma in midsection confocal images of MGAs collected at 10 min after transection (Fig.4E). These data suggested that many Ca2+-induced membranous structures originated from a plasmalemmal source (axolemma or glialemma). To test whether some of these Ca2+-induced membranous structures of plasmalemmal origin were axoplasmic vesicles not connected to the external saline, FITC-dextran (0.01%) was added to the bath at 1 min after transection and then removed 20 min later. We observed many disk-shaped patterns of FITC fluorescence (Fig. 4F) that fit within superposed ring-shaped patterns of FM 4-64 fluorescence (Fig. 4E). When FITC-dextran was removed from the bath, many of the FM-labeled ring-shaped structures continued to contain FITC-dextran fluorescence, suggesting that these structures were axoplasmic vesicles. To examine whether the axolemma or glialemma was the major source of injury-induced vesicular membrane, we pulse-labeled intact MGAs and GAs with 1 μm calcein AM for 10 min, followed by the removal of calcein AM from the bath. [Calcein AM is membrane-permeable, becomes fluorescent and membrane-impermeant after cleavage of the AM group by esterases, and much more rapidly labels the glial cytosol of giant axons than it labels the axoplasm (Eddleman et al., 1995)]. For example, after differentially labeling the glial cytosol of a GA with calcein, the axon was transected, and a hydrophilic dye (e.g., Texas Red-dextran) was added to the bath at 10 min after transection. Confocal images of Texas Red-dextran fluorescence often showed that invaginations of the axolemma were filled with Texas Red-dextran (Fig. 4G) at regions that lacked a calcein-labeled (fluorescence) evagination of the adaxonal glialemma (Fig. 4H). All these confocal data were consistent with the hypothesis that many Ca2+-induced membranous structures were vesicles that formed by endocytosis of the axolemma.
We obtained very similar data when the same experimental paradigms were applied to both GAs (n > 10) and MGAs (n > 10). The only observable difference between these axons was that Ca2+-induced endocytotic formation of larger vesicles or axolemmal invaginations was somewhat slower in GAs than in MGAs. That is, large membranous structures (>2 μm in diameter) usually were not observed in GAs for 15 min or more after transection, whereas >2 μm vesicles often occurred in MGAs within 5–10 min after transection.
In contrast to electron micrographs of control GAs (Fig.1A) and control MGAs (Fig. 1B), the axolemma and glialemma in severed GAs and MGAs were greatly disrupted near (within 20–50 μm of) the cut end when viewed at shorter (5–15 min) post-transection times (Fig. 1C,E). Gliaplasmic (including RER and glial nuclei) and axoplasmic contents intermingled in this region near cut axonal ends (Fig.1C,E). At shorter post-transection times, the axoplasm of GAs and MGAs severed in control external salines showed many single-walled structures (Fig.1D,F) but only a few double- or multi-walled structures near a cut axonal end (Fig.1C,E); gliaplasm showed many more double- or multi-walled structures near a cut axonal end. These double- or multi-walled structures did not appear to arise by microphagocytosis. For example, the axolemma near the cut end sometimes invaginated without accompanying glial evagination, and we saw no other convincing evidence of axonal microphagocytosis of adaxonal glia. Furthermore, we saw no glial invaginations closely associated with an axoplasmic evagination or other unambiguous evidence for microphagocytosis of the axon by the glia.
Although some of the double-walled structures in axoplasm or gliaplasm could have been swollen mitochondria (Fig. 1C, ∗1–∗3) sampled in an oblique plane at shorter post-transection times, other double- or multi-walled structures in gliaplasm very likely were not of mitochondrial origin, because they contained swollen mitochondria (Fig. 1E, ∗4) and/or remnants of RER (Fig. 1E, ∗5). These latter structures almost certainly arose from nonmitochondrial structures in gliaplasm, because axoplasm (like mitochondria) lacks RER in GAs (Villegas and Villegas, 1984) and MGAs (Ballinger and Bittner, 1980). Moreover, many of these double- or multi-walled structures in gliaplasm contained arrays of single-walled membranous structures that looked like disrupted portions of the transverse tubular lattice (Fig. 1C, ∗1). Examples of an intact transverse tubular lattice were easily found in glia surrounding intact axons (Fig. 1B) but were exceedingly difficult to find as intact structures in glia near the cut ends of severed axons at shorter post-transection times. Given all these data, we suggest that many of these double- and multi-walled structures in the gliaplasm at shorter post-transection times arose by disruptions of the glial endoplasmic reticulum (ER), transverse tubular lattice, and/or adjacent glialemma.
At shorter post-transection times, the frequency of single-walled structures in axoplasm and double- or multi-walled structures in gliaplasm and the extent of plasmalemmal discontinuities decreased with increasing distance from the cut axonal end. In regions farther (50–1000 μm) from the cut end at shorter post-transection times, we saw more single-walled structures (axoplasmic vesicles) in cortical than in core regions of the axoplasm. Very few double- or multi-walled structures were seen in cortical or core axoplasm farther from the cut end. We also saw no evidence of glial or axonal microphagocytosis or axolemmal–glialemmal disintegration in regions farther from the cut end at shorter post-transection times. Although mitochondria in the region near the cut end were generally swollen and lacked cristae (see above), mitochondria farther from the cut end were very similar to those in control axons (Fig. 1B).
At longer post-transection times (30–150 min), the region exhibiting a discontinuous axolemma and/or glialemma, a mixing of axoplasm and gliaplasm, and a distortion of mitochondria extended much farther (50–1000 μm) from the cut axonal ends of GA and MGAs (Fig.5) compared with shorter post-transection times. Many single-walled structures near the cut end could have originated from axoplasmic or glial sources at the cut end or could be axoplasmic vesicles that migrated to the cut end (see below). At longer post-transection times, the number of double- or multi-walled structures near the cut end was greatly increased (Fig.5E,F) compared with shorter post-transection times (Fig.4C,D,F). Many multi-walled structures were observed in regions with a disrupted plasmalemma and glialemma (Fig. 5C–F). The glialemma in GAs and MGAs was sometimes disrupted (or completely absent) across the entire adaxonal glial layer (bounded on its outer face by a layer of collagen in control axons) within 50–1000 μm of the cut end (Fig.5C–F), as previously reported for GAs by Gallant et al. (1995). At longer post-transection times, cytoskeletal elements easily observable in control axons (Fig.4A,B) were often no longer recognizable in the axoplasm or gliaplasm up to 200 μm from the cut end in MGAs and up to 1000 μm in GAs (Fig. 5C–F) at 30–150 min after transection.
Fig. 5.
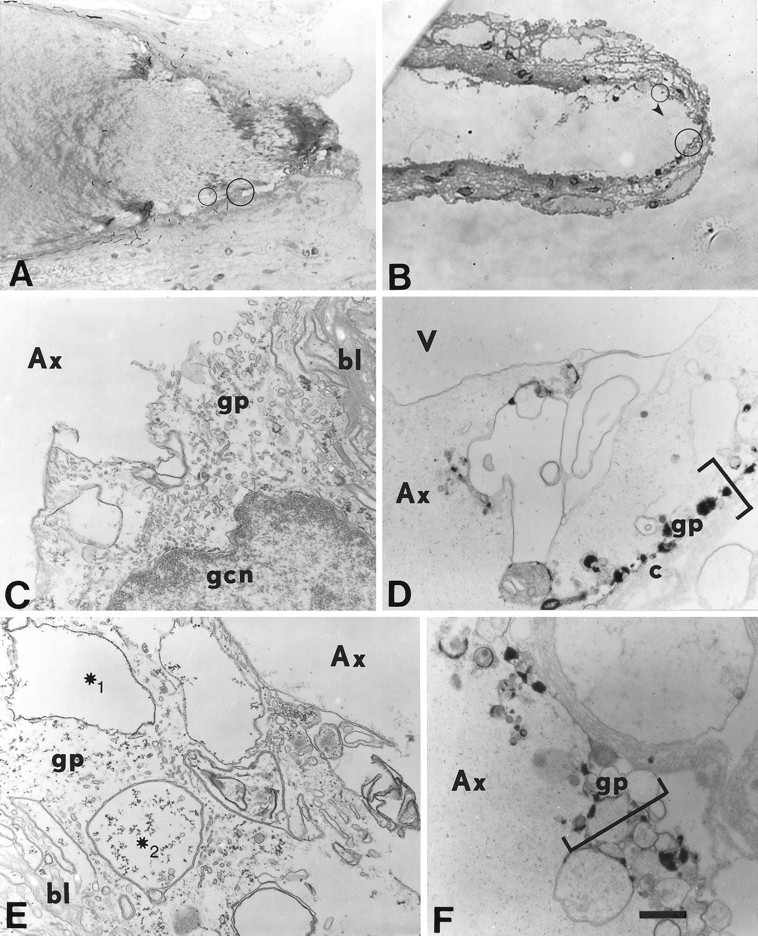
Light (A, B) and electron micrographs (C–F) of a transected squid GA (A, C,E) and a transected crayfish MGA (B, D, F).A–F, The cut axonal end is oriented to theright. Electron micrographs are enlargements of a representative portion of the circles inA and B, which in turn are representative of cortical axoplasmic and adaxonal glial regions within 200 μm of the cut axonal ends. Ax, Axoplasm; bl, basal lamina; c, collagen layer; gp, gliaplasm; gcn, glial cell nucleus. A, Midsagittal section of squid (Sepioteuthis) GA fixed at 150 min after transection. B, Midsagittal section of a crayfish MGA fixed at 60 min after transection. Thearrowhead marks a vesicle in the axoplasmic space near the cut end. C, Magnified view of the large circle in A where the axolemma and glialemma are no longer identifiable, and the gliaplasm mixes with the axoplasm ∼220 μm from the cut end. The axoplasm (Ax) contains no identifiable cytoplasmic structures. D, Magnified view of the large circle in B where the axolemma and glialemma are no longer present, and the adaxonal layer (brackets) has been replaced by electron-dense bodies and vesicles. V, Part of the large vesicle labeled with an arrowhead in B ∼15 μm from the cut end. E, Magnified view of the small circle in A ∼180 μm from the cut end. The axolemma and glialemma are no (Figure legend continued) longer identifiable. The gliaplasm contains many single-layered (∗1) and multi-layered (∗2) membranous structures. F, Magnified view of thesmall circle in B ∼30 μm from the cut end. The axolemma and glialemma are no longer identifiable. The adaxonal layer (brackets) contains single- and multi-layered membranous structures and electron-dense bodies, rather than its normal complement of cytoskeletal structures (e.g., microtubules, ER, and transverse tubular lattice). Scale bar (inF): A, 63 μm; B, 25 μm; C, E, 0.77 μm; D, F, 9.1 μm.
The most likely explanation for these confocal and ultrastructural data was that many Ca2+-induced membranous structures were single-walled vesicles that arose as axolemmal invaginations, some of which eventually pinched off to form axoplasmic vesicles and migrated to the cut end. Many other Ca2+-induced membranous structures that were double- or multi-walled probably arose from disruptions of the glial ER and transverse tubular lattice.
Time-lapse observations of injury-induced invaginations and vesicles forming by endocytosis
Fishman et al. (1990) suggested that injury-induced vesicles in GAs formed from single-walled axolemmal invaginations. To obtain more detailed data on the formation of Ca2+-induced membranous structures in GAs and MGAs, we used time-lapse confocal fluorescence microscopy to observe GAs (n = 5) or MGAs (n = 10) that were first pulse-labeled with FM 1-43 for 10 min in physiological saline and then transected in physiological saline that did not contain any dye. For the region near the cut end of the transected MGA shown in Figure 6, the FM 1-43 fluorescence was imaged at 11 min after transection in a longitudinal optical section through the axolemma, the glial sheath, and a portion of the extracellular space. An image of this optical section was recorded every 6 sec thereafter for 210 sec. These time-lapse images (Fig. 6) showed that the endocytotic formation of a vesicle from axolemma proceeded in a sequence of characteristic stages: (1) an initial invagination (Fig. 6A); (2) expansion of the invagination into a teardrop-shaped structure (Fig.6B); and (3) a sudden separation of the invagination from the axolemma at its point of attachment and simultaneous membrane closure (Fig. 6C). Immediately after separation of an invagination from the axolemma or on separation of two closely apposed invaginations, we often observed FM-labeled strands (tethers) connecting the two structures (Fig. 6F). Tethers eventually were broken as one structure moved farther away from the other. Similar tethers have been demonstrated artificially by pulling with laser tweezers on a coated bead in contact with a growth cone (Dai and Sheetz, 1995).
Fig. 6.
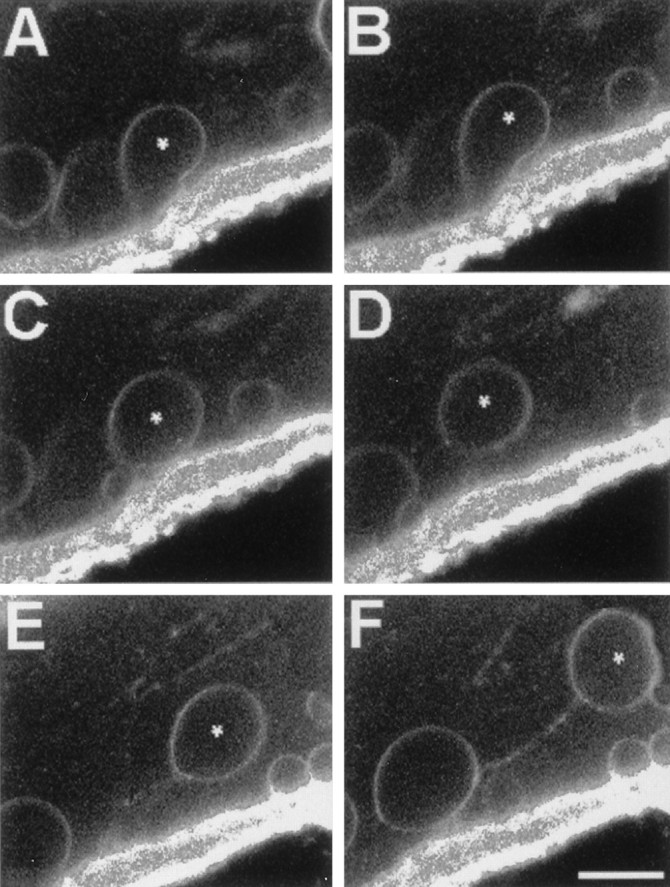
Series of time-lapse confocal fluorescence images in the same optical midsection ∼100 μm from the cut end showing stages of injury-induced endocytosis beginning with an invagination of FM-labeled membrane and ending with a vesicle moving in the axoplasm toward the cut end of a transected MGA. A–F, The axon is at the top, the bath is at the bottom, and the cut end is toward the right. The plasmalemmal membranes of an intact MGA were pulse-labeled with FM 1-43. The MGA was transected and imaged for FM 1-43 fluorescence starting at 11 min after transection without changing the confocal plane or the position of the micrometer stage. A–F, Successive images were acquired at 18 sec intervals and taken from a larger set of images acquired every 6 sec. Asterisks mark the same vesicle in every frame. Note that vesicles are sometimes joined by a fluorescent line, presumably a tether of membranous material. Scale bar (inF), 10 μm.
During imaging of vesicle formation, we also observed the formation of invaginations without subsequent formation of endocytotic vesicles. That is, structural development proceeded through stage 2, after which expansion of the invagination either stopped or continued without vesicle formation. We did not observe vesicle production without the preceding invagination stages 1 and 2. These observations suggested that all Ca2+-induced vesicles that formed by endocytosis began as invaginations, but that many Ca2+-induced invaginations did not become vesicles.
Movement, fusion, and accumulation of vesicles at cut axonal ends
To determine the fate of Ca2+-induced vesicles, we used time-lapse confocal fluorescence of an optical section at the cut end of a GA or MGA to follow the movement and accumulation of FM-labeled vesicles. For example, Figure7A–C shows three images (3, 8, and 13 min after transection) of a transected MGA pulse-labeled with FM 1-43. These three images were taken from a sequence of images acquired every 6 sec over a 13 min interval. The total number of vesicles in the region of axon imaged and the number of vesicles that accumulated at the cut end increased with time. Although the number of vesicles that formed also decreased with increasing distance from the cut end, we confirmed that formation of membranous structures was often substantial [∼20% of the axolemmal surface area up to 1 mm from the cut end of a GA (Fishman et al., 1990)].
Fig. 7.

Series of time-lapse fluorescence images in the same optical section of a severed MGA (A–C) and at the site of a micropuncture in the axolemma of an MGA (D–F) showing the movement toward, and accumulation at, the cut end and small hole, respectively, of vesicles formed by endocytosis (see Fig. 6) of the axolemma. The MGA was pulse-labeled with FM 1-43 and then transected. The cut end of the MGA is oriented to the left in A–C.A–C, Successive images at 3, 8, and 13 min after transection, respectively, representative of a larger set of images acquired every 6 sec. D–F, Successive images at the site of a micropuncture in an MGA at 5, 10, and 25 min, respectively, after the physiological saline, in which the MGA was punctured, was replaced by a saline containing Texas Red-dextran (1%) at 5 min after the micropuncture. The interior of the axon is at thetop of each panel. The black holes are confocal images of vesicles that contain no dye and are surrounded by Texas Red, which entered the axon through the micropuncture (arrow) after dye was added to the bath. Scale bar (in D):A–C, 40 μm; D–F, 20 μm.
We observed that vesicles usually moved toward the site of injury (Figs. 6, 7). The rate of movement of vesicles was quite variable, perhaps because of intermittent interactions with other membranous structures. For example, immediately after formation in cortical axoplasm (stage 3 above; Fig. 6C), some vesicles did not move until FM-labeled tethers (Fig. 6D–F) to neighboring anchored membranous structures were broken. After encountering another membranous structure, a moving vesicle sometimes fused with it or stopped moving. The advance of most vesicles toward the injury site was interrupted when vesicles moved around, became attached to, or fused with other membranous structures. Many vesicles accumulated rapidly (<15 min after transection) at the cut end of an MGA where they often fused with each other, other membranous structures, or the axolemma (Eddleman et al., 1997, their Fig. 2G,H).
Accumulations of vesicles at small axolemmal lesions
To determine whether Ca2+-induced vesicles accumulated at small axolemmal lesions as well as at cut ends, we micropunctured intact GAs and MGAs with the tip of a glass micropipette. These pipettes produced small plasmalemmal holes (lesions) of 1–40 μm diameter in the axolemma. After an MGA was penetrated by the tip of a micropipette, the injury response to small plasmalemmal lesions was similar to that of large lesions; vesicles formed by endocytosis of the axolemma and usually moved from all directions toward the small lesion (Fig. 7, compareA–C, D–F). Figure 7D–F shows the punctured portion of an MGA after replacement of the physiological saline 5 min after lesioning with a solution containing Texas Red-dextran (0.01%). A series of optical sections through the lesion site was acquired for Texas Red fluorescence at 5, 10, and 25 min after dye exposure. For temporal comparison, only the image at the same optical section is shown at each time. Entry of extracellular Texas Red into the hole before imaging at the specified times after puncture was sufficient to observe vesicles as black holes (Fig.7D–F) that formed before addition of the dye to the bath; these black holes were similar to the ones observed (Fig.3A,B) after transection of an axon injected with FITC-dextran. The increasing number of black holes in confocal images of fluorescence at the three times after lesioning showed that the number of accumulated vesicles increased with time at the lesion site. As observed for complete transections, the rates of vesicular movement toward the lesion site were quite variable. The extent of vesiculation in axons with small plasmalemmal holes was related to the size of the injury; i.e., the larger the plasmalemmal hole, the greater the amount of vesiculation and the greater the number of vesicles that accumulated at the micropuncture (compare Figs.7A–C, D–F,8E).
Fig. 8.
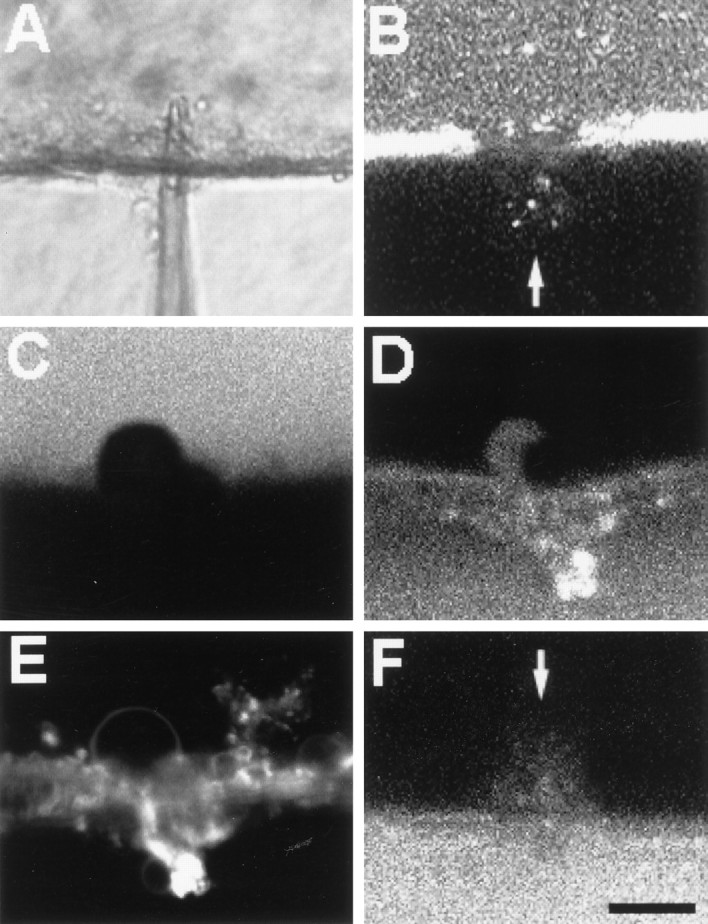
DIC (A) and confocal (B–F) images of MGAs micropunctured in Ca2+-containing salines (A–E) or Ca2+-free salines (F) showing dye exclusion amid an accumulation of Ca2+-induced vesicles and other membranous structures (C–E) or showing dye uptake (B, F). All MGAs were incubated in calcein AM for 5–30 min before being micropunctured. A–F, The axon is at the top, the bath is at thebottom, and the micropuncture is approximately in thecenter. A, Image of a micropipette penetrating an MGA. B, Image of calcein hydrophilic dye leaking out of an MGA at the micropuncture site (arrow) at 3 min after puncture, i.e., before formation of a barrier (seal) to hydrophilic dyes. C, Image of calcein fluorescence showing membranous structures identified with DIC (data not shown) at the micropuncture site in an MGA at 25 min after puncture.D, Same confocal plane as in C but imaged at 25 min after puncture for Texas Red-dextran, which was added to the bath at 20 min after puncture. E, Same confocal plane asC and D but imaged at 50 min after puncture for FM 1-43, which was added to the bath at 40 min after puncture. The styryl dye incorporated into the membranes of the structures at the injury site and did not label membranes in the interior of the MGA; i.e., a barrier to the FM dye formed amid a collection of vesicles at the micropuncture site. F, MGA micropunctured and maintained in Ca2+-free saline. Cy5-dextran hydrophilic dye was added to the bath at 20 min after puncture and then imaged at 25 min after puncture. Note uptake of Cy5-dextran into the axoplasm at the micropuncture site (arrow). Scale bar (in F): 15 μm.
A barrier to dye diffusion formed at the micropuncture site (Fig. 8) amid an accumulation of injury-induced vesicles and other membranous structures in crayfish MGAs (n > 10) similar to that reported at the cut end of earthworm or crayfish MGAs transected in physiological saline (Krause et al., 1994; Ballinger et al., 1997;Eddleman et al., 1997) or squid GAs transected in physiological saline containing exogenous calpain (Godell et al., 1997). Micropunctures 1–3 μm in diameter sealed within 10–60 sec to exclude entrance of Texas Red-dextran in the bath (data not shown), whereas larger micropunctures 10–40 μm in diameter took longer to exclude dye entry. For example, after an MGA was incubated in calcein AM, which first labels the gliaplasm and then the axoplasm as the AM group is cleaved by cytosolic esterases (Eddleman et al., 1995), the MGA received a 5- to 20-μm-diameter micropuncture (Fig. 8A). The location of the micropuncture site was indicated by a plume of outwardly diffusing calcein hydrophilic dye (Fig.8B). At 20 min after puncture, Texas Red-dextran hydrophilic dye was added to the bath saline. When imaged at 25 min after puncture, the MGA had formed a barrier to the outward diffusion of calcein or the inward diffusion of Texas Red-dextran (Fig.8C,D, respectively). This dye barrier was located at the same place for both dyes (Fig.8C,D) and formed amid a collection of membranous structures labeled by the addition of FM 1-43 at 40 min after puncture (Fig. 8E). When this same dye paradigm was applied to MGAs bathed in Ca2+-free physiological salines, intracellular calcein dye diffused out, and extracellular Cy5 hydrophilic dye diffused in; i.e., the MGA did not seal (Fig. 8F), consistent with previous reports that Ca2+ is necessary for the sealing of transected MGAs (Eddleman et al., 1997). All these data are consistent with the hypothesis that small holes, as well as complete transections, are repaired by Ca2+-induced vesicles and other membranous structures (Eddleman et al., 1997).
DISCUSSION
In this paper, we report that many single-walled, injury-induced vesicles arise by endocytosis from the axolemma when the axoplasmic free Ca2+ concentration exceeds 100 μm, fuse with other single- or multi-walled membranous structures of axoplasmic or glial origin, and migrate toward the lesion site where they interact with each other and other membranous structures to form a barrier (seal) to dye movement after complete axolemmal transections or micropunctures.
Ca2+ inflow primarily occurs at the lesion site to form injury-induced membranous structures
We find that the formation of injury-induced vesicles and other membranous structures does not occur when the Ca2+concentration in the physiological saline-bathing transected GAs and MGAs is buffered to submicromolar levels, but does occur in intact GAs when the axoplasmic Ca2+ concentration is raised to >100 μm by microdialysis. These data suggest that a 1000-fold increase in the concentration of intracellular Ca2+ above nominal intracellular levels (∼100 nm) is the factor that initiates the formation of injury-induced membranous structures. A threshold dependence on Ca2+, rather than a graded dependence, also suggests that the formation of injury-induced membranous structures is not solely produced by upregulation of an existing low-level process (e.g., endocytosis). These data are consistent with reports that extracellular Ca2+ must be raised to at least 100 μmto induce sealing of severed septal neurites in tissue culture (Xie and Barrett, 1991) and that intracellular Ca2+ reaches micromolar to millimolar levels after axotomy of Aplysiaaxons in vitro (Ziv and Spira, 1995).
The rise of intracellular Ca2+ concentration that induces the formation of membranous structures could be attributable to the entry of extracellular Ca2+ through depolarized ion channels in the plasmalemma (George et al., 1995), to the release of Ca2+ from internal stores, or to the entry of extracellular Ca2+ at the lesion site. Our data suggest that it is primarily the rise of intracellular Ca2+ resulting from Ca2+ entry at the cut axonal end that induces the formation of membranous structures. For example, we (Fishman et al., 1995) previously reported temporal changes in the pattern of luminescence of a Ca2+indicator (aequorin) that would be expected for Ca2+entry at the cut end of severed GAs, in contrast to Ca2+ entry through ion channels or through release of intracellular Ca2+. Furthermore, other reports of temporal variations of fluorescence intensity in severed axons loaded with fura-2 (Strautman et al., 1990) or mag-fura-2 (Ziv and Spira, 1995) are consistent with our data in that the greatest increase in intracellular Ca2+ after severance occurred first at the cut end and subsequently at sites more distant from the cut end.
Effectiveness of Ca2+ versus other ions in the formation of injury-induced membranous structures
In addition to increases in intracellular Ca2+concentration, plasmalemmal damage to eukaryotic cells maintained in physiological saline should increase the intracellular concentrations of Ca2+, Na+, and Cl− and should reduce K+. Previous extracellular measures have reported that Na+ and Ca2+ are the predominant ionic carriers of inwardly directed injury currents at the cut ends of lamprey spinal cords (Borgens et al., 1980) or squid GAs (Fishman et al., 1995). Our elevation by microdialysis of the axoplasmic concentrations of Na+ and Cl−induced the formation of membranous structures, but at much higher concentrations (to >100 mm) and on a reduced scale compared with elevation of axoplasmic Ca2+ (to >100 μm). Increases in Ca2+ are an appropriate cellular signal to trigger emergency repair of the plasmalemma by vesicles or other membranous structures, because in intact cells Ca2+ levels are so low (<100 nm), Ca2+ is in much greater electrochemical imbalance than any other ion, and changes in internal concentrations of Ca2+ effectively alter other cellular processes (e.g., endocytosis, exocytosis, and membrane fusion) involving vesicles or other membranous structures.
Requirement of the plasmalemma for the formation of injury-induced membranous structures
The formation of injury-induced membranous structures is not observed in desheathed axoplasm (axolemma and the glial sheath removed) bathed in an extracellular Ca2+-free physiological saline. Desheathed axoplasm does not vesiculate after addition of Ca2+ to the Ca2+-free physiological saline bathing the exposed axoplasm. Furthermore, GAs with almost all their axoplasm removed by KI perfusion form many vesicles or other injury-induced membranous structures after the GAs are transected in Ca2+-containing physiological saline. These observations are all consistent with the hypothesis that the axolemmal membrane is necessary for the injury-induced formation of substantial numbers of vesicles or other membranous structures.
Role of endocytosis in the formation of injury-induced membranous structures
All of our data (e.g., Figs. 6-8) are consistent with the hypothesis that many injury-induced membranous structures form from axolemmal infoldings that often produce axoplasmic vesicles by endocytosis. Injury-induced vesicles endocytosing from the axolemma may act as a seed in some preparations for fusion with membranous structures of other origin (see below). Injury-induced endocytosis of the axolemma may also aid sealing by reducing axolemmal surface area, thereby shortening and/or constricting the transected axon (Todora et al., 1994), which in turn may cause axoplasm to flow out of the lesion site. Such bulk axoplasmic flow may passively cause vesicles and other injury-induced membranous structures to move toward the lesion site where they could aggregate and interact with each other and the plasmalemma to form a diffusion barrier, i.e., a membranous seal, as previously described by Eddleman et al. (1997, their Fig. 4).
Injury-induced membranous structures also arise from sources other than the axolemma
In electron micrographs of GAs and MGAs, at >15 min after transection we observe some single-walled structures that appear to have a glial origin in regions where the axolemma and glialemma are no longer continuous, and gliaplasm and axoplasm are intermixed as evidenced by the presence of glial nuclei and rough endoplasmic reticulum near the cut axonal end (Figs. 1, 5). At >15 min after transection, we also see double- and multi-walled structures, especially in regions near the cut end where the axolemma and glialemma are no longer continuous. Some of these structures appear to arise from disrupted mitochondria, SER, RER, and the glial tubular lattice.
These data suggesting a nonaxolemmal origin of some injury-induced membranous structures are consistent with the following observations: (1) in transected squid GAs, some Ca2+-induced vesicles contain Ca2+ channels that are not found in the axolemma but are found in SER (Fishman and Tewari, 1990); (2) in transected pseudomyelinated earthworm MGAs, many injury-induced membranous structures appear to arise from delaminating pseudomyelin layers in the glial sheath (Ballinger et al., 1997); and (3) in micropunctured sea urchin oocytes, a barrier (seal) to dye diffusion forms amid many preformed vesicles, some of which accumulate and fuse or otherwise interact at the lesion site (Terasaki et al., 1997).
All these data from squid, crayfish, and earthworm axons or sea urchin oocytes suggest that membranous structures, which seal plasmalemmal damage, can arise from the damaged cell or adjacent undamaged cells (e.g., glia) as injury-induced membranous structures of plasmalemmal origin (e.g., axolemma, glialemma, and pseudomyelin) or of preformed membranous structures in the cytoplasm (e.g., smooth endoplasmic reticulum, membranes of the glial transverse tubular lattice, and predocked vesicles). That is, these data suggest that neurons and other eukaryotic cells have evolved the ability to use any available membranous structures to seal plasmalemmal damage.
Conservative evolution of molecular mechanisms for vesicular involvement in plasmalemmal sealing and other cell processes
Injury-induced vesiculation is a widely reported phenomenon that occurs in cells of various tissues from animals of different phylogenetic origin (for references, see Eddleman et al., 1997; McNeil and Steinhardt, 1997). A conservative evolution of vesicles and/or other membranous structures to repair plasmalemmal damage in eukaryotes may share some molecular mechanisms with the conserved vesicular mechanisms responsible for intracellular protein transport (Rothman and Wieland, 1996) (e.g., axonal transport and vesicular traffic in the Golgi apparatus), release of transmitters at nerve synapses (Südhof, 1995), growth of regenerating axons (Cheng and Reese, 1988; Ashery et al., 1996), etc. In support of this hypothesis, we note that botulinum toxin B, which disrupts docking of vesicles to the presynaptic membrane (Südhof, 1995), also prevents vesicular sealing of plasmalemmal damage in sea urchin eggs (Steinhardt et al., 1994) and crayfish MGAs (our unpublished observations).
Footnotes
This work was supported by grants from National Institutes of Health (NIH; NS31256) and the State of Texas (Advanced Technology 3658-446). We thank Dr. Raymond J. Lipicky, Dr. Michael Tytell, and Lee DeForke for technical assistance and suggestions. We thank the National Resource Center for Cephalopods at the Marine Biochemical Institute (University of Texas Medical Branch, Galveston, TX) for squid (NIH Grant RR01024), Carl Zeiss, Inc. for the use of equipment, and the Marine Biological Laboratory (Woods Hole, MA) and the Cell Research Institute (University of Texas at Austin, Austin, TX) for the use of facilities.
Correspondence should be addressed to Dr. George D. Bittner, Department of Zoology, The University of Texas at Austin, Austin, TX 78712.
REFERENCES
- 1.Ashery U, Penner R, Spira ME. Acceleration of membrane recycling by axotomy of cultured Aplysia neurons. Neuron. 1996;16:641–651. doi: 10.1016/s0896-6273(00)80083-5. [DOI] [PubMed] [Google Scholar]
- 2.Ballinger ML, Bittner GD. Ultrastructural studies of severed giant and other CNS axons in crayfish. Cell Tissue Res. 1980;208:123–133. doi: 10.1007/BF00234178. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger ML, Blanchette AR, Krause TL, Smyers ME, Fishman HM, Bittner GD. Delaminating myelin membranes help seal the cut ends of severed earthworm giant axons. J Neurobiol. 1997;33:945–960. doi: 10.1002/(sici)1097-4695(199712)33:7<945::aid-neu6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Baumgold J, Terakawa S, Iwasa K, Gainer H. Membrane-associated cytoskeletal proteins in squid giant axons. J Neurochem. 1981;36:759–764. doi: 10.1111/j.1471-4159.1981.tb01653.x. [DOI] [PubMed] [Google Scholar]
- 5.Betz WJ, Mao F, Bewick GS. Activity-dependent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bi G, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131:1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borgens RB, Jaffe LJ, Cohen MJ. Large and persistent electrical currents enter the transected lamprey spinal cord. Proc Natl Acad Sci USA. 1980;77:1209–1213. doi: 10.1073/pnas.77.2.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown ER, Abbott NJ. Ultrastructure and permeability of the Schwann cell layer surrounding the giant axon of the squid. J Neurocytol. 1993;22:283–298. doi: 10.1007/BF01187127. [DOI] [PubMed] [Google Scholar]
- 9.Brown ER, Bone Q, Ryan KP, Abbott NJ. Morphology and electrical properties of Schwann cells around the giant axon of the squids Loligo forbesi and Loligo vulgaris. Proc R Soc London B Biol Sci. 1991;243:255–262. doi: 10.1098/rspb.1991.0039. [DOI] [PubMed] [Google Scholar]
- 10.Cheng TP, Reese TS. Compartmentalization of anterogradely and retrogradely transported organelles in axons and growth cones from chick optic tectum. J Neurosci. 1988;8:3190–3199. doi: 10.1523/JNEUROSCI.08-09-03190.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai J, Sheetz MP. Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys J. 1995;68:988–996. doi: 10.1016/S0006-3495(95)80274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Délèze J. The recovery of resting potential and input resistance in sheep heart injured by knife or laser. J Physiol (Lond) 1970;208:547–562. doi: 10.1113/jphysiol.1970.sp009136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeMello WC. Membrane sealing in frog skeletal muscle fibers. Proc Natl Acad Sci USA. 1973;70:982–984. doi: 10.1073/pnas.70.4.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eddleman CS, Godell CM, Fishman HM, Tytell M, Bittner GD. Fluorescent labeling of the glial sheath of giant nerve fibers. Biol Bull. 1995;189:218–219. doi: 10.1086/BBLv189n2p218. [DOI] [PubMed] [Google Scholar]
- 15.Eddleman CS, Ballinger ML, Smyers ME, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci USA. 1997;94:4759–4764. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 17.Fishman HM, Metuzals J. Ca2+-induced axosome formation in internally dialyzed giant axons of Loligo pealei. Biol Bull. 1993;185:292–293. doi: 10.1086/BBLv185n2p292. [DOI] [PubMed] [Google Scholar]
- 18.Fishman HM, Tewari KP. Single Ca2+ channels in patches of axosomes from transected squid axon. Biophys J. 1990;57:523a. [Google Scholar]
- 19.Fishman HM, Tewari KP, Stein PH. Injury-induced vesiculation and membrane redistribution in squid giant axon. Biochim Biophys Acta. 1990;1023:421–435. doi: 10.1016/0005-2736(90)90135-b. [DOI] [PubMed] [Google Scholar]
- 20.Fishman HM, Krause TL, Miller AL, Bittner GD. Retardation of the spread of extracellular Ca2+ into transected, unsealed squid giant axons. Biol Bull. 1995;189:208–209. doi: 10.1086/BBLv189n2p208. [DOI] [PubMed] [Google Scholar]
- 21.Foissner I. The relationship of echinate inclusions and coated vesicles on wound healing in Nitella flexilis (Characeae). Protoplasma. 1988;142:164–175. [Google Scholar]
- 22.Gallant PE, Hammar K, Reese TS. Cytoplasmic constriction and vesiculation after axotomy in the squid giant axon. J Neurocytol. 1995;24:943–954. doi: 10.1007/BF01215644. [DOI] [PubMed] [Google Scholar]
- 23.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6449–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert DL, Adelman WJ, Jr, Arnold JM. Squid as experimental animals. Plenum; New York: 1990. [Google Scholar]
- 25.Gingell D. Contractile responses at the surface of an amphibian egg. J Embryol Exp Morphol. 1970;23:583–609. [PubMed] [Google Scholar]
- 26.Godell CM, Smyers ME, Eddleman CS, Ballinger ML, Fishman HM, Bittner GD. Calpain activity is essential for the sealing of severed giant axons. Proc Natl Acad Sci USA. 1997;94:4765–4770. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon K, Jeon M. Cytoplasmic filaments and cellular wound healing in amoeba proteus. J Cell Biol. 1975;67:243–249. doi: 10.1083/jcb.67.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krause TL, Fishman HM, Ballinger ML, Bittner GD. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodish H, Baltimore D, Berk A, Zipursky SL, Matsudaira P, Darnell J. Molecular cell biology, Ed 3. Freeman; New York: 1995. [Google Scholar]
- 30.McNeil PL, Steinhardt RA. Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol. 1997;137:1–4. doi: 10.1083/jcb.137.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miyake K, McNeil PL. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol. 1995;131:1737–1745. doi: 10.1083/jcb.131.6.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullins LJ, Brinley FJ., Jr . Internal dialysis in the squid giant axon. In: Gilbert DL, Adelman WJ Jr, Arnold JM, editors. Squid as experimental animals. Plenum; New York: 1990. pp. 133–152. [Google Scholar]
- 33.Nishiye H. The mechanism of Ca2+ action on the healing-over process in mammalian cardiac muscles: a kinetic analysis. Jpn J Physiol. 1977;27:451–466. doi: 10.2170/jjphysiol.27.451. [DOI] [PubMed] [Google Scholar]
- 34.Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 35.Severs NJ, Slade AM, Powel T, Twist VW, Green CR. Integrity of the dissociated adult cardiac myocyte: gap junction tearing and the mechanism of plasma membrane resealing. J Muscle Res Cell Motil. 1990;11:154–166. doi: 10.1007/BF01766494. [DOI] [PubMed] [Google Scholar]
- 36.Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- 37.Strautman AF, Cork RJ, Robinson KR. The distribution of free calcium in transected spinal axons and its modulation by applied electrical fields. J Neurosci. 1990;10:3564–3575. doi: 10.1523/JNEUROSCI.10-11-03564.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 39.Tanner SL, Storm EE, Bittner GD. Protein transport in intact and severed (anucleate) crayfish giant axons. J Neurochem. 1995;64:1491–1501. doi: 10.1046/j.1471-4159.1995.64041491.x. [DOI] [PubMed] [Google Scholar]
- 40.Terasaki M, Miyake K, McNeil PL. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J Cell Biol. 1997;139:63–74. doi: 10.1083/jcb.139.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Todora MA, Fishman HM, Krause TL, Bittner GD. Shortening of a severed squid giant axon is non-uniform and occurs in two phases. Neurosci Lett. 1994;179:57–59. doi: 10.1016/0304-3940(94)90934-2. [DOI] [PubMed] [Google Scholar]
- 42.Viancour TA, Seshan KR, Bittner GD, Sheller RA. Organization of axoplasm in crayfish giant axons. J Neurocytol. 1987;16:557–566. doi: 10.1007/BF01668508. [DOI] [PubMed] [Google Scholar]
- 43.Villegas GM, Villegas R. Squid axon ultrastructure. Curr Top Membr Transp. 1984;22:3–37. [Google Scholar]
- 44.Xie X, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca2+-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziv NE, Spira ME. Axotomy induces a transient and localized elevation of the free intracellular calcium concentration to the millimolar range. J Neurophysiol. 1995;74:2625–2637. doi: 10.1152/jn.1995.74.6.2625. [DOI] [PubMed] [Google Scholar]