

Abstract
Prepulse inhibition (PPI), a phenomenon in which a weak prestimulus decreases the startle response to an intense stimulus, provides an operational measure of sensorimotor gating (a process by which an organism filters sensory information) and is diminished in schizophrenia and schizotypal patients. The psychotomimetic phencyclidine and its potent congener dizocilpine are noncompetitive antagonists of the NMDA receptor complex, and they disrupt PPI in rodents, mimicking the clinically observed PPI deficit. The neuroanatomical substrates mediating the PPI-disruptive effects of noncompetitive NMDA antagonists are unknown. The present study sought to identify brain regions subserving the disruption of PPI produced by noncompetitive NMDA antagonists in rats. PPI was measured in startle chambers immediately after bilateral infusion of dizocilpine (0, 0.25, 1.25, and 6.25 μg/0.5 μl/side) into one of six brain regions: amygdala, dorsal hippocampus, medial prefrontal cortex, nucleus accumbens, ventral hippocampus, and dorsomedial thalamus. Dizocilpine significantly decreased PPI after infusion into the amygdala or dorsal hippocampus. A trend toward PPI disruption was observed with administration into medial prefrontal cortex. In contrast, no change in PPI was produced by dizocilpine infusion into nucleus accumbens, ventral hippocampus, or dorsomedial thalamus. Startle reactivity was increased by dizocilpine infusion into amygdala, dorsal hippocampus, nucleus accumbens, and dorsomedial thalamus, but not medial prefrontal cortex. These findings indicate that multiple limbic forebrain regions mediate the ability of noncompetitive NMDA antagonists to disrupt PPI and that the PPI-disruptive and the startle-increasing effects of dizocilpine are mediated by different central sites.
Keywords: startle, prepulse inhibition, schizophrenia, dizocilpine, MK-801, glutamate, phencyclidine, amygdala, nucleus accumbens, medial prefrontal cortex, hippocampus, dorsomedial thalamus
Noncompetitive NMDA antagonists, such as phencyclidine (PCP), are psychotomimetic, producing in healthy humans many of the cognitive and perceptual disturbances that are characteristic of schizophrenia (Javitt and Zukin, 1991; Krystal et al., 1994). Identifying the mechanism by which PCP and related compounds produce their behavioral effects could lead to a better understanding of the neural substrates underlying drug-induced psychotic states and perhaps schizophrenia. To this end, the behavioral effects of PCP-like compounds have been studied extensively in animals. One of the cardinal effects of noncompetitive NMDA antagonists in rodents is the disruption of prepulse inhibition (PPI) (Mansbach and Geyer, 1989, 1991; Dulawa and Geyer, 1996). PPI refers to the normal reduction in the magnitude of the startle response to an intense stimulus when that stimulus is immediately preceded by a prepulse (Hoffman and Ison, 1980). PPI provides an operational measure of sensorimotor gating, one mechanism by which an organism filters information from its surroundings (Braff and Geyer, 1990; Geyer et al., 1990). Several psychiatric populations, including schizophrenia, schizotypal, and obsessive–compulsive disorder patients, that exhibit symptoms that are consistent with deficient central inhibitory processes have lower levels of PPI than healthy control subjects (Braff et al., 1992; Grillon et al., 1992; Cadenhead et al., 1993; Swerdlow et al., 1993; Bolino et al., 1994). Therefore, understanding the mechanism by which PCP-like drugs disrupt PPI might provide insight into the mechanisms underlying deficient sensorimotor gating in humans.
The neuroanatomical substrates mediating the PPI-disruptive effects of noncompetitive NMDA antagonists are unknown. To date, the only study that examined the effects of centrally administered noncompetitive NMDA antagonists on PPI found that low doses of dizocilpine failed to disrupt PPI when infused into the nucleus accumbens (NAcc) “core” (Reijmers et al., 1995). The purpose of the present investigation was to identify the neuroanatomical sites that contribute to the PPI-disruptive effects of the channel-blocking NMDA antagonists in rats by directly microinfusing the noncompetitive NMDA antagonist dizocilpine into several different brain regions and testing for potential decreases in PPI. Six sites were selected for study on the basis of their previously reported involvement in the modulation of PPI, as well as their high levels of PCP binding sites (Suzuki et al., 1995). The amygdala (AM) was chosen because it has been reported that either lesions of this region or infusion of the competitive NMDA antagonist AP-5 into this region disrupts PPI (Wan and Swerdlow, 1997). The dorsal hippocampus (DH) was selected because it contains perhaps the highest concentration of NMDA receptors in the brain and has been found to mediate the disruption of PPI produced by the cholinergic agonist carbachol (Caine et al., 1991). Similarly, multiple lines of evidence implicate the NAcc in the dopaminergic regulation of PPI (Swerdlow et al., 1992). Manipulations of the ventral hippocampus (VH) (Wan et al., 1996), the dorsomedial thalamus (DMT) (Kodsi and Swerdlow, 1997), or medial prefrontal cortex (MPFC) (Koch and Bubser, 1994) can alter PPI, suggesting that these sites might also be good candidates for mediating the PPI-disruptive effects of PCP-like NMDA antagonists.
MATERIALS AND METHODS
Subjects. Sixty-eight experimentally naive male Sprague Dawley rats (Harlan Laboratories, San Diego, CA) were used in the present studies. Rats weighed ∼300 gm at the time of surgery and 320–360 gm at the time of behavioral testing. Animals were housed in clear plastic cages in groups of two to three and were allowed access to food (Lab Chow; Harlan Teklab, Madison, WI) and waterad libitum throughout the experiment. The animal colony was temperature- and humidity-controlled and operated on a reversed 12 hr light/dark cycle (lights on at 7:00 P.M., lights off at 7:00 A.M.). On arrival in the colony, all rats were handled gently by the experimenter every day for 1–2 min each.
Surgery. Four to 7 d after the animals arrived, surgeries were performed to implant bilateral chronic indwelling cannulas aimed at the brain regions of interest. Animals were anesthetized by continuous inhalation of vaporized halothane delivered via a nose cone attached to the stereotaxic apparatus (Kopf Instruments, Tujunga, CA). Six different brain regions were targeted in separate groups of rats (n = 7–10 for each site): AM, DH, MPFC, NAcc, DMT, and VH. The coordinates for DH, NAcc, and DMT were based on the atlas of Pellegrino et al. (1979) and were as follows: DH, anteroposterior (AP), −1.6 mm from bregma; lateromedial (LM), ±1.5 mm from midline; dorsoventral (DV), −2.5 mm from skull surface; NAcc, AP, +3.5 mm from bregma; LM, ±1.7 mm from midline; DV, −5.7 mm from skull surface; DMT, AP, −1.2 mm from bregma; LM, ±0.8 mm from midline; DV, −5.2 mm from skull surface. The coordinates for AM, MPFC, and VH were as follows: AM, AP, −2.6 mm from bregma; LM, ±4.9 mm from midline; DV, −5.4 mm from skull surface; MPFC, AP, +3.0 mm from bregma; LM, ±0.8 mm from midline; DV, −2.2 mm from skull surface; VH, AP, −5.2 mm from bregma; LM, ±5.3 mm from midline; DV, −5.6 mm from skull surface. Coordinates for these latter three sites were derived from the atlas of Paxinos and Watson (1986). The cannulas (stainless steel, 23 gauge; Small Parts, Miami Lakes, FL) were affixed to the skull with stainless steel screws (Small Parts) and light-curable dental cement (Henry Schein, Port Washington, NY). Stainless steel stylets were placed into the cannulas to prevent occlusion. After surgery, animals were given daily health checks in which the experimenter gently handled the rats and replaced missing stylets.
Drug infusion. On all test days, animals were wrapped loosely in a cotton dish towel, and stylets were removed and placed into 70% ethanol. Cannulas were cleaned with a dental broach, and stainless steel injectors (30 gauge; Small Parts) were lowered so that they extended 1.5–3.0 mm below the tips of the cannulas. Thus, the final DV coordinates for the various sites were (in mm below skull surface): AM, 8.4; DH, 4.5; MPFC, 4.2; Nacc, 8.2; DMT, 6.7; VH, 7.6. The injectors were attached to polyethylene tubing, which was attached to 10 μl Hamilton microsyringes (Fisher Scientific, Pittsburgh, PA) that were mounted on a motorized pump. A total of 0.5 μl/side of saline or drug was delivered over 93 sec. The pump was then shut off, and injectors were kept in place for an additional 60 sec to allow for absorption of the injection bolus into the tissue. Injectors were then removed, stylets were replaced, and animals were placed immediately into startle chambers for behavioral testing.
Experimental design. One week after surgery, behavioral testing began. All animals were tested initially in a short baseline startle session (20 trials) immediately after receiving sham infusions (injectors were lowered, but no fluid was delivered) to acclimate them to the infusion and testing procedure. Two to 3 d later, drug infusions commenced. Dizocilpine (MK-801) was used as the central probe, because it has been found upon systemic administration to produce the same effects on PPI as the classic psychotomimetic phencyclidine but is more potent than PCP (Mansbach and Geyer, 1989;Bakshi et al., 1994; Bakshi and Geyer, 1995). In each experiment, several doses of dizocilpine (0, 0.25, 1.25, or 6.25 μg, all in a volume of 0.5 μl/side) were given in a counterbalanced order (Latin Square design) over 4 test days. A minimum of 4 d separated successive tests. Dizocilpine was purchased from Research Biochemicals (Natick, MA) and dissolved in warm isotonic saline.
Behavioral testing. All testing occurred within four startle chambers (San Diego Instruments, San Diego, CA), each consisting of a clear nonrestrictive Plexiglas cylinder resting on a platform inside of a ventilated and illuminated chamber. A high-frequency loudspeaker inside the chamber produced both a continuous background noise of 65 dB and the various acoustic stimuli. As described previously (Mansbach et al., 1988), the whole-body startle response of the animal caused vibrations of the Plexiglas cylinder, which were converted into analog signals by a piezoelectric unit attached to the platform. These signals were then digitized and stored by a microcomputer and interface unit. Weekly calibrations were performed on the chambers to ensure the accuracy of the sound levels and measurements. Sound levels were measured as described previously (Mansbach et al., 1988) using the decibel(A) scale. In the test session, a background noise (65 dB) was presented alone for 5 min and then continued throughout the remainder of the session. The test session used in all of the experiments contained five different trial types and had a duration of 20 min: a “pulse-alone” trial in which a 40 msec 120 dB broadband burst was presented; three “prepulse plus pulse” trials in which 20 msec noises that were either 3, 6, or 12 dB above the background noise were presented 100 msec before the onset of the 120 dB pulse; and a “no stimulus” trial, which included only the background noise. All trial types were presented several times in a pseudorandom order for 52 trials (20 pulse-alone trials and eight each of the remaining trial types). In addition, four pulse-alone trials, which were not included in the calculation of PPI values, were presented at the beginning of the test session to achieve a relatively stable level of startle reactivity for the remainder of the session [based on the observation that the most rapid habituation of the startle reflex occurs within the first few presentations of the startling stimulus (Geyer et al., 1990)]. An average of 15 sec (ranging from 9 to 21 sec) separated consecutive trials. The baseline session used to familiarize rats with the testing procedure consisted of 20 trials (17 120 dB pulse-alone and three prepulse plus pulse trials with a 12 dB prepulse intensity).
Histology. After completion of the experiments, all animals were perfused transcardially with isotonic saline, followed by 10% formalin (Fisher Scientific) to fix the brain tissue. Brains were removed, stored in 10% formalin, and subsequently sliced into 60 μm sections using a sliding microtome with a freezing stand (Leica, Deerfield, IL). Slices were mounted onto slides, stained with cresyl violet, and examined for injector tip placement under a microscope. The experimenter was blind to the behavioral data at the time of histological analysis. Only animals whose injector tip placements fell within the targeted areas were used for data analysis.
Data analysis. The startle response to the 120 dB burst was recorded for each pulse-alone and prepulse plus pulse trial. Two measures were calculated from these data for each animal. First, the amount of PPI was calculated as a percentage score for each prepulse plus pulse trial type: PPI = 100 - [(startle response for prepulse plus pulse trial)/(startle response for pulse-alone trial)] × 100. Because the prepulse intensity factor did not interact significantly with any other factor in any of the experiments, this factor was collapsed by averaging the PPI values for the different prepulse intensities and thereby creating a global PPI score for each animal. Second, startle magnitude was calculated as the average response to all of the pulse-alone trials. Only the data from the first half of the test session were used, because nonspecific diffusion-related effects were noted occasionally in the second half (which occurred ∼15 min after infusion). All PPI and startle magnitude data were analyzed with separate one-factor ANOVA with treatment as a repeated measure. Post hoc analyses were performed using the Newman–Keul’s test. α level was set to 0.05.
RESULTS
Prepulse inhibition
Figure 1, A–F, depicts the effects of dizocilpine infusion into the various brain regions of interest. ANOVA of data from the AM experiment revealed a significant main effect of drug treatment (F(3,15) = 4.26;p < 0.023). Post hoc analyses indicated that this effect was attributable to the highest dose of dizocilpine, which markedly reduced PPI from saline values (p< 0.05) (Fig. 1A). Similarly, a main effect of drug treatment was observed after dizocilpine infusion into the DH (F(3,21) = 3.80; p < 0.026) (Fig. 1B). Further analysis showed that PPI values for the 6.25 μg dose were significantly lower than those for saline (p < 0.05). For the MPFC, a trend toward a main effect of drug treatment was indicated by ANOVA (F(3,27) = 2.07; p < 0.128). Although this trend did not reach statistical significance, examination of Figure 1C reveals a tendency for the high dose of dizocilpine to decrease PPI. In contrast to these findings, ANOVA of PPI data from the VH (F(3,27) = 0.38; NS), NAcc (F(3,15) = 0.51; NS), and DMT (F(3,27) = 1.13; NS) experiments failed to show significant effects of drug treatment. Thus, dizocilpine infusion into the AM, the DH, and perhaps the MPFC decreased PPI, whereas dizocilpine infusion into the VH, the NAcc, or the DMT failed to affect PPI.
Fig. 1.

Effects on prepulse inhibition of dizocilpine microinfusion into AM (A), DH (B), MPFC (C), VH (D), NAcc (E), and DMT (F). Error bars represent SE for each experiment. Doses are in μg/0.5 μl/side. *p < 0.05, compared with saline vehicle.
Startle magnitude
The effects of intracranial dizocilpine infusion on startle magnitude are illustrated in Figure 2,A–F. When infused into the AM, dizocilpine produced a main effect on startle magnitude (F(3,15) = 4.76;p < 0.016). Although this effect appeared to be dose-dependent, Newman–Keul’s test indicated that only at the highest dose did dizocilpine significantly elevate startle magnitude (p < 0.05) (Fig. 2A). Figure2B depicts a smaller but statistically significant increase in startle magnitude after dizocilpine infusion into the DH (F(3,21) = 4.05; p < 0.021).Post hoc analyses revealed again that the highest dose of dizocilpine produced this effect (p < 0.05). In contrast, no effects on startle magnitude were observed after dizocilpine infusion into the MPFC (F(3,27) = 1.24; NS) (Figure 2C). ANOVA of data from the VH experiment indicated a strong trend toward a main effect of drug treatment on startle magnitude (F(3,27) = 2.98;p < 0.052). As had been observed with the AM, ANOVA revealed a significant main effect of dizocilpine treatment on startle magnitude for the NAcc (F(3,15) = 12.32;p < 0.001). Further analysis indicated that this effect was attributable to a marked increase in startle magnitude by the 6.25 μg dose (p < 0.05) (Fig.2E). Finally, infusion of dizocilpine into the DMT also increased startle magnitude, as indicated by a significant main effect of treatment (F(3,27) = 10.32;p < 0.001), as well as post hoc comparisons of means, which indicated that both the medium and high doses elevated startle magnitude (p < 0.05) (Fig.2F). Thus, startle magnitude was increased after dizocilpine infusion into all sites except for the MPFC.
Fig. 2.

Effects on startle magnitude of dizocilpine microinfusion into AM (A), DH (B), MPFC (C), VH (D), NAcc (E), and DMT (F). Error bars represent SE for each experiment. Doses are in μg/0.5 μl/side. *p < 0.05, compared with saline vehicle.
Histology
Figure 3, A–F, depicts the location of injector tip placements in the six different brain regions studied. Although it is possible that subtle neuropathological changes might be produced by relatively high doses of dizocilpine (Olney et al., 1989; Ellison, 1995), it does not appear that the dizocilpine infusions in the present studies produced lesions or excessive necrosis in any of the brain regions examined, as illustrated in the photomicrograph of a representative Nissl-stained section of the AM (Fig. 4). Thus, multiple dizocilpine infusions into the brain may be a functionally viable protocol for studying the behavioral effects of centrally administered noncompetitive NMDA antagonists.
Fig. 3.
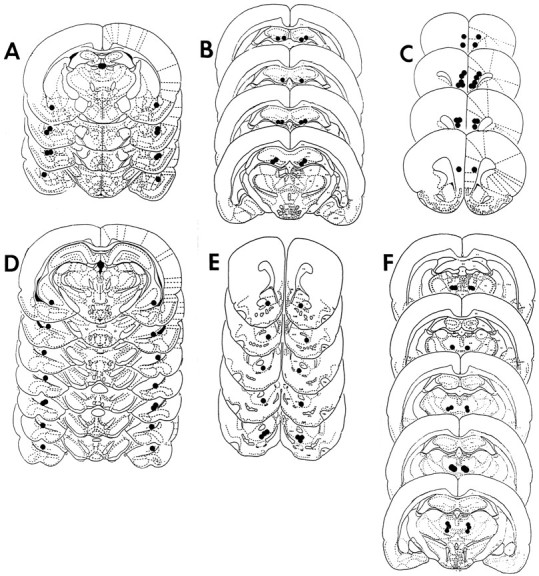
Reconstructions of coronal cross sections depicting the location of injector tips in AM (A), DH (B), MPFC (C), VH (D), NAcc (E), and DMT (F). Eachfilled circle represents injector tip placement for a different animal. Sketches were adapted from either the atlas ofPellegrino et al. (1979) or the atlas of Paxinos and Watson (1986).
Fig. 4.

Photomicrograph of a Nissl-stained coronal section through the AM. Note the lack of necrosis or lesioning at the injection sites after dizocilpine infusion. The tissue integrity of this section is representative of that in all the other brain regions studied.
DISCUSSION
It was found in the present studies that central administration of dizocilpine, a PCP-like noncompetitive NMDA antagonist, disrupts PPI in rats. A significant decrease in PPI was observed after infusion into AM or DH. In contrast, microinfusion of dizocilpine into NAcc, VH, or DMT had no effect on PPI. The lack of effect after infusion into these regions, however, cannot be attributed simply to a paucity of PCP binding sites, because there are high levels of tritiated PCP binding in these sites in quantitative autoradiographic studies (Suzuki et al., 1995). A near-significant decrease in PPI was found after dizocilpine infusion into MPFC. Startle magnitude was increased by dizocilpine infusion into all brain regions except for MPFC, with the largest increases in this measure being noted after AM and NAcc infusions. Together, these results indicate that multiple forebrain limbic regions, including AM, DH, and possibly MPFC, contribute to the disruption of sensorimotor gating produced by noncompetitive NMDA antagonists and that different anatomical sites underlie the PPI-disruptive and startle magnitude-increasing effects of dizocilpine.
It could be argued that the loss of PPI after dizocilpine infusion into AM was an artifact of increased startle reactivity, because startle magnitude was markedly increased by the same dose of dizocilpine that decreased PPI. It is unlikely, however, that such a ceiling effect would fully account for the deficit in PPI, because startle magnitude was increased to a comparable level after dizocilpine infusion into NAcc, but no effect on PPI was observed with dizocilpine infusion into this site. Similarly, intra-DMT infusions produced a smaller but dose-dependent increase in startle magnitude but did not decrease PPI. Thus, increases in startle magnitude, even if they are large, cannot completely explain the disruption of PPI produced by central infusion of dizocilpine. Moreover, a decrease in PPI that was comparable to that seen after intra-AM dizocilpine infusion was observed after dizocilpine administration into DH, but the increase in startle magnitude in this experiment was much smaller than that for the AM experiment. Thus, even in the absence of a large increase in startle magnitude, a significant decrease in PPI was seen with intracranial dizocilpine infusion. The effects on PPI of centrally administered noncompetitive NMDA antagonists can therefore be dissociated from the effects of these compounds on startle magnitude. Moreover, the finding that dizocilpine infusion into some brain regions decreases PPI without markedly increasing startle magnitude (DH) and that dizocilpine infusion into other regions increases startle reactivity without disrupting PPI (NAcc) indicates that perhaps different brain regions subserve these two behavioral effects of systemically administered noncompetitive NMDA antagonists (Mansbach and Geyer, 1989). Dizocilpine administration also has been reported to increase locomotor activity, most likely via a dopaminergic mechanism (French, 1986; Tricklebank et al., 1989;Lehmann-Masten and Geyer, 1991; Bubser et al., 1992; Ouagazzal et al., 1993; Ouagazzal and Amalric, 1995; Narayan et al., 1996). Although locomotor activity was not measured in the present study, it should be noted that the doses of dizocilpine required to elicit hyperactivity are generally appreciably higher than those needed to disrupt PPI, either with systemic or central administration (Mansbach and Geyer, 1989; Lehmann-Masten and Geyer, 1991; Bakshi et al., 1994; Ouagazzal and Amalric, 1995; Narayan et al., 1996). A few studies indicate that dizocilpine infusion into the NAcc increases locomotor activity (Ouagazzal and Amalric, 1995; Narayan et al., 1996); future studies that provide a detailed anatomical mapping of brain regions that mediate dizocilpine-induced hyperactivity will be important in clarifying the degree of homology between the neural substrates underlying dizocilpine-induced PPI deficits and hyperactivity.
The present findings indicate that systemic administration of compounds such as dizocilpine or PCP might result in PPI deficits via actions within AM and/or DH. A possible role for MPFC is also indicated, given the strong trend toward a disruption in PPI after intra-MPFC dizocilpine infusion. Previous work suggests that MPFC is involved in PPI, because lesions of this region disrupt PPI (Bubser and Koch, 1994). The decreases in PPI after dizocilpine infusion into AM or DH provide further evidence for the involvement of these regions in the modulation of PPI, because previous studies have reported disrupted PPI after AM lesions (Wan and Swerdlow, 1997) or intra-DH infusion of the cholinergic agonist carbachol (Caine et al., 1991). Similarly, the lack of disruption in PPI after dizocilpine infusion into VH is consistent with the finding that competitive NMDA antagonists do not disrupt PPI when administered into this region (Wan et al., 1996). VH, however, does influence PPI, because glutamatergic stimulation or septal activation of this site reduces PPI (Koch, 1996; Wan et al., 1996). Thus, it may be that stimulation of VH may be important for PPI, but blockade of intrinsic NMDA receptors within this site is not.
The failure to find a disruption of PPI after dizocilpine infusion into NAcc is consistent with a previous report that no loss of PPI was caused by intra-accumbens infusion of dizocilpine (Reijmers et al., 1995). Interestingly, these same authors reported that high doses of AP-5 decreased PPI; this finding is consistent with a more recent study by Kretschmer and Koch (1997) in which intra-accumbens infusions of AP-5 or the glycine-site antagonist 7-chloro-kynurenate disrupted PPI. Given this apparent discrepancy between the effects of dizocilpine and the other NMDA antagonists, a separate study was conducted in naive animals in which dizocilpine, AP-5, or 7-chloro-kynurenate were infused into the NAcc in a counterbalanced order over several test days. Although the doses used were similar to those used by Kretschmer and Koch (1997), we failed to find a significant deficit in PPI after infusion of any of the three NMDA antagonists (data not shown), although the dizocilpine-induced increase in startle magnitude seen in the present studies was replicated. Perhaps methodological and/or parametric differences [a lower “pulse” intensity and higher “prepulse” intensity was used by Kretschmer and Koch (1997)] contributed to these different results. It should be noted that a difference in the placement of injector tips is not likely to account for the discrepancy between our results and those of Kretschmer and Koch (1997), because in both studies, placements fell approximately within the core of the NAcc.
Alternatively, it may be that different populations of NMDA receptors are targeted by the channel-blocking NMDA antagonists such as dizocilpine and the competitive and glycine-site antagonists such as AP-5 and 7-chloro-kynurenate. Indeed, evidence for the existence of NMDA receptor subtypes has been found recently (Moriyoshi et al., 1991;Monyer et al., 1992; Monaghan and Buller, 1994). Thus, it may be that the channel blocker dizocilpine disrupts PPI via an NMDA receptor subtype located within AM and DH, whereas the glutamate- and glycine-site antagonists produce their effects via actions at a different set of NMDA receptors located within NAcc. Clearly, both the role of NAcc in NMDA-mediated changes in PPI and the possible existence of dizocilpine-preferring NMDA receptors need further investigation. It should be noted, however, that in the present studies, dizocilpine did appear to be behaviorally active within NAcc, because the high dose, which disrupted PPI and increased startle magnitude after intra-AM infusion, markedly increased startle magnitude after infusion into NAcc.
It could be argued that observed decreases in PPI in AM and DH rats might reflect nonspecific diffusion-related effects of dizocilpine, because the dose required to disrupt PPI was very high. The finding, however, that no decrease in PPI was noted after dizocilpine infusion into the VH, which is located in close apposition to the AM, argues strongly against this diffusion hypothesis. Moreover, it was noted that in the second half of the test session (15 min after drug infusion), decreases in PPI were observed in the DMT animals in response to the high dose of dizocilpine (data not shown). This delayed loss of site-specificity could be attributable to diffusion of the drug from the DMT into the lateral ventricle [because intracerebroventricular dizocilpine infusion also disrupts PPI (Bakshi and Geyer, 1994)] or the DH and suggests that diffusion-related nonspecific effects may be possible but occur at a later time point than the one reported in the present results. The dizocilpine-induced decrease in PPI in AM and DH rats was immediate in onset, further arguing against the notion that these decreases were attributable to nonspecific diffusion-related effects.
The amount of PPI reduction produced by dizocilpine infusion into any of these sites was small relative to systemic administrations of noncompetitive NMDA antagonists (Bakshi et al., 1994). It is possible that systemically administered NMDA antagonists reach multiple limbic regions and produce a small disruption of PPI at each site, which additively results in the larger effect observed with systemic administration of these compounds. It has been hypothesized that the regulation of PPI takes place within forebrain regions which, via a complex circuit involving multiple cortical, striatal, pallidal, and thalamic sites (Kodsi and Swerdlow, 1994, 1995, 1997), connect to the primary startle circuit (Davis et al., 1982) at the level of the pons. Manipulations that disrupt transmission within the pedunculopontine tegmentum decrease PPI (Koch et al., 1993; Swerdlow and Geyer, 1993). The small decreases in PPI seen with separate infusions of dizocilpine into AM, DH, or MPFC may summate within this circuitry, perhaps at the level of the pons and thus result in the large PPI disruption seen with systemic administration. Future studies manipulating the neurochemical substrates of PCP-induced PPI deficits within the anatomical regions indicated in the present studies will aid in clarifying the precise neural circuitry mediating the ability of noncompetitive NMDA antagonists to disrupt PPI. Preliminary work along these lines indicates that the antipsychotic Seroquel (quetiapine) or the α-1 adrenergic antagonist prazosin prevent the disruption of PPI that is produced by intra-AM or intra-DH dizocilpine infusion (Bakshi and Geyer, 1998).
The present results clearly point to the involvement of multiple forebrain limbic regions, including AM, DH, and perhaps MPFC, in the regulation of PPI deficits that are produced by channel-blocking NMDA antagonists such as dizocilpine or PCP. A recent study examined the uptake of 2-deoxyglucose after systemic administration of dizocilpine in rats, in a dose range identical to that which disrupts PPI (Bakshi and Geyer, 1995), and found that glucose utilization was markedly elevated in DH and basolateral AM but not affected in anteromedial thalamus or NAcc (Sharkey et al., 1996). This distribution of brain activation almost perfectly mirrors the pattern of results in the present studies, providing converging lines of evidence for the notion that systemic administration of dizocilpine has functional effects in the AM and DH and that these sites could contribute significantly to the disruption of sensorimotor gating produced by noncompetitive NMDA antagonists. The precise mechanism by which AM and DH become activated (as indicated by increased glucose metabolism in these regions) by dizocilpine remains to be determined. It is possible that the NMDA receptors that are blocked by dizocilpine are located on GABAergic interneurons, resulting in a disinhibition of neuronal activity in these areas. Alternatively, it has been found recently that noncompetitive NMDA antagonists can actually induce the release of glutamate within the forebrain, resulting in the activation of non-NMDA glutamatergic receptors (Moghaddam et al., 1997); it may be that dizocilpine administration indirectly activates AM and DH via the release of glutamate in these regions. Clearly, it will be important in future studies to determine whether the PPI-disruptive effects of noncompetitive NMDA antagonists are prevented by non-NMDA glutamate receptor antagonists. It has been suggested previously that limbic regions, including sites indicated in the present studies, contribute to the psychotomimetic effects of noncompetitive NMDA antagonists in humans (Lahti et al., 1995). The present studies constitute an important step toward the elucidation of the neural circuits responsible for the PPI-disruptive effects of psychotomimetics such as PCP and perhaps to the circuits responsible for the similar deficits in sensorimotor gating seen in schizophrenia.
Footnotes
This work was supported by National Institute on Drug Abuse Grant R02-DA02925 and National Institute of Mental Health Grant R37-MH42228. M.A.G. was supported by Research Scientist Award K05-MH01223, and V.P.B. was supported by National Institute of Mental Health Grant F31-MH11636. M.A.G. holds an equity position with San Diego Instruments (San Diego, CA).
Correspondence should be addressed to Mark A. Geyer, Department of Psychiatry, P.O. Box 0804, University of California at San Diego, La Jolla, CA, 92093-0804.
REFERENCES
- 1.Bakshi VP, Geyer MA. Central infusion of MK-801 disrupts prepulse inhibition of the startle response. Soc Neurosci Abstr. 1994;20:1774. [Google Scholar]
- 2.Bakshi VP, Geyer MA. Antagonism of phencyclidine-induced deficits in prepulse inhibition by the putative atypical antipsychotic olanzapine. Psychopharmacology. 1995;122:198–201. doi: 10.1007/BF02246096. [DOI] [PubMed] [Google Scholar]
- 3.Bakshi VP, Geyer MA (1998) Alpha-1 adrenergic receptors mediate sensorimotor gating deficits produced by intracerebral dizocilpine administration in rats. Neuroscience, in press. [DOI] [PubMed]
- 4.Bakshi VP, Swerdlow NR, Geyer MA. Clozapine antagonizes phencyclidine-induced deficits in sensorimotor gating of the startle response. J Pharmacol Exp Ther. 1994;271:787–794. [PubMed] [Google Scholar]
- 5.Bolino F, Di Michele V, Di Dicco L, Manna V, Daneluzzo E, Cassachia M. Sensorimotor gating and habituation evoked by electrocutaneous stimulation in schizophrenia. Biol Psychiatry. 1994;36:670–679. doi: 10.1016/0006-3223(94)91176-2. [DOI] [PubMed] [Google Scholar]
- 6.Braff DL, Geyer MA. Sensorimotor gating and schizophrenia: human and animal model studies. Arch Gen Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- 7.Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- 8.Bubser M, Koch M. Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology. 1994;113:487–492. doi: 10.1007/BF02245228. [DOI] [PubMed] [Google Scholar]
- 9.Bubser M, Keseberg U, Notz PK, Schmidt WJ. Differential behavioural and neurochemical effects of competitive and non-competitive NMDA receptor antagonists in rats. Eur J Pharmacol. 1992;229:75–82. doi: 10.1016/0014-2999(92)90288-f. [DOI] [PubMed] [Google Scholar]
- 10.Cadenhead KS, Geyer MA, Braff DL. Impaired startle prepulse inhibition and habituation in patients with schizotypal personality disorder. Am J Psychiatry. 1993;150:1862–1867. doi: 10.1176/ajp.150.12.1862. [DOI] [PubMed] [Google Scholar]
- 11.Caine SB, Geyer MA, Swerdlow NR. Carbachol infusion into the dentate gyrus disrupts sensorimotor gating of startle in the rat. Psychopharmacology. 1991;105:347–354. doi: 10.1007/BF02244429. [DOI] [PubMed] [Google Scholar]
- 12.Davis M, Gendelman DS, Tischler MD, Gendelman PM. A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci. 1982;2:791–805. doi: 10.1523/JNEUROSCI.02-06-00791.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dulawa SC, Geyer MA. Psychopharmacology of prepulse inhibition in mice. Chin J Physiol. 1996;39:139–146. [PubMed] [Google Scholar]
- 14.Ellison G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res Rev. 1995;20:250–267. doi: 10.1016/0165-0173(94)00014-g. [DOI] [PubMed] [Google Scholar]
- 15.French ED. Effects of N-allylnormetazocine (SKF 10,047), phencyclidine, and other psychomotor stimulants in the rat following 6-hydroxydopamine lesion of the ventral tegmental area. Neuropharmacology. 1986;25:447–450. doi: 10.1016/0028-3908(86)90241-8. [DOI] [PubMed] [Google Scholar]
- 16.Geyer MA, Swerdlow NR, Mansbach RS, Braff DL. Startle response models of sensorimotor gating and habituation deficits in schizophrenia. Brain Res Bull. 1990;25:485–498. doi: 10.1016/0361-9230(90)90241-q. [DOI] [PubMed] [Google Scholar]
- 17.Grillon C, Ameli R, Charney DS, Krystal J, Braff DL. Startle gating deficits occur across prepulse intensities in schizophrenic patients. Biol Psychiatry. 1992;32:939–943. doi: 10.1016/0006-3223(92)90183-z. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman HS, Ison JR. Reflex modification in the domain of startle. I. Some empirical findings and their implications for how the nervous system processes sensory input. Psychol Rev. 1980;87:175–189. [PubMed] [Google Scholar]
- 19.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 20.Koch M. The septohippocampal system is involved in prepulse inhibition of the acoustic startle response in rats. Behav Neurosci. 1996;110:468–477. doi: 10.1037//0735-7044.110.3.468. [DOI] [PubMed] [Google Scholar]
- 21.Koch M, Bubser M. Deficient sensorimotor gating after 6-hydroxydopamine lesion of the rat medial prefrontal cortex is reversed by haloperidol. Eur J Neurosci. 1994;6:1837–1845. doi: 10.1111/j.1460-9568.1994.tb00576.x. [DOI] [PubMed] [Google Scholar]
- 22.Koch M, Kungel M, Herbert H. Cholinergic neurons in the pedunculopontine tegmental nucleus are involved in the mediation of prepulse inhibition of the acoustic startle response in the rat. Exp Brain Res. 1993;97:71–82. doi: 10.1007/BF00228818. [DOI] [PubMed] [Google Scholar]
- 23.Kodsi MH, Swerdlow NR. Quinolinic acid lesions of the ventral striatum reduce sensorimotor gating of acoustic startle in rats. Brain Res. 1994;643:59–65. doi: 10.1016/0006-8993(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 24.Kodsi MH, Swerdlow NR. Prepulse inhibition in the rat is regulated by ventral and caudodorsal striato-pallidal circuitry. Behav Neurosci. 1995;109:912–928. doi: 10.1037//0735-7044.109.5.912. [DOI] [PubMed] [Google Scholar]
- 25.Kodsi MH, Swerdlow NR. Regulation of prepulse inhibition by ventral pallidal projections. Brain Res Bull. 1997;43:219–228. doi: 10.1016/s0361-9230(96)00440-6. [DOI] [PubMed] [Google Scholar]
- 26.Kretschmer BD, Koch M. Role of the strychnine-insensitive glycine binding site in the nucleus accumbens and anterodorsal striatum in sensorimotor gating: a behavioral and microdialysis study. Psychopharmacology. 1997;130:131–138. doi: 10.1007/s002130050220. [DOI] [PubMed] [Google Scholar]
- 27.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 28.Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. NeuroReport. 1995;6:869–872. doi: 10.1097/00001756-199504190-00011. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann-Masten VD, Geyer MA. Spatial and temporal patterning distinguishes the locomotor activating effects of dizocilpine and phencyclidine in rats. Neuropharmacology. 1991;30:629–636. doi: 10.1016/0028-3908(91)90083-n. [DOI] [PubMed] [Google Scholar]
- 30.Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:299–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- 31.Mansbach RS, Geyer MA. Parametric determinants in pre-stimulus modification of acoustic startle: interaction with ketamine. Psychopharmacology. 1991;105:162–168. doi: 10.1007/BF02244303. [DOI] [PubMed] [Google Scholar]
- 32.Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology. 1988;94:507–514. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- 33.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monaghan DT, Buller AL. Anatomical, pharmacological, and molecular diversity of native NMDA receptor subtypes. In: Collingridge GL, Watkins JC, editors. The NMDA receptor. Oxford UP; New York: 1994. pp. 158–176. [Google Scholar]
- 35.Monyer H, Sprengel R, Schopfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sackman B, Seeburg P. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 36.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterisation of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 37.Narayan S, Willins D, Dalia A, Wallace L, Uretsky N. Role of dopaminergic mechanisms in the stimulatory effects of MK-801 injected into the ventral tegmental area and the nucleus accumbens. Pharmacol Biochem Behav. 1996;54:565–573. doi: 10.1016/0091-3057(95)02220-1. [DOI] [PubMed] [Google Scholar]
- 38.Olney JW, Labruyere J, Price MT. Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science. 1989;244:1360–1362. doi: 10.1126/science.2660263. [DOI] [PubMed] [Google Scholar]
- 39.Ouagazzal A, Amalric M. Competitive NMDA receptor antagonists do not produce locomotor hyperactivity by a dopamine-dependent mechanism. Eur J Pharmacol. 1995;294:137–146. doi: 10.1016/0014-2999(95)00518-8. [DOI] [PubMed] [Google Scholar]
- 40.Ouagazzal A, Neoullon A, Amalric M. Effects of D1 and D2 receptor blockade on MK-801-induced hyperlocomotion in rats. Psychopharmacology. 1993;111:427–434. doi: 10.1007/BF02253532. [DOI] [PubMed] [Google Scholar]
- 41.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Marrickville, Australia: 1986. [Google Scholar]
- 42.Pellegrino LJ, Pellegrino AS, Cushman AJ. A stereotaxic atlas of the rat brain. Plenum; New York: 1979. [Google Scholar]
- 43.Reijmers LGJE, Vanderheyden PML, Peeters BWMM. Changes in prepulse inhibition after local administration of NMDA receptor ligands in the core region of the rat nucleus accumbens. Eur J Pharmacol. 1995;272:131–138. doi: 10.1016/0014-2999(94)00629-l. [DOI] [PubMed] [Google Scholar]
- 44.Sharkey J, Ritchie IM, Butcher SP, Kelly JS. Comparison of the patterns of altered cerebral glucose utilisation produced by competitive and non-competitive NMDA receptor antagonists. Brain Res. 1996;735:67–82. doi: 10.1016/0006-8993(96)00574-4. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki T, Yamamoto T, Hori T, Abe S, Moroji T, Shiraishi H, Ito T, Ho IK. Quantitative autoradiographic localization of [3H]3-OH-PCP (1-(1(3-hydroxyphenyl)cyclohexyl)piperidine) binding sites in rat brain. Brain Res Bull. 1995;37:431–435. doi: 10.1016/0361-9230(95)00005-4. [DOI] [PubMed] [Google Scholar]
- 46.Swerdlow NR, Geyer MA. Prepulse inhibition of acoustic startle in rats after lesions of the pedunculopontine nucleus. Behav Neurosci. 1993;107:104–117. doi: 10.1037//0735-7044.107.1.104. [DOI] [PubMed] [Google Scholar]
- 47.Swerdlow NR, Caine SB, Braff DL, Geyer MA. The neural substrates of sensorimotor gating of the startle reflex: a review of recent findings and their implications. J Psychopharmacol. 1992;6:176–190. doi: 10.1177/026988119200600210. [DOI] [PubMed] [Google Scholar]
- 48.Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL. A preliminary assessment of sensorimotor gating in patients with OCD. Biol Psychiatry. 1993;33:298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- 49.Tricklebank MD, Singh L, Oles RJ, Preston C, Iversen SD. The behavioural effects of MK-801: a comparison with antagonists acting non-competitively and competitively at the NMDA receptor. Eur J Pharmacol. 1989;167:127–135. doi: 10.1016/0014-2999(89)90754-1. [DOI] [PubMed] [Google Scholar]
- 50.Wan FJ, Swerdlow NR. The basolateral amygdala regulates sensorimotor gating of acoustic startle in the rat. Neuroscience. 1997;76:715–724. doi: 10.1016/s0306-4522(96)00218-7. [DOI] [PubMed] [Google Scholar]
- 51.Wan FJ, Caine SB, Swerdlow NR. The ventral subiculum modulation of prepulse inhibition is not mediated via dopamine D2 or nucleus accumbens non-NMDA glutamate receptor activity. Eur J Pharmacol. 1996;314:9–18. doi: 10.1016/s0014-2999(96)00535-3. [DOI] [PubMed] [Google Scholar]