

Abstract
A great many animal models for audiogenic seizures have been described. The extent to which these models may provide insight into neuroscience fields such as abnormal locomotor behavior (wild running), seizures and anticonvulsants, and neuroinsults and neuroprotectors is examined here by our study of magnesium deficiency-dependent audiogenic seizures (MDDASs) in adult mice. MDDASs were induced in all of the eight tested adult murine strains and are presented as a sequence of four successive components (latency, wild running, convulsion, and recovery phase periods). Compared with several classic seizure tests, the nutritional MDDAS model responded to low doses of prototype antiepileptic drugs (AEDs), including phenytoin (PHT), carbamazepine (CBZ), phenobarbital (PB), valproic acid (VPA), ethosuximide (ESM), and diazepam (DZP). Modulation by AEDs of the four components of MDDAS indicated that this seizure test was discriminatory, distinguishing between phenytoinergic (PHT, CBZ), GABAergic (PB, VPA, DZP), and ethosuximide (ESM) compounds. Suitability of the MDDAS test for evaluation of neuroprotective compounds was also examined: it showed partial (melatonin) and complete (WEB2170, an anti-PAF agent) reduction of recovery phase by non-anticonvulsant doses of test compounds. These neuroprotective responses were compared with neuroprotective potentials determined in a model of neonatal cerebral injury induced by focal injection of ibotenate (a glutamate analog). WEB2170 and melatonin reduced the size of lesions in white matter, but only WEB2170 protected cortical plate against ibotenate-induced lesions. In addition to the original neuroprotective behavior of WEB2170, studies on the neuroprotectors also supported GABAergic anticonvulsant activity of melatonin in the MDDAS test.
Keywords: antiepileptic drugs, phenytoin, carbamazepine, phenobarbital, ethosuximide, valproic acid, diazepam, nutrition, magnesium deficiency, anticonvulsants, audiogenic seizures, electroshock, MES, pentylenetetrazol, bicuculline, neuroprotectors, picrotoxin, thieno-triazolodiazepine, benzodiazepine, hetrazepine, melatonin, ibotenate, PAF, anti-PAF, WEB2170, GABA, NMDA receptor, discriminatory screening, seizure test
Seizures triggered by an acoustic stimulus (i.e., audiogenic seizures) have been described in laboratory animals in a great many circumstances, including genetic models (Chapman et al., 1984; Neumann and Collins, 1992) and acquired susceptibility. Audiogenic susceptibility may be secondary to auditive alterations caused by noise (Pierson and Liebmann, 1992), acoustic deprivation (Pierson and Swann, 1991), and kanamycin (Pierson and Swann, 1988). This susceptibility may also result from focal infusion in the inferior colliculus of bicuculline (Millan et al., 1986; Bagri et al., 1989; Terra and Garcia-Cairasco, 1992; Tsutsui et al., 1992) and cyclic AMP analogs (Ludvig and Moshe, 1987), or from exposure to H13/04 catecholamide (a tryptophan hydroxylase inhibitor) (Stine and Kellogg, 1982), metaphit (a phencyclidine analog) (Debler et al., 1993;Lipovac et al., 1993), and α-difluoromethylornithine (Gerrish et al., 1993). Withdrawals in alcohol (Faingold and Riaz, 1994, 1995; Riaz and Faingold, 1994) and progesterone (Hom et al., 1993; Voiculescu et al., 1994) have been well documented to favor audiogenic seizures. Other animal models for acquired audiogenic susceptibility include electrolytic lesions of substantia nigra (Doretto and Garcia-Cairasco, 1995), ischemia (Kawai et al., 1995; Schurr et al., 1995), and dietary manipulations such as magnesium deprivation (Bac et al., 1993a,b).
Cerebral imbalance of the major regulatory antagonistic systems, i.e., decreased GABAergic activity associated with increased excitatory aminoacidergic pathways, has been found to play a key role in initiating audiogenic seizures (Engstrom and Woodbury, 1988; Faingold et al., 1992, 1994; Wieraszko and Seyfried, 1993; Cordero et al., 1994;Terra and Garcia-Cairasco, 1994). Mechanisms for audiogenic susceptibility also include reduction of Ca2+ ATPase activity (Palayoor and Seyfried, 1984; Palayoor et al., 1986; Seyfried et al., 1986), cyclic AMP responsiveness (Ludvig et al., 1985), and histaminergic and adrenergic neuronal efficiencies (Jobe et al., 1986;Tuomisto et al., 1987), as well as enhancement of glutamate decarboxylase mRNA expression (Ribak et al., 1993), and, as an adaptive mechanism for defective anion transport, of carbonic anhydrase activity (Woodbury et al., 1984; Engstrom et al., 1986; Faingold et al., 1986;Yu et al., 1986). Abnormal serotonin physiology such as impaired metabolism (Peters and Lack, 1985), receptor distribution (increased density of cortical 5HT2 binding sites) (Jazrawi and Horton, 1989), and binding (Statnick et al., 1996) have also been described.
Circumstances or mechanisms responsible for audiogenic seizures have been the topic of many investigations, and the search for novel conditions or mechanisms is not specifically the subject of this paper. In fact, a previously described condition for audiogenic seizure, i.e., magnesium deficiency-dependent audiogenic seizures (MDDASs) in adult mice, is considered here from the strict point of view of a system for which we document general characteristics and multiple facets. In this respect, it is shown that the MDDAS model represents a seizure test responsive to low doses of antiepileptic drugs (AEDs), displays discriminatory properties toward anticonvulsant activities of AEDs, and provides an original assessment of neuroprotective properties of test compounds. In addition to these interesting multiple facets of the MDDAS model, the present work further supports the GABAergic anticonvulsant activity of melatonin (a neurohormonal indol compound) and originally reports neuroprotective properties of WEB2170 (an anti-PAF agent). Discriminatory effects of AEDs on the abnormal locomotor behavior component of MDDASs, namely wild running, are also pointed out.
MATERIALS AND METHODS
Animals. For the MDDAS tests, eight murine strains, including AKR, BALB/C, B6D2F1, CBA, C57BL/6, C3 Hr/He, DBA/2, and OF1 strains [4-month-old (adult) mice], were purchased from IFFA Credo (Arbresle, France). For each strain, adult mice were randomly divided into groups of 20 mice per cage. Mice were given either magnesium-deficient or standard control magnesium-containing food, i.e., 50 ± 5 mg/kg of Mg or 930 ± 30 mg/kg of Mg synthetic diets, respectively. Diet content in magnesium was monitored by a previously established procedure (Maurois et al., 1989). Body weight gain was similar in control and magnesium-deficient mice. For classic animal seizure tests, albino mice (CF-1 strain, 18–25 gm; Charles River, Wilmington, MA) given a standard animal chow (S/L Custom Lab Diet-7) were used as experimental animals.
Seizure tests. For the MDDAS tests, individual animals were placed in a 9-dm3-volume test chamber (30, 20, and 15 cm for length, width, and height, respectively) and exposed for 15 sec to an auditory signal of 10 ± 0.1 kHz frequency and 100 ± 1 dB intensity. This acoustic stimulus signal was produced by a signal generator and projected via a high-frequency speaker mounted on the roof of the chamber. The noise level was measured close to the animal’s ear by an external decibel-meter probe. Each audiogenic seizure test used only one test chamber. Each animal was subjected to only one audiogenic seizure test during the experimentation. Audiogenic seizure tests were videotaped.
The classic animal seizure tests were conducted according to the anticonvulsant drug development (ADD) program protocol (Krall et al., 1978; Porter et al., 1984; Kupferberg, 1989), including the maximal electroshock seizure (MES) pattern test and three chemical tests: subcutaneous pentylenetetrazol seizure threshold test (sc. Ptz), subcutaneous bicuculline seizure threshold test (sc. Bic), and subcutaneous picrotoxin seizure threshold test (sc. Pic) (White et al., 1995b). The chemical tests measured the capacity of a test compound to provide complete protection against threshold seizures induced by the subcutaneous injection of the CD97 (dose estimated to elicit convulsions in 97% of animals) of each chemical convulsant. The subcutaneous CD97 values of pentylenetetrazol, bicuculline, and picrotoxin in mice were 85, 2.7, and 3.15 mg/kg, respectively (White et al., 1995b).
Antiepileptic drugs. Phenobarbital (PB), ethosuximide (ESM), valproate (VPA), phenytoin (PHT), and diazepam (DZP) were prepared in 0.9% sodium chloride solution. Carbamazepine (CBZ) was administered in a 15% polyethylene glycol (PEG) 400/water mixture (MDDAS test) or in a 0.5% methylcellulose mixture (classic seizure tests). The drugs were administered intraperitoneally in a volume of 0.01 ml/gm body weight to mice at the previously defined time-to-peak drug effect. To determine anticonvulsant potency and neurotoxicity, groups of 10 (MDDAS test) or 8 (classic seizure tests) mice were tested with various doses of the compounds between the limits of 100 and 0% protection or toxicity. Doses of drug required to produce complete prevention of seizures in 50% of mice (ED50) or minimal neurotoxicity in 50% of animals (TD50), and respective 95% confidence intervals, were calculated on a computer program by probit analysis. The protection index (PI) was calculated as the ratio between TD50 and ED50 values.
Minimal neurotoxicity determinations. The median minimal neurotoxic dose (TD50) in mice was determined by the rotorod procedure. The test animal is placed on a 1-inch-diameter knurled plastic rod rotating at 6 rpm. Normal mice can remain on a rod rotating at this speed without any difficulty. Animals are evaluated for neurological deficit (e.g., ataxia, sedation, hyperexcitability) by observing their balance on the device for at least 1 min (CF1 mice) or 2 min (magnesium-deficient mice) in each of three trials.
Ibotenate-induced cerebral injury. The ibotenate-induced cerebral injury model has been described previously (Marret et al., 1995; Gressens et al., 1997). Essentially, at postnatal day 5, Swiss mice pups were anesthetized with ether and kept under a warming lamp. A 26 gauge needle mounted on a 50 μl Hamilton syringe equipped with a calibrated microdispenser was placed intracerebrally according to precise stereotaxic coordinates. Two boluses of 1 μl (10 μg ibotenate/1 μl PBS) were injected at a 30 sec interval, one in the white matter and the other in the cortical plate. The test neuroprotector compounds (i.e., WEB2170 from Boehringer Ingelheim, Ingelheim am Rein, Germany, and melatonin from Sigma, St. Louis, MO) were given by intraperitoneal route within the 3 min after intracerebral ibotenate injection. At postnatal day 10, formol was infused intracardially in killed animals. Brains were removed and post-fixed before histological evaluation on serial coronal sections of ibotenate-induced cerebral lesions was performed.
Statistical analyses. Statistical analyses for classic anticonvulsant tests (Porter et al., 1984) and for ibotenate-induced lesion measurements (Gressens et al., 1997) were performed as previously described. In the MDDAS tests, normal distribution was assessed according to the Shapiro Wilk’s test. Statistical significance was calculated by ANOVA. The difference between the groups was calculated using the Kruskal–Wallis test.
RESULTS
MDDAS: a nutritionally induced seizure model with various components
Adult mice from AKR, BALB/C, B6D2F1, CBA, C57BL/6, C3 Hr/He, DBA/2, and OF1 strains were given either a magnesium-deficient diet or a standard control magnesium-containing diet. For the duration of the treatment period, none of the adult mice (all strains considered) given the standard control diet developed audiogenic seizures. This contrasted with animals receiving magnesium-deficient diets and attested to the magnesium-deficiency dependence of the audiogenic seizure model. Determinations of time periods of magnesium-deficient diet administration that induced susceptibility to audiogenic seizures in 50 and 100% of treated animals are documented in Figure1. DBA/2 mice needed the shortest period (20 d) for induction of the audiogenic seizure susceptibility in all animals, whereas both C57BL/6 and BALB/C mice comparatively required approximately twice the administration period (42 d) of magnesium-deprivation to become completely sensitive (100%) to the audiogenic seizure (Fig. 1). Intermediary periods of magnesium deprivation were necessary for OF1 mice; 22 and 36 d of treatment induced audiogenic seizure susceptibility in 50 and 100% of animals.
Fig. 1.

The MDDAS model: time periods of magnesium deprivation required to induce susceptibility to audiogenic seizures in various strains of mice. Normal time periods of magnesium-deficient diet administration leading to audiogenic seizure susceptibility in 50 and 100% of tested animals are given.
The timing of events characterizing the audiogenic seizure episode was studied in detail in magnesium-deficient adult mice. The general course corresponded exactly to that described in the literature by Seyfried et al. (1986) for the genetic models of audiogenic seizures and consisted of a progressive sequence of phases. There was a short latency period from the initiation of sound stimulation to the onset of an explosive burst of wild, frenzied running. The wild running phase progressed into a clonic sequence, starting with the fall of the mouse on its side, characterized by violent kicking movements, followed by a tonic sequence during which forelegs and subsequently hindlegs become rigidly extended to the rear. After this clonic–tonic phase, a recovery phase takes place before the animal recovers its normal locomotor activity. The chronology of these events is given in Figure2, with a detailed videotaped illustration of a time frame that includes the end of the wild running, the convulsive phase, and the early recovery phase. Four components of the overall seizure episode induced by sound stimulation were individualized: the latency period, the wild running phase, the convulsive phase, and the recovery phase. Standard values for each of these four components in audiogenic seizures performed in magnesium-deficient adult DBA/2 and OF1 mice are given in Figure3. Moreover, this provides a comparison of values recorded in these two nutritional models with those obtained in the genetic audiogenic seizure model represented by the young (3- to 4-weeks-old) nonmagnesium-deprived DBA/2 mice. Little or no differences were found between the three groups of mice.
Fig. 2.
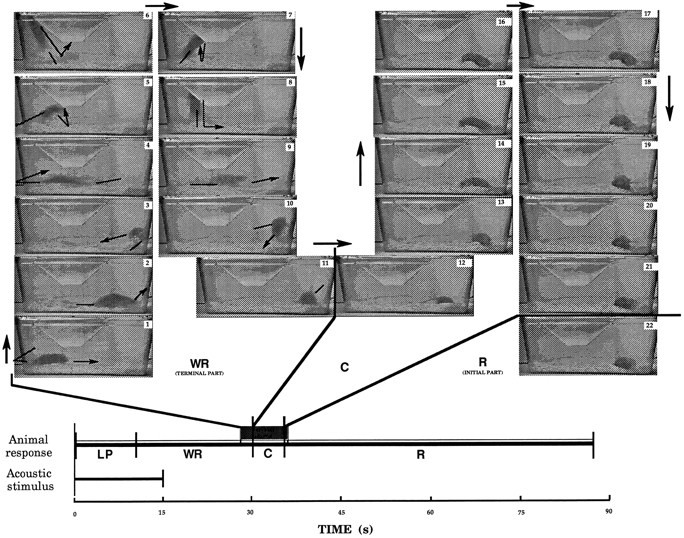
The MDDAS model: chronology of the overall audiogenic seizure episode in magnesium-deficient adult mice. Durations of acoustic stimulus (10 kHz frequency and 100 dB intensity) and the four components of the animal response (LP, latency period; WR, wild running; C, convulsive phase; R, recovery phase) are given at thebottom of the figure and provide an overview of the chronology of the audiogenic seizure in a magnesium-deficient DBA/2 adult mouse (similar data were recorded with magnesium-deficient mice from other strains such as the OF1 strain). The top of the figure shows pictures taken from a videotaped experiment and a time segment including the end of the wild running phase (1–11, with a time span between each of these pictures equal to 0.2 sec), the convulsive phase (12–21, with time spans of ∼0.5 sec between pictures), and the early recovery phase (22). Arrows have been added to help the understanding of presented documents. Arrowsoutside the animal cages refer to the chronological order of events;arrows inside the cages refer to the movements expressed by the mouse during the wild running. A standard laboratory animal cage of 9 dm3 was used. Observe that this cage volume does not restrict the locomotor activity of the mouse during wild running. Note also the remarkable excess of physical energy expressed by the animal during wild running. Other comments are in Results.
Fig. 3.

The MDDAS model: time period values for the four components of the audiogenic seizure episode—comparison with the 21-d-old DBA/2 mice genetic model. The components of the audiogenic seizure episode have been determined in the nutritional MDDAS models (magnesium-deficient adult OF1 and DBA/2 mice) and the genetic model provided by the normally magnesium-fed 21-d-old DBA/2 mice.
MDDAS: a seizure test responsive to low doses of AEDs and endowed with discriminatory properties
Responses of MDDAS to prototype AEDs were studied according to two distinct lines of experimentation. In the first approach (referred to as the MDDAS prevention model), the ability of AEDs to prevent audiogenic seizures in magnesium-deficient animals was studied (protected nonconvulsing animals were considered). In the second approach (referred to as the MDDAS modulation model), effects of AEDs on each of the four components of MDDAS were investigated (convulsing animals were studied).
The MDDAS prevention model (prevention by AEDs of seizures in the MDDAS model)
Various prototype AEDs including PHT, CBZ, PB, VPA, ESM, and DZP were tested for their ability to protect magnesium-deficient mice against audiogenic seizures. Their neurotoxicity in the rotorod test was also measured in the magnesium-deficient animals. Percentages of either anticonvulsant protection (percentage of animals with prevention of audiogenic seizures) or neurotoxicity induced by prototype AEDs as a function of the intraperitoneal dose of drug administered to magnesium-deficient adult DBA/2 and OF1 mice were determined. From these determinations, ED50, TD50, and PI values of AEDs were calculated in the MDDAS model to allow comparison with pharmacological characteristics of the tested drugs in four classic animal seizure tests, including MES, scPTZ, scBic, and scPic tests (Table 1).
Table 1.
Anticonvulsant activities and neurotoxicities of prototype AEDs in MDDAS and classic animal seizure tests
| MDDAS model | Classical animal seizure test models | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drugs | Pharmacological data | Adult OF1 mice | MDDAS | Adult DBA2 mice | MDDAS | Drugs | Pharmacological data | Adult CF1 mice | MES | scPTZ | scBic | scPic |
| PHT | ED50 | 24 | 23 | PHT | ED50 | 9.5 | >300 | >100 | >100 | |||
| TD50 | 70 | 73 | TD50 | 65 | ||||||||
| PI | 3.0 | 3.2 | PI | 6.9 | <0.22 | <0.7 | <0.7 | |||||
| CBZ | ED50 | 25 | 23 | CBZ | ED50 | 8.8 | 102 | 100 | 37 | |||
| TD50 | 90 | 87 | TD50 | 72 | ||||||||
| PI | 3.6 | 3.8 | PI | 8.1 | 0.7 | 0.7 | 1.9 | |||||
| PB | ED50 | 13 | 12 | PB | ED50 | 22 | 13 | 38 | 27 | |||
| TD50 | 76.6 | 72 | TD50 | 69 | ||||||||
| PI | 6.1 | 6.0 | PI | 3.2 | 5.2 | 1.8 | 2.5 | |||||
| VPA | ED50 | 70 | 73 | VPA | ED50 | 272 | 149 | 360 | 387 | |||
| TD50 | 418 | 440 | TD50 | 426 | ||||||||
| PI | 6.0 | 6.0 | PI | 1.6 | 2.9 | 1.2 | 1.1 | |||||
| ESM | ED50 | 73 | 78 | ESM | ED50 | >1000 | 130 | 459 | 243 | |||
| TD50 | 435 | 420 | TD50 | 441 | ||||||||
| PI | 6.0 | 5.4 | PI | <0.44 | 3.4 | 1.0 | 1.8 | |||||
| DZP | ED50 | 0.09 | 0.12 | DZP | ED50 | 19.1 | 0.17 | 1.2 | 1.2 | |||
| TD50 | 2.4 | 2.6 | TD50 | 7.3 | ||||||||
| PI | 26.7 | 21.7 | PI | 0.4 | 43.0 | 6.1 | 6.1 | |||||
ED50 (dose ensuring an anticonvulsant protection in 50% of tested animals) and TD50 (dose eliciting minimal neurotoxicity signs in 50% of animals submitted to the rotorod test) values are expressed as milligrams per kilogram. PI is equal to the ratio between TD50 and ED50 values. For other determinations on the response of classical convulsant animal models (CF1 mice) to AEDs, see White et al. (1995b).
In the MDDAS test, prevention of seizures induced by AEDs in adult OF1 and DBA/2 mice were essentially similar (Table 1). DZP was by far the most active AED, presenting with an ED50 value of 0.09–0.12 mg/kg and a PI of 22–27. PB was also relatively potent in this test, giving an ED50 of 12–13 mg/kg and a PI of 6.0–6.1. PHT and CBZ gave similar ED50 values (23–25 mg/kg) and comparable PI (3.0–3.2 and 3.6–3.8, respectively). VPA and ESM were less active, behaving roughly similarly in their anticonvulsant protection offered in the MDDAS test, with ED50 values of 70–73 and 73–78 mg/kg, respectively.
The MDDAS model essentially distinguished from classic animal models the low levels of AEDs required to produce anticonvulsant, whereas it maintained good protection indices.
The MDDAS modulation model (modulation by AEDs of the various components of the MDDAS model)
The effect of AEDs—at a dose close to the ED50, exactly between ED40 and ED45—on the various components of the seizure episode was studied in the MDDAS model (Table 2).Grosso modo, the effects of AEDs on the time periods of these components (latency for wild running, wild running, convulsion, and recovery phases) occurred according to different profiles, i.e., those expressed by (1) ESM, (2) DZP, PB, and VPA, and (3) CBZ and PHT, respectively. In fact, with the exception of ESM, all of AEDs tested induced modifications in the various components of the observed seizures (Table 2). The convulsions observed in animals on ESM were indistinguishable from those in animals having received no drugs, despite the fact that the actual number of convulsing animals was reduced by ESM (at a dose preventing seizures in 40–45% of animals). For the other AEDs, the four components of the seizure episode were altered as follows (Table 2 shows statistical significance of variations vs controls). Latency periods for wild running were increased by 1.5-fold for PB, VPA, and DZP, and fivefold for CBZ and PHT. Duration of wild running was increased by PB, VPA, and DZP (133, 137 and 155% of control values, respectively) and, by contrast, was reduced by CBZ and PHT (74 and 62% of control values, respectively) (Table 2). Duration of convulsions was reduced by 28, 35, and 41% for VPA, PB, and DZP, respectively, and by 61 and 67% for PHT and CBZ, respectively. Duration of the recovery phases was also decreased either by 50% for PB, VPA, and DZP or by 90% for CBZ and PHT (Table 2).
Table 2.
Modulation by AEDs of the various components of the seizure episode in the MDDAS model
| Administration of drugs | Latency for wild running | Duration of wild running | Convulsions phase time-period | Recovery phase time-period |
|---|---|---|---|---|
| No drugs | 10.8 ± 0.8 | 20.1 ± 1.5 | 5.4 ± 0.7 | 52.9 ± 1.9 |
| PHT (20 mg/kg) | 52.4 ± 2.9** | 7.6 ± 0.4** | 2.1 ± 0.5** | 6.4 ± 0.7** |
| CBZ (20 mg/kg) | 56.8 ± 2.5** | 5.3 ± 0.6** | 1.8 ± 0.3** | 5.2 ± 0.5** |
| PB (10 mg/kg) | 16.5 ± 1.1* | 26.8 ± 1.8* | 3.5 ± 0.3* | 27.6 ± 1.3** |
| VPA (70 mg/kg) | 15.4 ± 1.5* | 27.5 ± 2.1* | 3.9 ± 0.5* | 28.2 ± 1.6** |
| ESM (70 mg/kg) | 13.5 ± 0.6 | 19.5 ± 1.3 | 5.8 ± 0.6 | 54.3 ± 1.4 |
| DZP (0.10 mg/kg) | 17.3 ± 1.3* | 31.2 ± 1.6* | 3.2 ± 0.3* | 25.4 ± 1.1** |
The various time period determinations are expressed as seconds in convulsing animals at drug dosing, ensuring an anticonvulsant protection in 40–45% and no protection in 55–60% of tested animals. Adult DBA2 mice receiving a magnesium-deficient (50 ppm for 22 d) were used. Essentially, similar results and significant resolutions were obtained on adult OF1 mice given a magnesium-deficient (50 ppm for 42 d) diet.
* p < 0.05 (vs magnesium-deficient mice having received no drugs).
** p < 0.01 (vs magnesium-deficient mice having received no drugs).
MDDAS: a neuroinsult model for the study of neuroprotective properties of compounds
The suitability of the MDDAS test for evaluation of neuroprotective properties resulted from a serendipitous observation made during the screening of drugs for evaluation of anticonvulsant properties. One compound, i.e., WEB2170, presented with a relatively striking behavior in the MDDAS model. Although at the doses tested (5, 10, and 15 mg/kg) this compound was devoid of anticonvulsant activity (neither significant change in the number of convulsing animals nor significant modulatory effect on the latency, wild running, and convulsion time periods was evident), it completely abolished the recovery phase period; all of the test animals actually recovered normal locomotor activity as soon as their convulsions were terminated. As described above, a mean recovery phase of ∼53 sec is normally taking place between the end of seizures and recovery of normal locomotor activity (Figs. 2, 3; Table 2).
WEB2170 is a thieno-triazolodiazepine previously developed by the pharmaceutical industry for anti-PAF properties (Weber and Heuer, 1989). Neuroprotective properties of WEB2170 were not previously reported. Consequently, it was imperative to confirm that the effect observed on WEB2170 in the MDDAS model was compatible with neuroprotection. First, melatonin, a known neuroprotector agent with free radical-scavenging properties, was investigated in the MDDAS model. Second, neuroprotective properties of WEB2170, along with melatonin, were evaluated further in a standardized test using ibotenate focal injection as the cerebral injury.
Evaluation of melatonin in the MDDAS model showed that the neurohormonal indol compound was capable of reducing the recovery phase time period (<25% reduction at 10 mg/kg). Another phenomenon was that increasing the dose of melatonin resulted in the appearance of an anticonvulsant activity, with an ED50 approximated to be 60 mg/kg (TD50 >300 mg/kg). In convulsing animals submitted to that dose, melatonin influenced the various MDDAS components according to the GABAergic (Table 2) profile, with a ∼1.5-fold increase in both latency and wild running time periods and a 1.5-fold decrease in the convulsion time period. The recovery time period was relatively short, with reduction of this component being the combined results from anticonvulsant and neuroprotective properties of melatonin.
Evaluations of neuroprotective properties of five doses (0.002, 0.02, 0.2, 2, and 20 μg) of WEB2170 and melatonin in the ibotenate-induced cerebral injury model are given in Figure4. The sizes of ibotenate-induced lesions were reduced by WEB2170 in both cortical plate (gray matter) and white matter; these two neuroprotective effects occurred for doses higher than 2 and 0.2 μg, respectively. Melatonin was efficient in reducing ibotenate-induced lesions in white matter at doses similar to the efficacious doses of WEB2170. By contrast with the latter compound, at doses tested melatonin was devoid of neuroprotective effect on lesions induced in cortical plate.
Fig. 4.
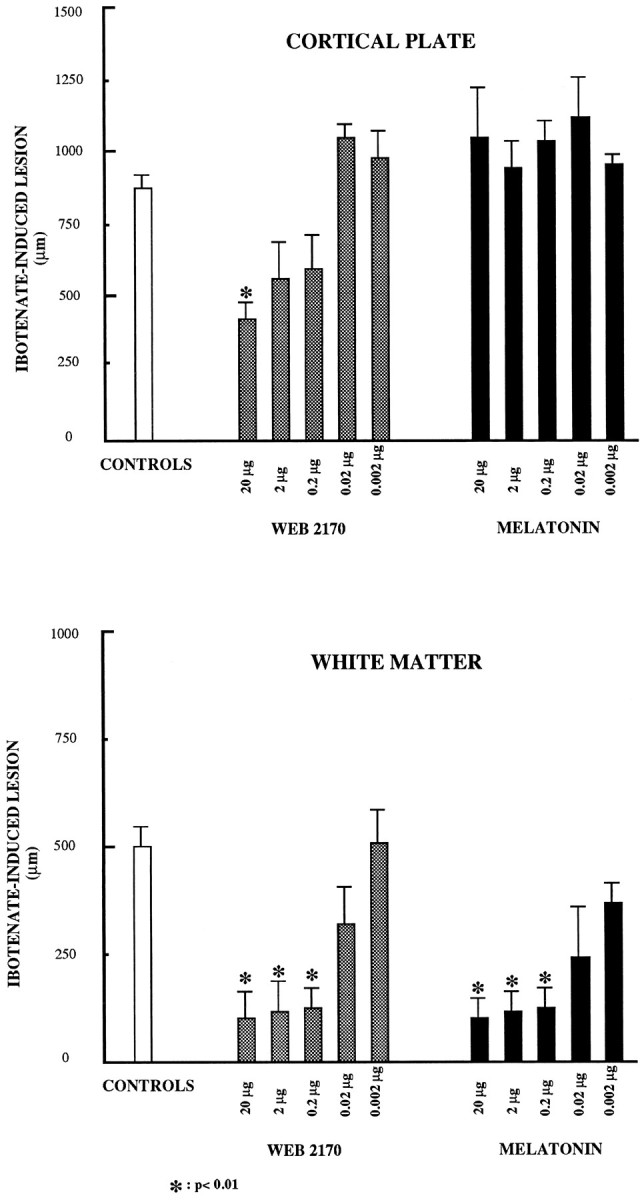
Evaluation of the neuroprotective potentials of WEB2170 and melatonin in a model of cerebral injury induced by focal injection of ibotenate. Cerebral focal injections of ibotenate were performed in mice at postnatal day 5 in the absence (CONTROLS) or the presence of concomitant neuroprotective treatment (either WEB2170 orMELATONIN given by intraperitoneal route). Five doses of neuroprotectors were screened to allow determination of neuroprotection potency offered by test compounds. Reduction of the size of lesions was neuroprotection. Each result is mean value of six separate experimental determinations (±SEM) and is illustrated by histogram representation. Other comments are in Results.
MDDASs: neuroprotective potential of AEDs
AEDs were evaluated in the MDDAS modulation test at the anticonvulsant ED0 (i.e., highest dose tested that was unable to reduce the number of convulsing animals) to determine whether AEDs were endowed with neuroprotective properties in this test. At this dose, GABAergic compounds (PB, VPA, DZP) and ESM had no effect on MDDAS components. Phenytoinergic AEDs (PHT, CBZ) doubled the latency and reduced by 15–20% the wild running period. PHT but not CBZ reduced the convulsion phase time period by 20%. Recovery phase period was reduced by only 8% by PHT, but relatively unexpectedly by >30% by CBZ, an observation that might suggest some neuroprotective potential for the latter compound in the MDDAS test.
MDDASs: miscellaneous
Table 3 provides an overview for major MDDAS data generated by the present experimentation and includes discriminatory properties of the seizure test and suitability for neuroprotector activity evaluation. Table 3 also documents the results obtained with AEDs tested in the MDDAS test at the ED0 and ED100 anticonvulsant doses. At the ED100anticonvulsant dose (i.e., lowest dose tested that reduced by 100% the number of convulsing animals), all four MDDAS components were prevented by phenytoinergic (PHT, CBZ) and ethosuximide AEDs. By contrast, GABAergic compounds (PB, VPA, DZP) were unable to abolish completely the wild running behavior, despite the fact that neither convulsions nor recovery phase were recorded in tested animals. At the ED100 dose, the wild running period was actually increased by GABAergic compounds, and higher doses were required for the abolishment of the wild running component of MDDASs.
Table 3.
Discriminatory properties toward AEDs and neuroprotectors of the MDDAS modulation model: an overview
| Compounds | Main mode of action | MDDAS prevention test (anticonvulsant efficace doses) | MDDAS modulation test (modulation of MDDAS components) | ||||
|---|---|---|---|---|---|---|---|
| Latency | Wild running | Convulsion | Recovery | ||||
| AEDs | |||||||
| Phenytoinergic PHT | Neuronal voltage-dependent sodium channels | ED50 (24 mg/kg) | ⇑⇑ | ⇓⇓ | ⇓⇓ | ⇓⇓⇓ | |
| CBZ | ED50 (25 mg/kg) | ||||||
| GABAergic PB | Interaction with GABAA receptor– chloride ionophore | ED50 (13 mg/kg) | ⇑ | ⇑ | ⇓ | ⇓ | |
| DZP | complex or | ED50 (0.09 mg/kg) | |||||
| VPA | GABA metabolism | ED50 (70 mg/kg) | |||||
| Ethosuximide ESM | Interaction with calcium channels of type T | ED50 (73 mg/kg) | No effect | No effect | No effect | No effect | |
| Neuroprotectors | |||||||
| WEB2170 | Anti-PAF | 10 mg/kg (no anticonvulsant activity at doses tested) | No effect | No effect | No effect | Suppression (100% reduction) | |
| Melatonin | Free radical scavenger melatoninergic receptors | At non-anticonvulsant dose: 10 mg/kg | No effect | No effect | No effect | ⇓ | |
| GABAergic mechanisms(?) | At the anticonvulsant ED50: 60 mg/kg | ⇑ | ⇑ | ⇓ | ⇓⇓ | ||
| Neuroprotective potential of AEDs3-a | |||||||
| GABAergic | |||||||
| PB | ED0 (2.5 mg/kg) | ||||||
| DZP | ED0 (0.01 mg/kg) | ||||||
| VPA | ED0 (30 mg/kg) | ||||||
| ESM | ED0 (30 mg/kg) | No effect | No effect | No effect | No effect | ||
| PHT | ED0 (5 mg/kg) | ⇑⇑ | ↓ | ↓ | • | ||
| CBZ | ED0 (5 mg/kg) | ⇑⇑ | ↓ | No effect | ⇓ | ||
| Miscellaneous3-1002 | |||||||
| Phenytoinergic | |||||||
| PHT | ED100 (80 mg/kg) | ||||||
| CBZ | ED100 (100 mg/kg) | No effect | No effect | No effect | No effect | ||
| ESM | ED100 (100 mg/kg) | ||||||
| GABAergic | |||||||
| PB | ED100 (50 mg/kg) | ⇑ | ⇑ | No effect | No effect | ||
| DZP | ED100 (0.5 mg/kg) | ||||||
| VPA | ED100 (100 mg/kg) | ||||||
Anticonvulsant efficaces doses (ED) in the MDDAS prevention test are defined as follows: ED50 values refer to doses of compounds preventing the convulsive component of MDDASs in 50% of test animals; ED100 values refer to the lowest tested doses preventing the seizure component of MDDAS in 100% of test animals; and ED0 values refer to the highest tested doses of compounds that do not reduce the percentage of convulsing animals. For general information on the main mode of action of AEDs, see MacDonald and Kelly (1995). Note that in practice in MDDAS modulation tests, ED40–ED45 instead of ED50 doses of AEDs were used (see Table 2).
The symbols •, ↓, ⇓, ⇓⇓, and ⇓⇓⇓ signify a decrease in periods of <10%, between 10 and 25%, between 25 and 200%, between 200 and 500%, and >500%, respectively. The symbols ⇑ and ⇑⇑ signify an increase in time periods between 25 and 200% and between 200 and 500%.
MDDAS testing of AEDs at non-anticonvulsant doses.
F3-1002: MDDAS testing of AEDs at dose-preventing seizures in 100% of test animals.
DISCUSSION
MDDASs: biological significance of data
MDDASs represent a nutritional model of neurological disturbances, including abnormal locomotor behavior (wild running), seizures, and cerebral injury. Moreover, MDDASs can be considered a relatively universal model in view of successful induction by magnesium deficiency of audiogenic susceptibility in all adult murine strains tested. Studies using folic acid as a normally excluded systemic agent (data not shown) suggest further that, functionally, the blood–brain barrier (BBB) in magnesium deficiency would be relatively intact. This view is corroborated by the absence of increased neurotoxicity induced in magnesium-deficient animals by AEDs (compare TD50 values, Table 2). These preliminary data, however, could not absolutely rule out limited anatomical lesions of BBB or development of BBB disruption with repetition of seizures. In the present work, no repetition of seizures was performed in the same animal: each animal underwent only one MDDAS test during the whole experimentation.
The responses to low AED doses of MDDAS prevention testing suggest that the procedure is potentially suitable for initial anticonvulsant screening with yes–no type information on the test compounds, but the most important interest in the MDDAS prevention model might rely on the fact that daily doses of AEDs recommended for chronic therapy in human epilepsy are directly proportional, and for each AED they are approximately threefold less than the ED50 determined for acute control of seizures in the nutritional animal model. Therefore, values obtained in this test might well predict therapeutic efficacy in humans.
Discriminatory properties of the MDDAS modulation model toward phenytoinergic (PHT, CBZ), GABAergic (PB, VPA, DZP), and ethosuximide (ESM) AEDs are well supported by the present experimental data. The MDDAS initiation component, i.e., wild running, is generated from the inferior colliculus (IC). IC neurons exhibit extensive firing increases before and during wild running, and silence during the convulsion and recovery phases (Faingold and Randall, 1995). Wild running responds well to AEDs, which inhibit the spread of seizures such as neuronal voltage-dependent sodium channel blockers (phenytoinergic compounds) (Table 2), and consistent with the impairment of GABA inhibition reported in audiogenic seizure models (Faingold et al., 1994), it is not reduced by GABAergic compounds. By contrast, the convulsive phase responds well to GABAergic activity. This might suggest that in the MDDAS model, convulsion and wild running would require distinct cerebral sites or cellular pathways or both. Physiological connections, however, would not be excluded, as suggested by the observation that wild running inhibition through severe locomotor activity reduction succeeded in preventing audiogenic seizures (data not shown). As concluded previously by others (Wada et al., 1995), excessive physical activity might be a step favoring the development of certain forms of seizures.
The suitability of MDDASs for studying neuroprotection is an additional interesting facet. Many neuroinsult models have been developed, among which are manipulations leading to reduced brain oxygenation and perfusion, and local cerebral traumatism or chemicals infusion. In the MDDAS test performed in the absence of specific neuroprotectors, the recovery phase was always taking place after the animals had convulsed, and no recovery phase was observed when animals did not convulse, even if these animals presented with wild running (this particular profile is observed with GABAergic AEDs at the anticonvulsant ED100and adjacent higher doses) (Table 3). Use of the convulsive episode as a cerebral insult is of direct relevant interest in human clinics, especially for cerebral injuries generated by severe convulsive episodes (e.g., status epilepticus), which despite adequate control by therapy and apparently normal brain function may progress secondarily within 24–48 hr into important cerebral damages. This explains in such circumstances the importance of the prognosis made 48 hr after control of the initial cerebral insult.
Exact underlying mechanisms leading to the recovery phase after ictus are not fully understood. Because these mechanisms may be partially (melatonin) or completely (WEB2170) neutralized by neuroprotector compounds, common cerebral injury events such as increased oxidative stress caused by excessive free radical generation may be reasonably hypothesized along with transient edema, a possible resultant of seizure episodes. The intrinsic mode of action of the two neuroprotective compounds shown here to be efficacious in this model should also be taken into account.
Melatonin-induced neuroprotection evidenced by partial neutralization of the recovery phase after ictus of MDDASs and reduction of ibotenate cerebral toxicity might be accounted for by the well known free radical scavenging properties of this compound (Poegeller et al., 1994), although other mechanisms could be involved (Acuna-Castroviejo et al., 1995). Either proconvulsant (Sandik et al., 1992) or anticonvulsant (Champney and Champney, 1992) activities of melatonin have been reported previously. Present data obviously argue in favor of anticonvulsant potential expressed by melatonin. Very importantly, our work provides valuable circumstantial evidence for confirming the recent view supported by neurochemical and behavioral data that melatonin interacts with the GABAergic system (Acuna-Castroviejo et al., 1995; Golombek et al., 1996). Neurological pathological states other than epilepsy may be associated with abnormal GABA physiology (e.g., some forms of depression are characterized by reduced body fluid levels of GABA) (Petty, 1995), and the present results should encourage the use of melatoninergic compounds as potentially efficacious drugs in these disorders.
Neuroprotective properties of WEB2170 were discovered through evaluation in the MDDAS test. This effect was spectacular and led to a complete suppression of the recovery phase. Evaluation in the well standardized ibotenate injury model confirms the potency and interest in this compound by providing evidence of protection not only for white matter but also, by contrast with melatonin, for cortical plate matter. The major generally known mode of action of WEB2170 is anti-PAF activity (Weber and Heuer, 1989). It may be stressed that PAF is an ether phospholipid for which receptors have now been identified in brain on synaptic endings and intracellular membranes (Marcheselli et al., 1990). Very importantly, cerebral PAF concentrations increase during trauma, leading to increased intracellular Ca2+ concentrations (Kumar et al., 1988; Kornecki and Ehrlich, 1991). In fact, PAF can stimulate excitatory amino acid neurotransmission and has no effect on GABA-mediated inhibition (Clark et al., 1992). Moreover, PAF antagonism has neuroprotective potential (Kornecki and Ehrlich, 1988). Neuroprotection by PAF antagonists includes enhanced availability of short ACTH fragments (Maclennan et al., 1995), reduction of PAF-induced increases in intracellular Ca2+ concentrations, and reduced function of excitatory amino acid receptors (Lindsberg et al., 1991). The last mechanisms are consistent with the excellent response to WEB2170 of ibotenate-induced lesions and the MDDAS recovery phase component. Ibotenate is a glutamate analog (originating from the muscarina amanita mushroom) that produces cerebral damages through potent activation of NMDA and metabotropic receptors (Marret et al., 1995). NMDA receptor activation of lesser importance is suggested for the MDDAS model as a result of systemic magnesium deficiency and plausible reduction in magnesium bioavailability for the receptor. Removal (or reduction) of magnesium located in a voltage-dependent site of the ionic channel of NMDA receptor is known to act as a second key; the first key is the agonist (glutamate) of the receptor at the same time that several receptor subsites, such as the glycine site, also contribute to activation of the NMDA receptor (Monoghan et al., 1989).
Study of the neuroprotective potential of AEDs indicates that the MDDAS neuroprotectant evaluation test would be specific for neuroprotectors and distinct from MDDAS anticonvulsant evaluation. CBZ excepted, none of the test AEDs (PHT, PB, VPA, DZP, ESM) gave neuroprotective responses at the anticonvulsant ED0. Regarding CBZ, it is noteworthy that this compound was found to be more efficient as a neuroprotectant than in blocking Na+ currents in the veratridine-induced cell death model (Lakics et al., 1995). In the last experimentation and in the present MDDAS model, the intrinsic neuroprotective potential of CBZ may be concluded to be relatively better than that of PHT.
The mechanisms and experimental conditions leading to susceptibility to audiogenic seizures were reviewed in the introductory remarks. For several of these audiogenic seizures, control by AEDs has been documented previously. During the present experimentation, the multiple facets of the MDDAS model and their biological significance have been extensively developed. The results are encouraging, and subsequent neuroscience research should determine the extent to which the above-cited audiogenic seizure models might compare favorably with the MDDAS test in terms of discriminatory screening of AEDs and suitability for the study of neuroprotective compounds.
Footnotes
This work was supported by the Comité Directeur de la Recherche (University Hospital of Lille and Faculty of Medicine from University of Lille II). P.M. and J.V. are sponsored by the French Institut National de la Santé et de la Recherche Médicale. Experimental data on classic animal seizure models were obtained from the laboratories of Drs. H. Wolfe and S. White (University of Utah), contracted by National Institutes of Health.
Correspondence should be addressed to Dr. J. Vamecq, Institut National de la Santé et de la Recherche Médicale, Domaine du Centre d’Etude et de Recherche Technologique des Industries Alimentaires, 369, Rue Jules Guesde, BP 39, F-59651 Villeneuve d’Ascq Cedex, France.
REFERENCES
- 1.Acuna-Castroviejo D, Escames G, Macias M, Hoyos AM, Carballo AM, Arauzo M, Montes R, Vives F. Cell protective role of melatonin. J Pineal Res. 1995;19:57–63. doi: 10.1111/j.1600-079x.1995.tb00171.x. [DOI] [PubMed] [Google Scholar]
- 2.Bac P, Herrenknecht C, Binet P, Durlach J. Audiogenic seizures in magnesium-deficient mice: effects of magnesium pyrrolidone-2-carboxylate, magnesium acetyltaurinate, magnesium chloride and vitamin B6. Magnes Res. 1993a;6:11–19. [PubMed] [Google Scholar]
- 3.Bac P, Tran G, Paris M, Binet P. Characteristics of audiogenic seizure induced by magnesium deficiency in the mice. C R Acad Sci III. 1993b;316:676–81. [PubMed] [Google Scholar]
- 4.Bagri A, Sandner G, Di Scala G. Effects of unilateral microinjections of GABAergic drugs into the inferior colliculus on auditory evoked potentials and on audiogenic seizure susceptibility. Exp Neurol. 1989;104:82–87. doi: 10.1016/0014-4886(89)90012-5. [DOI] [PubMed] [Google Scholar]
- 5.Champney TH, Champney JCA. Novel anticonvulsant action of chronic melatonin in gerbils. NeuroReport. 1992;3:1152–1154. doi: 10.1097/00001756-199212000-00031. [DOI] [PubMed] [Google Scholar]
- 6.Chapman AG, Croucher MJ, Meldrum BS. Evaluation of anticonvulsant drugs in DBA/2 mice with sound-induced seizures. Arzneimittelforsch. 1984;34:1261–1264. [PubMed] [Google Scholar]
- 7.Clark GD, Happel LT, Zorumski CF, Bazan NG. Enhancement of hippocampal excitatory synaptic transmission by platelet-activating factor. Neuron. 1992;9:1211–1216. doi: 10.1016/0896-6273(92)90078-r. [DOI] [PubMed] [Google Scholar]
- 8.Cordero ML, Ortiz JG, Santiago G, Negron A, Moreira JA. Altered GABAergic and glutamatergic transmission in audiogenic seizure-susceptible mice. Mol Neurobiol. 1994;9:253–258. doi: 10.1007/BF02816124. [DOI] [PubMed] [Google Scholar]
- 9.Debler EA, Lipovac MN, Lajtha A, Zlokovic BV, Dunlop DS, Jacobson AE, Rice KC, de Costa B, Reith ME. Metaphit-induced audiogenic seizures in mice: I. Pharmacologic characterization. Epilepsia. 1993;34:201–210. doi: 10.1111/j.1528-1157.1993.tb02400.x. [DOI] [PubMed] [Google Scholar]
- 10.Doretto MC, Garcia-Cairasco N. Differential audiogenic seizure sensitization by selective unilateral substantia nigra lesions in resistant Wistar rats. Physiol Behav. 1995;58:273–282. doi: 10.1016/0031-9384(95)00050-s. [DOI] [PubMed] [Google Scholar]
- 11.Engstrom FL, Woodbury DM. Seizure susceptibility in DBA and C57 mice: the effects of various convulsants. Epilepsia. 1988;29:389–395. doi: 10.1111/j.1528-1157.1988.tb03736.x. [DOI] [PubMed] [Google Scholar]
- 12.Engstrom FL, White HS, Kemp JW, Woodbury DM. Acute and chronic acetazolamide administration in DBA and C57 mice: effects of age. Epilepsia. 1986;27:19–26. doi: 10.1111/j.1528-1157.1986.tb03496.x. [DOI] [PubMed] [Google Scholar]
- 13.Faingold CL, Randall ME. Pontine reticular formation neurons exhibit a premature and precipitous increase in acoustic responses prior to audiogenic seizures in genetically epilepsy-prone rats. Brain Res. 1995;704:218–226. doi: 10.1016/0006-8993(95)01116-1. [DOI] [PubMed] [Google Scholar]
- 14.Faingold CL, Riaz A. Increased responsiveness of pontine reticular formation neurons associated with audiogenic seizure susceptibility during ethanol withdrawal. Brain Res. 1994;663:69–76. doi: 10.1016/0006-8993(94)90463-4. [DOI] [PubMed] [Google Scholar]
- 15.Faingold CL, Riaz A. Ethanol withdrawal induces increased firing in inferior colliculus neurons associated with audiogenic seizure susceptibility. Exp Neurol. 1995;132:91–98. doi: 10.1016/0014-4886(95)90062-4. [DOI] [PubMed] [Google Scholar]
- 16.Faingold CL, Gehlbach G, Travis MA, Caspary DM. Inferior colliculus neuronal response abnormalities in genetically epilepsy-prone rats: evidence for a deficit of inhibition. Life Sci. 1986;39:869–878. doi: 10.1016/0024-3205(86)90368-1. [DOI] [PubMed] [Google Scholar]
- 17.Faingold CL, Naritoku DK, Copley CA, Randall ME, Riaz A, Anderson CA, Arneric SP. Glutamate in the inferior colliculus plays a critical role in audiogenic seizure initiation. Epilepsy Res. 1992;13:95–105. doi: 10.1016/0920-1211(92)90064-z. [DOI] [PubMed] [Google Scholar]
- 18.Faingold CL, Marcinczyk MJ, Casebeer DJ, Randall ME, Arneric SP, Browning RA. GABA in the inferior colliculus plays a critical role in control of audiogenic seizures. Brain Res. 1994;640:40–47. doi: 10.1016/0006-8993(94)91855-4. [DOI] [PubMed] [Google Scholar]
- 19.Golombek DA, Pevet P, Cardinali DP. Melatonin effects on behavior: possible mediation by the central GABAergic system. Neurosci Biobehav Rev. 1996;20:403–412. doi: 10.1016/0149-7634(95)00052-6. [DOI] [PubMed] [Google Scholar]
- 20.Gerrish KE, Fuller DJ, Gerner EW, Gensler HL. Inhibition of DMFO-induced audiogenic seizures by chlordiazepoxide. Life Sci. 1993;52:1101–1108. doi: 10.1016/0024-3205(93)90431-2. [DOI] [PubMed] [Google Scholar]
- 21.Gressens P, Marret S, Hill JM, Brenneman DE, Gozes I, Fridkin I, Evrard P. Vasoactive intestinal peptide prevents excitotoxic hypoxic-like cell death in the murine developing brain. J Clin Invest. 1997;100:390–397. doi: 10.1172/JCI119545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hom AC, Leppik IE, Rask CA. Effects of estradiol and progesterone on seizure sensitivity in oophorectomized DBA/2J mice and C57/EL hybrid mice. Neurology. 1993;43:198–204. doi: 10.1212/wnl.43.1_part_1.198. [DOI] [PubMed] [Google Scholar]
- 23.Jazrawi SP, Horton RW. 5-HT2 receptor binding and 5-HT uptake in mouse brain: developmental changes and the relationship to audiogenic seizure susceptibility in DBA/2J mice. Brain Res Dev Brain Res. 1989;45:257–263. doi: 10.1016/0165-3806(89)90043-6. [DOI] [PubMed] [Google Scholar]
- 24.Jobe PC, Dailey JW, Reigel CE. Noradrenergic and serotonergic determinants of seizure susceptibility and severity in genetically epilepsy-prone rats. Life Sci. 1986;39:775–782. doi: 10.1016/0024-3205(86)90455-8. [DOI] [PubMed] [Google Scholar]
- 25.Kawai K, Penix LP, Kawahara N, Ruetzler CA, Klatzo I. Development of susceptibility to audiogenic seizures following cardiac arrest cerebral ischemia in rats. J Cereb Blood Flow Metab. 1995;15:248–258. doi: 10.1038/jcbfm.1995.31. [DOI] [PubMed] [Google Scholar]
- 26.Kornecki E, Ehrlich YH. Neuroregulatory and neuropathological actions of the ether-phospholipid platelet activating factor. Science. 1988;240:1792–1794. doi: 10.1126/science.3381103. [DOI] [PubMed] [Google Scholar]
- 27.Kornecki E, Ehrlich YH. Calcium ion mobilization in neuronal cells induced by PAF. Lipids. 1991;26:1243–1246. doi: 10.1007/BF02536540. [DOI] [PubMed] [Google Scholar]
- 28.Krall RL, Penry JK, White BG, Kupferberg HJ, Swinyard EA. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia. 1978;19:409–428. doi: 10.1111/j.1528-1157.1978.tb04507.x. [DOI] [PubMed] [Google Scholar]
- 29.Kumar R, Harvey SAK, Ester MK, Hanahan DJ, Olson MS. Production and effects of platelet-activating factor in the rat brain. Biochim Biophys Acta. 1988;963:375–383. doi: 10.1016/0005-2760(88)90304-9. [DOI] [PubMed] [Google Scholar]
- 30.Kupferberg HJ. Antiepileptic drug development program: a cooperative effort of government and industry. Epilepsia. 1989;30[Suppl 1]:S51–S56. doi: 10.1111/j.1528-1157.1989.tb05815.x. [DOI] [PubMed] [Google Scholar]
- 31.Lakics V, Molnar P, Erdo SL. Protection against veratridine toxicity in rat cortical cultures: relationship to sodium channel blockade. NeuroReport. 1995;7:89–92. [PubMed] [Google Scholar]
- 32.Lindsberg PJ, Hallenbeck JM, Feuerstein G. Platelet-activating factor in stroke and brain injury. Ann Neurol. 1991;30:117–129. doi: 10.1002/ana.410300202. [DOI] [PubMed] [Google Scholar]
- 33.Lipovac MN, Debler EA, Zlokovic BV, Jacobson AE, Rice KC, de Costa B, Selmeci G, Chi L, Reith ME. Metaphit-induced audiogenic seizures in mice: II. Studies on N-methyl-d-aspartic acid, GABA, and sodium channel receptors and on the disposition of metaphit in the brain. Epilepsia. 1993;34:211–219. doi: 10.1111/j.1528-1157.1993.tb02401.x. [DOI] [PubMed] [Google Scholar]
- 34.Ludvig N, Moshe SL. Cyclic AMP derivatives injected into the inferior colliculus induce audiogenic seizure-like phenomena in normal rats. Brain Res. 1987;437:193–196. doi: 10.1016/0006-8993(87)91545-9. [DOI] [PubMed] [Google Scholar]
- 35.Ludvig N, Harsing LG, Hideg J, Vizi ES. Reduced cyclic AMP responsiveness in the colliculus inferior of audiogenic seizure-prone rats. Biochem Pharmacol. 1985;34:2042–2044. doi: 10.1016/0006-2952(85)90333-8. [DOI] [PubMed] [Google Scholar]
- 36.Macdonald RL, Kelly KM. Antiepileptic drug mechanisms of action. Epilepsia. 1995;36[Suppl 2]:S2–S12. doi: 10.1111/j.1528-1157.1995.tb05996.x. [DOI] [PubMed] [Google Scholar]
- 37.Maclennan K, Smith PF, Darlington CL. Ginkgolide B accelerates vestibular compensation of spontaneous ocular nystagmus in guinea pig following unilateral labyrinthectomy. Exp Neurol. 1995;131:272–278. doi: 10.1016/0014-4886(95)90049-7. [DOI] [PubMed] [Google Scholar]
- 38.Marcheselli VL, Rossowska MJ, Domingo M-T, Braquet P, Bazan NG. Distinct platelet-activating factor binding sites in synaptic endings and in intracellular membranes of rat cerebral cortex. J Biol Chem. 1990;265:9140–9145. [PubMed] [Google Scholar]
- 39.Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Maurois P, Gueux E, Rayssiguier Y. Protective effect of severe magnesium deficiency on Plasmodium chabaudi infection. Magnes Res. 1989;2:183–187. [PubMed] [Google Scholar]
- 41.Millan MH, Meldrum BS, Faingold CL. Induction of audiogenic seizure susceptibility by focal infusion of excitant amino acid or bicuculline into the inferior colliculus of normal rats. Exp Neurol. 1986;91:634–639. doi: 10.1016/0014-4886(86)90059-2. [DOI] [PubMed] [Google Scholar]
- 42.Monoghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptor. Their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- 43.Neumann PE, Collins RL. Confirmation of the influence of chromosome 7 locus on susceptibility to audiogenic seizures. Mamm Genome. 1992;3:250–253. doi: 10.1007/BF00292152. [DOI] [PubMed] [Google Scholar]
- 44.Palayoor ST, Seyfried TN. Genetic association between Ca2+-ATPase activity and audiogenic seizures in mice. J Neurochem. 1984;42:1171–1174. doi: 10.1111/j.1471-4159.1984.tb12771.x. [DOI] [PubMed] [Google Scholar]
- 45.Palayoor ST, Seyfried TN, Bernard DJ. Calcium ATPase activities in synaptic plasma membranes of seizure-prone mice. J Neurochem. 1986;46:1370–1375. doi: 10.1111/j.1471-4159.1986.tb01749.x. [DOI] [PubMed] [Google Scholar]
- 46.Peters RI, Lack DB. Audiogenic seizures and brain serotonin after l-tryptophan and p-chlorophenylalanine. Exp Neurol. 1985;90:540–548. doi: 10.1016/0014-4886(85)90151-7. [DOI] [PubMed] [Google Scholar]
- 47.Petty F. GABA and mood disorders: a brief review and hypothesis. J Affect Disord. 1995;34:275–281. doi: 10.1016/0165-0327(95)00025-i. [DOI] [PubMed] [Google Scholar]
- 48.Pierson M, Liebmann SL. Noise exposure-induced audiogenic seizure susceptibility in Sprague Dawley rats. Epilepsy Res. 1992;13:35–42. doi: 10.1016/0920-1211(92)90005-e. [DOI] [PubMed] [Google Scholar]
- 49.Pierson MG, Swann JW. The sensitive period and optimum dosage for induction of audiogenic seizure susceptibility by kanamycin in the Wistar rat. Hear Res. 1988;32:1–10. doi: 10.1016/0378-5955(88)90142-6. [DOI] [PubMed] [Google Scholar]
- 50.Pierson MG, Swann J. Ontogenic features of audiogenic seizure susceptibility induced in immature rats by noise. Epilepsia. 1991;32:1–9. doi: 10.1111/j.1528-1157.1991.tb05602.x. [DOI] [PubMed] [Google Scholar]
- 51.Poeggeler B, Saarela S, Reiter RJ, Tan DX, Chen LD, Manchester LC, Barlow-Walden LR. Melatonin: a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro. Ann NY Acad Sci. 1994;738:419–420. doi: 10.1111/j.1749-6632.1994.tb21831.x. [DOI] [PubMed] [Google Scholar]
- 52.Porter RJ, Cereghino JJ, Gladding GD, Hessie BJ, Kupferberg HJ, Scoville B, White BG. Antiepileptic drug development program. Cleve Clin J Med. 1984;51:293–305. doi: 10.3949/ccjm.51.2.293. [DOI] [PubMed] [Google Scholar]
- 53.Riaz A, Faingold CL. Seizures during ethanol withdrawal are blocked by focal microinjection of excitant amino acid antagonists into the inferior colliculus and pontine reticular formation. Alcohol Clin Exp Res. 1994;18:1456–1462. doi: 10.1111/j.1530-0277.1994.tb01450.x. [DOI] [PubMed] [Google Scholar]
- 54.Ribak CE, Lauterborn JC, Navetta MS, Gall CM. The inferior colliculus of GEPRs contains greater numbers of cells that express glutamate decarboxylase (GAD67) mRNA. Epilepsy Res. 1993;14:105–113. doi: 10.1016/0920-1211(93)90015-y. [DOI] [PubMed] [Google Scholar]
- 55.Sandik R, Tsagas N, Anninos PA. Melatonin as a proconvulsive hormone in humans. Int J Neurosci. 1992;63:125–135. doi: 10.3109/00207459208986662. [DOI] [PubMed] [Google Scholar]
- 56.Schurr A, Payne RS, Reid KH, Iyer V, Tseng MT, Li MM, Chan SA, Young C, Miller JJ, Rigor BM. Cardiac arrest-induced global ischemia studied in vitro. Life Sci. 1995;57:2425–2430. doi: 10.1016/0024-3205(95)02238-7. [DOI] [PubMed] [Google Scholar]
- 57.Seyfried TN, Glaser GH, Yu RK, Palayoor ST. Inherited convulsive disorders in mice. Advances in neurology, Vol 44 (Delgado-Escueta AV, Ward AA, Woodbury DM, Porter RJ, eds), pp 115–133. Raven; New York: 1986. [PubMed] [Google Scholar]
- 58.Statnick MA, Dailey JW, Jobe PC, Broning RA. Abnormalities in 5-TH1A and 5-HT1B receptor binding in severe-seizure genetically epilepsy-prone rats (GEPR-9 sec). Neuropharmacology. 1996;35:111–118. doi: 10.1016/0028-3908(95)00141-7. [DOI] [PubMed] [Google Scholar]
- 59.Stine S, Kellogg C. Inhibition of tryptophan hydroxylase: neurochemical action of catecholamide seizure-induced agent. Neurochem Res. 1982;7:87–98. doi: 10.1007/BF00965072. [DOI] [PubMed] [Google Scholar]
- 60.Terra VC, Garcia-Cairasco N. Neuroethological evaluation of audiogenic seizures and audiogenic-like seizures induced by microinjection of bicuculline into the inferior colliculus. II. Effects of nigral clobazam microinjections. Behav Brain Res. 1992;52:19–28. doi: 10.1016/s0166-4328(05)80321-3. [DOI] [PubMed] [Google Scholar]
- 61.Terra VC, Garcia-Cairasco N. NMDA-dependent audiogenic seizures are differentially regulated by inferior colliculus subnuclei. Behav Brain Res. 1994;62:29–39. doi: 10.1016/0166-4328(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 62.Tsutsui J, Terra VC, Oliveira JA, Garcia-Cairasco N. Neuroethological evaluation of audiogenic seizures and audiogenic-like seizures induced by microinjection of bicuculline into the inferior colliculus. I. Effects of midcollicular knife cuts. Behav Brain Res. 1992;52:7–17. doi: 10.1016/s0166-4328(05)80320-1. [DOI] [PubMed] [Google Scholar]
- 63.Tuomisto L, Tacke U, Willman A. Inhibition of sound-induced convulsions by metoprine in the audiogenic seizure susceptible rat. Agents Actions. 1987;20:252–254. doi: 10.1007/BF02074683. [DOI] [PubMed] [Google Scholar]
- 64.Voiculescu V, Hategan D, Manole E, Ulmeanu A. Increased susceptibility to audiogenic seizures following withdrawal of progesterone. Rom J Neurol Psychiatry. 1994;32:131–133. [PubMed] [Google Scholar]
- 65.Wada Y, Mogi T, Inoue H, Koizumi A. A mouse model of a sudden death induced by noise exposure is useful to investigate human responses to physical stress. Ind Health. 1995;33:29–33. doi: 10.2486/indhealth.33.29. [DOI] [PubMed] [Google Scholar]
- 66.Weber KH, Heuer HO. Hetrazepines as antagonists of platelet activating factor. Med Res Rev. 1989;9:181–218. doi: 10.1002/med.2610090204. [DOI] [PubMed] [Google Scholar]
- 67.Wieraszko A, Seyfried TN. Influence of audiogenic seizures on synaptic facilitation in mouse hippocampal slices is mediated by N-methyl-d-aspartate receptor. Epilepsia. 1993;34:979–984. doi: 10.1111/j.1528-1157.1993.tb02122.x. [DOI] [PubMed] [Google Scholar]
- 68.White HS, Johnson M, Wolf HH, Kupferberg HJ. The early identification of anticonvulsant activity: role of the maximal electroshock and subcutaneous pentylenetetrazol seizure models. Ital J Sci. 1995a;16:73–77. doi: 10.1007/BF02229077. [DOI] [PubMed] [Google Scholar]
- 69.White HS, Woodhead JH, Franklin MR. General principles: experimental selection, quantification, and evaluation of anticonvulsant drugs. In: Levy R, Mattson R, Meldrum BS, editors. Antiepileptic drugs, Ed 4. Raven; New York: 1995b. pp. 99–110. [Google Scholar]
- 70.Woodbury DM, Engstrom FL, White HS, Chen CF, Kemp JW, Chow SY. Ionic and acid-base regulation of neurons and glia during seizures. Ann Neurol [Suppl] 1984;16:S135–S144. doi: 10.1002/ana.410160720. [DOI] [PubMed] [Google Scholar]
- 71.Yu O, Ito M, Chiu TH, Rosenberg HC. GABA-gated chloride ion influx and receptor binding studies in C57BL6J and DBA2J mice. Brain Res. 1986;399:374–378. doi: 10.1016/0006-8993(86)91531-3. [DOI] [PubMed] [Google Scholar]