

Abstract
We have shown previously that the synergistic interaction of acidic fibroblast growth factor (aFGF) and a coactivator (dopamine, protein kinase A, or protein kinase C activator) will induce the novel expression of tyrosine hydroxylase (TH) in neurons of the developing striatum. In this study we sought to determine whether, concomitant with TH expression, there were unique changes in transcription factors binding the AP-1 regulatory element on the TH gene. Indeed, we found a significant recruitment of proteins into TH–AP-1 complexes as well as a shift from low- to high-affinity binding. Supershift experiments further revealed dramatic changes in the proteins comprising the AP-1 complexes, including recruitment of the transcriptional activators c-Fos, a novel Fos protein, Fos-B, and Jun-D. Concomitantly, there was a decrease in repressor-type factors ATF-2 and CREM-1. aFGF appeared to play a central but insufficient role, requiring the further participation of at least one of the coactivating substances. Experiments examining the signal transduction pathway involved in mediating these nuclear events demonstrated that the presence of only an FGF (1, 2, 4, 9) competent to induce TH caused the phosphorylation of mitogen-activated protein kinase (MAPK). Moreover, the treatment of cells with MEK/ERK inhibitors (apigenin or PD98059) eliminated TH expression and the associated AP-1 changes, suggesting that MAPK was a critical mediator of these events. We conclude that, during transdifferentiation, signals may be transmitted via MAPK to the TH–AP-1 site to increase activators and reduce repressors, helping to shift the balance in favor of TH gene expression at this and possibly other important regulatory sites on the gene.
Keywords: acidic fibroblast growth factor, dopamine, protein kinase A, tyrosine hydroxylase, striatal neurons, AP-1
The catecholamine (CA) neurotransmitters play a pivotal role in mediating normal brain function. Because CA production depends, in large measure, on synthesis by the rate-limiting enzyme tyrosine hydroxylase (TH), understanding TH regulatory mechanisms has become a major enterprise in contemporary neurobiology. The discovery in our laboratory of a novel way in which to trigger expression of the TH gene in developing neurons has provided us with a unique approach. Some years ago we demonstrated that striatal neurons remain phenotypically plastic for a brief period in their differentiation. During this window of opportunity, striatal neurons, which are normally GABAergic, can be coaxed to transdifferentiate into TH-expressing/dopamine (DA)-producing cells if they are exposed to the appropriate cues in culture (Du et al., 1994; Max et al., 1996). Although these cues were identified first in muscle extract (Iacovitti et al., 1989), their effects later were attributed to the synergistic interaction of a specific growth factor and a second obligatory coactivating substance. Thus, the coadministration of acidic fibroblast growth factor (aFGF), which was essential for the effect, and either a CA (e.g., dopamine) or a protein kinase A (PKA) [e.g., isobutylmethylxanthine (IBMX)/forskolin] or protein kinase C (PKC) [e.g., phorbol 12-myristate 13-acetate (TPA; Sigma)] activator initiated expression of the TH gene from the quiescent state (Du and Iacovitti, 1995, 1997a,b).
Whether the agents that promote the transdifferentiation of striatal neurons in culture also govern the differentiation of DA neuronsin vivo is not yet known. Certainly, aFGF is found locally in the brainstem, and circulating CAs have free access to the brain during the period when DA neurons first differentiate (Fu et al., 1991;Schnürch and Risau, 1991; Wilcox and Unnerstall, 1991; Nurcombe et al., 1993). However, their mere presence in the embryonic brain is not proof of their physiological role in DA differentiation. It is possible that these agents simply mimic physiological processes by activating pathways in common with the relevant endogenous substances. Regardless of their role in development, defining what is needed to express the TH gene in our system may provide critical insight into reproducing that expression in neuronal stem cells for therapeutic use (Iacovitti and Stull, 1997).
In this paper we therefore began by exploring where on the TH gene aFGF and the coactivators produce their crucial effects and the signaling pathways that are traveled to reach these gene targets. One possibility is that all TH-inducing agents produce a common transcriptional response after cytosolic convergence of their individual signaling pathways. One likely mediator for such a union is mitogen-activated protein kinase (MAPK), which serves as a major relay station for the merging of intracellular traffic (Ray and Sturgill, 1987; L’Allemain et al., 1991) and which, in other systems, transmits signals initiated by aFGF, kinases, etc. (Sutherland et al., 1993; Zhan et al., 1994). Regardless of the path traveled, the signals ultimately must mediate their effects by modifying the transcriptional machinery of the TH gene. Although there are many regulatory elements that may be involved in transcriptional activation, we began our studies with the AP-1 site, which has been critically implicated in cell-specific and growth factor/kinase-regulated expression of TH in PC12 cells (Gizang-Ginsberg and Ziff, 1990, 1994; Carroll et al., 1991; Fung et al., 1992; Yoon and Chikaraishi, 1992; Kim et al., 1993a,b, 1994; Best et al., 1995;Lazaroff et al., 1995). Our goals in this study, therefore, were twofold. First, we sought to determine whether, coincident with the novel expression of TH in striatal neurons, differentiation agents (aFGF, coactivators) produced unique changes in the transcription factors binding to the AP-1 site of the TH gene. Second, we wondered whether those exogenous signals (or their intermediaries) reached the AP-1 site after relay through the MAPK cascade.
MATERIALS AND METHODS
Materials. aFGF was a kind gift of Rhône-Poulenc Rorer (Collegeville, PA). All other FGFs were purchased from R & D Systems (Minneapolis, MN). MAPK inhibitors were supplied by Calbiochem (La Jolla, CA). Antibodies to transcription factors (with no overlapping specificities) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) as follows: c-Fos (catalog number 52), Fos-B (number 48), Fra-1 (number 183), Fra-2 (number 171), c-Jun (number 822), p-c-Jun (number 1694), Jun-B (number 73), Jun-D (number 74), CREB-1 (number 186), CREB-2 (number 200), CBP (number 369), CREM-1 (number 440), ATF-1 (number 243), ATF-2 (number 187), ATF-3 (number 188), and ATF-4 (number 244). All tissue culture reagents were purchased from Life Technologies (Gaithersburg, MD).
Cell culture. Pregnant Sprague Dawley rats were purchased from Taconic Lab Animals at 9 d gestation ± 12 hr fertilization day, equal to embryonic day 0 (E0). Pregnant dams were anesthetized with pentobarbital on E14, and the embryos were removed. The developing striatum was isolated as described previously (Iacovitti, 1991); meninges were removed and incubated in Ca2+, Mg2+-free HBSS (CMF-HBSS) for 8 min at 37°C in a clinical rotator (40 rpm). The incubation mixture was replaced with 0.01% trypsin in CMF-HBSS. Trypsinization was halted after 8 min, and the tissue was rinsed twice in Leibovitz medium (L-15) and placed in culture medium containing DMEM, 10% fetal calf serum (Irvine Scientific, Santa Ana, CA), glucose (6 mg/ml), glutamine (204 μg/ml), and penicillin/streptomycin (100 U/ml). Cells were dissociated by trituration and plated onto plastic tissue culture dishes previously coated with 0.01 mg/ml polymerized polyornithine. The cellular plating density was ∼2.5–5.0 × 104cells/cm2. After a 1 hr stabilization period in standard serum-containing media, the cultures were rinsed and incubated overnight in a chemically defined serum-free medium (DM) containing 50% DMEM, 50% Ham’s F12 media, 1% ITS+ (Life Technologies), glucose (6 mg/ml), glutamine (204 μg/ml), and penicillin/streptomycin (100 U/ml). The next day the cultures were treated with one or all of the following: 10 ng/ml aFGF, 200 nm TPA, 20 μm DA, and 0.25 mmIBMX plus 50 μm forskolin or as otherwise indicated in the text. For critical period experiments the cultures of either E18 cells or E14 cells aged 5 d in vitro were used; both yielded identical gel shift patterns.
Immunocytochemistry. The day after treatment the cultures were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and processed with antibodies to TH (1:5000 dilution; kind gift of Dr. T. H. Joh, Cornell University Medical College, New York, NY), using the immunoperoxidase/ABC method of staining (Elite Vectakit, Vector Laboratories, Burlingame, CA). TH expression was determined by counting positively stained cells in 50% of the microscopic fields on the culture dish, using an eyepiece reticle at 10× magnification. Cultures were scored for TH induction as described previously (Iacovitti et al., 1989; Du et al., 1995), and the results were expressed as the percentage of total neurons counted in the same microscopic fields.
Nuclear protein extraction. After various treatments as indicated in the text, the cells were rinsed twice with 10 ml of 10 mm PBS, pH 7.4, and were scraped gently from the plates into 1.5 ml of homogenization buffer [0.25 m sucrose plus (in mm) 15 Tris-HCl, pH 7.9, 60 KCl, 15 NaCl, 5 EDTA, 1 EGTA, 0.15 spermine, 0.5 spermidine, 1 DTT, and 0.1 PMSF with 2 μg/ml leupeptin and 5 μg/ml aprotinin]. Cells were homogenized with a Dounce homogenizer. Nuclei were pelleted by centrifugation at 4000 × g for 10 min at 4°C. The nuclear pellet was resuspended in 1 ml of nuclear extraction buffer [0.5 m HEPES, pH 7.9, and 0.5 m KCl plus (in mm) 0.75 MgCl2, 0.5 EDTA, 1 DTT, and 0.1 PMSF with 12.5% glycerol, 2 μg/ml leupeptin, and 5 μg/ml aprotinin] and incubated at 4°C with shaking for 30 min. After 30 min of salt extraction the nuclei were collected by centrifugation at 14,000 × gfor 45 min at 4°C, and the supernatant was dialyzed against dialyzing buffer [(in mm) 10 Tris-HCl, pH 7.9, 1 EDTA, 5 MgCl2, 10 KCl, 1 DTT, and 0.1 PMSF plus 10% glycerol, 2 μg/ml leupeptin, and 5 μg/ml aprotinin] at 4°C for 4 hr. Nuclear proteins were quantitated via the Bradford method at 595 nm.
Gel shift, competition, and supershift assays. Gel shift assays were performed with the following specific DNA probes (only the sense DNA strand is shown) from the rat TH gene: TH–AP-1, 5′ GCTGAGGGTGATTCAGAGG 3′; TH–Hept, 5′ TTGATCTAATGGGACGGAG 3′; and TH–CRE, 5′ GAGGGGCTTTGACGTCAGCCTGG 3′.
Sense and antisense strands of oligonucleotide were annealed into double-stranded oligonucleotides and were 5′ end-labeled with T 4 polynucleotide kinase and [δ32P] ATP. Protein–DNA binding reactions (10 μl) contained 3 μg of nuclear proteins and (in mm) 10 Tris-HCl, pH 7.5, 50 NaCl, 0.5 EDTA, 1 MgCl2, and 0.5 DTT plus 4% glycerol and 0.05 mg/ml poly (dI–dC). After 10 min preincubation at room temperature, 1 μl of 32P-labeled TH–AP-1, etc. probe (0.07 pmol) was added and incubated at room temperature for 20 min. Then the DNA–protein complexes were resolved on a 4% nondenaturing polyacrylamide gel in 0.5× TBE running buffer [(in mm) 44.5 Tris-HCl, pH 8.0, 44.5 boric acid, and 1 EDTA]. All gels were dried and exposed to x-ray film. For competition experiments the amount of nonradioactive oligonucleotide as indicated in the figures was added to each reaction before the addition of labeled probe in buffer. Supershift experiments were performed by using antibodies of established specificity for only one transcription factor, with no cross-reactivity with any other members of a given gene family on Western analysis (see Tables, pp 81 and 85 of Santa Cruz Biotechnology 1998 catalog). Assays were performed by preincubating 1 μl of 1 μg/μl transcription factor antibodies with 3 μg of nuclear extracts for 1 hr on ice in binding reaction buffer before labeled TH-specific probe was added.
Western blotting. Cultures of striatal cells were rinsed, harvested (10 × 106/dish), and homogenized with a Dounce homogenizer in 0.2 ml lysis buffer [containing 1% (w/w) NP-40, 0.5% (w/v) sodium deoxycholate, 0.5% (w/v) sodium vanadate, 0.1% SDS, 0.15 m NaCl and (in mm) 10 PBS, pH 7.2, 2 EDTA, 50 sodium fluoride, 1 DTT, and 0.1 PMSF plus 2 μg/ml leupeptin and 5 μg/ml aprotinin]. The cells were centrifuged at 11,750 × g for 10 min at 4°C. The supernatant was collected and the pellet discarded. Samples containing 20 μg of protein were analyzed by electrophoresis on 15% SDS-polyacrylamide gels and transferred to an Amersham (Arlington Heights, IL) ECL nitrocellulose membrane, using an electroblotting apparatus. The membranes were blocked with Blotto (TBS, 0.5% Tween, and 5% powdered milk) and then incubated in primary antibodies (Fos/Jun and CREB family antibodies; 1:1000 dilution of 1 μg/μl antibodies), followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1000). Immunoreactivity was visualized via enhanced chemiluminescence techniques (Amersham).
RESULTS
Induction of distinct TH–AP-1 complexes in striatal neurons by aFGF and coactivators
The AP-1 like element (TGATTCA) on the TH gene promoter, which differs from the AP-1 consensus sequence by one nucleotide (T), is found at −206 to −200 bp (Fig.1A). To determine whether this AP-1 site indeed was involved in the initiation of TH gene expression in naive (non-TH-expressing) neurons, we performed gel shift assays with TH-specific AP-1 oligonucleotides (indicated byunderlining in Fig. 1A). Thus, E14 rat striatal neurons were grown in DM or in DM containing 10 ng/ml aFGF, 200 nm TPA, 20 μm DA, 0.25 mmIBMX, and 50 μm forskolin. One hour later the cultures were harvested for the gel shift assay. Sister cultures were carried overnight and processed for TH immunocytochemistry for verification of TH induction. As reported previously in mice (Du and Iacovitti, 1997a,b), the combined treatment of striatal neurons with aFGF plus DA, TPA, IBMX, and forskolin resulted in a striking induction of TH expression in E14 rat striatal neurons in 80% of neurons on the dish as compared with controls in which only a few intrinsic TH cells were observed (Fig. 1B).
Fig. 1.

Induction of distinct TH–AP-1 binding protein complexes in E14 striatal neurons after treatment with aFGF and its coactivators. A, Nucleotide sequence of the rat TH gene promoter. The cis-acting DNA sequences for various transcription factors (AP-1, AP-2, E-Box, etc.) are printed inbold. The TATA box is represented in boldand is underlined. The transcription start site is indicated by the arrow. The TH-specific AP-1 and Hept oligonucleotides used in the gel shift and supershift assays are indicated by underlining. Note that the rat TH sequences are derived from Cambi et al. (1989). B, Immunocytochemical localization of TH in cultured striatal neurons stimulated by aFGF and its coactivators. Neurons were established in culture as described previously (Iacovitti et al., 1989, 1991) 1 d before incubation in control media (left panel) or in media supplemented with 10 ng/ml aFGF, 200 nm TPA, 20 μm DA, 0.25 mm IBMX, and 50 μmforskolin (right panel). The next day the cultures were fixed, and TH was localized immunocytochemically. Note that aFGF plus the coactivators produced a striking induction of TH in many cultured striatal neurons. C, Autoradiogram of a representative gel shift of TH–AP-1 binding in rat E14 striatal neurons fed DM (Control) or DM containing aFGF and the coactivators. An end-labeled TH–AP-1 oligonucleotide duplex from −214 to −196 bp of the TH promoter sequences was incubated with 3 μg of nuclear extracts from rat E14 striatal neurons treated with aFGF, DA, TPA, IBMX, and forskolin (concentrations as above) or with control media only. The protein–DNA complexes were resolved on a 4% native polyacrylamide gel, as described in Materials and Methods. There were two bands of AP-1 binding complexes present in both control and induced striatal neurons (lanes 1, 8). The lower band was found predominantly in control cultures (lane 1), whereas the upper band was the primary band in stimulated cultures (lane 8). Oligonucleotide competition assays (lanes 2–7, 9–14) were performed with unlabeled double-stranded oligonucleotides corresponding to TH–AP-1 and TH–Hept (−170 to −152 bp, as shown in A). The lower bands were easily competed by specific TH–AP-1 oligonucleotides, but not by nonspecific TH–Hept oligonucleotides. The bands decreased in intensity with the addition of increasing amounts (50-, 100-, and 200-fold excess) of specific TH–AP-1 oligonucleotides, whereas their intensity remained the same with the addition of excess nonspecific TH–Hept oligonucleotides.
Gel shift assays revealed that two protein complexes (upper and lower bands) bound to the AP-1 site in both control cultures of striatal neurons (Fig. 1C, lane 1) and in those stimulated by aFGF plus the coactivators (Fig. 1C, lane 8). Importantly, however, in control striatal neurons the lower band of AP-1 complexes predominated, whereas the upper band was more striking in striatal neurons stimulated by aFGF and coactivators. To confirm the specificity of both AP-1 complexes, we performed competition experiments. In these studies the lower band of AP-1 complexes was readily competed by 50- to 200-fold molar excess of specific TH–AP-1 oligonucleotides (Fig. 1C, lanes 2–4, 9–11). In contrast, the upper band of AP-1 complexes (Fig. 1C,lanes 2–4, 9–11) could be decreased but not eliminated by 50- to 200-fold molar excess of TH–AP-1 oligonucleotides. This may reflect a higher affinity of these proteins for the AP-1 element or alternatively that some portion of the proteins binds nonspecifically to this site and does not mediate TH differentiation. As anticipated, nonspecific oligonucleotides (TH–Hept) were ineffective competitors of both upper and lower bands (Fig. 1C, lanes 5–7, 12–14), signifying that these were AP-1 sequence-specific protein complexes.
Fos/Jun composition of TH–AP-1 complexes changes after TH induction in striatal neurons
We next wondered whether the changes in AP-1 complexes observed after maximal stimulation reflected differences in their protein composition. To identify the individual components of the TH–AP-1 complexes in control and stimulated neurons, we performed supershift experiments, using specific antibodies to discrete members of the Fos/Jun family that are known to bind to the AP-1 site. Cultures of striatal neurons were grown either in DM or in DM containing 10 ng/ml aFGF, 200 nm TPA, 20 μm DA, 0.25 mm IBMX, and 50 μm forskolin. Neurons were harvested 1 or 6 hr later, and the nuclear extracts were prepared. Then extracts were preincubated with specific Fos/Jun antibodies for 1 hr before analysis of AP-1 binding activity. These supershift experiments (Fig. 2A) revealed that, in controls, the TH–AP-1 protein complexes contained the transcription factors Fos-B, c-Jun, and Jun-D, as evidenced by a shift in their electrophoretic mobility after incubation with their respective antibodies (Fig. 2A, lanes 3, 6, 8). After stimulation by aFGF and the coactivators for 1 hr, there was a dramatic increase in the amount of protein binding to the AP-1 site, including marked increases in Fos-B and Jun-D binding (Fig2A, lanes 11, 16), whereas c-Jun binding remained near constitutive levels (Fig.2A, lanes 6, 14). Importantly, two additional transcription factors (Fig.2A, lane 10, a andb) were recruited into the AP-1 complexes that supershifted with the specific antibody to c-Fos (Santa Cruz Biotechnology catalog number 52). By 6 hr after stimulation, however, c-Fos a and b binding had decreased greatly, returning to nearly control levels, whereas Fos-B and Jun-D binding remained elevated (Fig. 2B). Although control levels of c-Jun were increased greatly at 6 hr, stimulation with aFGF and the coactivators did not influence binding levels further. Absent from the AP-1 complexes at 1 hr (Fig.2A) and 6 hr (Fig. 2B) were Fra-1, Fra-2, and Jun-B in both control (lanes 4, 5, 7) and stimulated striatal cultures (lanes 12, 13, 15).
Fig. 2.
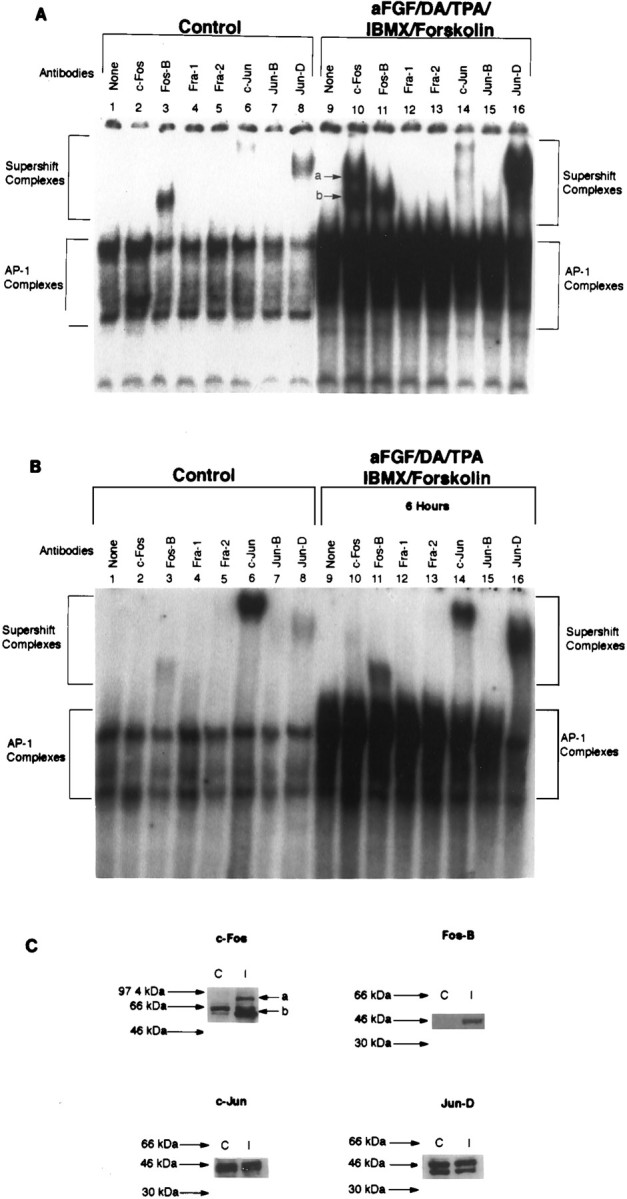
Fos/Jun composition of TH–AP-1 binding protein complexes after TH induction in E14 striatal neurons. A, Autoradiogram of a representative supershift assay of TH–AP-1 binding in rat E14 striatal neurons that were fed DM (Control) or DM containing aFGF and the coactivators. Cultures were generated as described in the Figure 3legend. At 1 hr before the addition of radiolabeled AP-1 probe, either no antibody (None) or 1 μl of antibody to the individual Fos and Jun family members (Santa Cruz Biotechnology) was added to 3 μg of nuclear extract proteins obtained from rat E14 striatal neurons treated with 10 ng/ml aFGF, 20 μm DA, 200 nm TPA, 0.25 mm IBMX, 50 μmforskolin, or control media at 4°C for 1 hr. The protein–DNA complexes were resolved as described in the Figure 3 legend.B, Autoradiogram of a supershift assay of the TH–AP-1 protein complexes formed 6 hr after E14 striatal neurons were stimulated by aFGF and the coactivators. Experiments were conducted with procedures identical to those described in A.C, Immunoblot analysis of control (C) and induced (I) striatal neurons 1 hr after treatment, using the same antibodies as above. Molecular mass (kDa) is indicated by arrows. Two bands of c-Fos immunoreactivity were seen at 62 (band b) and 84 (band a) kDa. c-Jun immunoreactivity was unchanged, whereas Fos-B was increased by stimulation with aFGF and coactivators.
Interestingly, Western blot analysis of these cells at the 1 hr post-treatment time (Fig. 2C) revealed that classic c-Fos (60–62 kDa; band b) was indeed present in control cultures although it had not been detected in the complexes present at the TH–AP-1 site under nonstimulated conditions. As was seen in supershift experiments, however, stimulation by aFGF and the coactivators caused a significant increase in classic c-Fos (bandb) as well as a novel c-Fos-like protein band of ∼84 kDa (band a). Western analysis further revealed that c-Jun expression was unchanged, whereas Fos-B was increased by stimulation with aFGF and coactivators. Interestingly, we did not see a rise in Jun-D expression after stimulation despite the dramatic increase in its binding to the TH–AP-1 site (supershift assay), possibly indicating changes instead in its phosphorylation.
Contribution of aFGF and individual coactivators to the Fos/Jun composition of the TH–AP-1 protein complexes
The studies described above reveal marked changes in Fos/Jun binding at the TH–AP-1 site after maximal induction in TH expression (aFGF plus all coactivators). The next series of supershift experiments was designed to study the individual contributions made by each of these TH-inducing agents to the AP-1 protein complex. Thus, cultures of striatal neurons were grown either in control media or in media containing only one of the following: 10 ng/ml aFGF, 200 nm TPA, 20 μm DA, or 0.25 mm IBMX plus 50 μm forskolin. Neurons were harvested 1 hr later, and nuclear extracts were prepared as described above for supershift analysis or processed for TH immunocytochemistry the next day. As reported previously (Du and Iacovitti, 1995, 1997a,b), neither aFGF nor individual coactivators induced the appearance of immunoreactive TH on their own.
Acidic fibroblast growth factor (aFGF)
Despite the fact that aFGF alone has no TH-inducing capacity, we found, quite remarkably, that it produced all of the same qualitative changes in supershifted TH–AP-1 complexes that were seen when the gene was expressed (i.e., after aFGF plus the coactivators). Consequently, we observed in cultures treated with aFGF alone (Fig.3A) the formation of two supershifted complexes containing c-Fos-like proteins (Fig.3A, lane 10, a and b) as well as increased amounts of Fos-B and Jun-D bound to TH–AP-1 site (lanes 11, 16; control, lanes 3, 8). As seen previously, c-Jun remained unchanged as compared with control (lanes 6, 14), whereas Fra-1, Fra-2, and Jun-B were undetected in both control and stimulated cultures (lanes 4, 5, 7, 12, 13, 15). Although aFGF alone induced the the appearance of the same components in the TH–AP-1 complexes, importantly, the amount of c-Fos, Fos-B, and Jun-D present in the complexes was considerably less than in striatal neurons stimulated by aFGF and all of the coactivators. Therefore, consistent with our previous results (Du et al., 1994; Du and Iacovitti, 1995, 1997a,b), aFGF appears to play a central but insufficient role in initiating TH gene expression, requiring the further participation of other coactivators.
Fig. 3.

Supershift experiments at TH–AP-1 site, using Fos/Jun family antibodies after the treatment of E14 striatal neurons with aFGF alone or individual coactivators. A, Autoradiogram showing the results of a supershift experiment with E14 striatal neurons fed DM (Control) or DM containing 10 ng/ml aFGF. The stimulation of striatal neurons by aFGF resulted in the appearance of two new supershifted c-Fos bands (lane 10a,b) and increased binding of Fos-B (lane 11) and Jun-D (lane 16) at the TH–AP-1 site; c-Jun (lane 14) remained the same intensity as compared with the control. Experiments were conducted as described in the Figure 4A legend. B, Autoradiogram showing the results of a supershift experiment with striatal neurons fed DM (Control) or DM containing 20 μm DA. Only the supershifted complex containing Jun-D (lane 16) increased binding to the AP-1 site in contrast to the control (lane 8). The binding of Fos-B (lane 11) and c-Jun (lane 14) was the same as for control (lanes 3, 6). No c-Fos was detected in either control (lane 2) or DA-stimulated striatal neurons (lane 10).C, Autoradiogram showing the results of a supershift experiment with striatal neurons fed DM (Control) or DM containing 0.25 mm IBMX and 50 μmforskolin. After stimulation the binding of Fos-B and Jun-D to the TH–AP-1 site increased as compared with control (lanes 3, 8, 11, 16), whereas c-Jun remained the same as control (lanes 6, 14). c-Fos, Fra-1, Fra-2, and Jun-B were not detected in both stimulated (lanes 10, 12, 13) and control neurons (lanes 2, 4, 5). Note that the IBMX/forskolin treatment produced a preponderance of proteins in the high-affinity upper AP-1 complex. D, Autoradiogram showing the results of a supershift experiment with striatal neurons fed DM (Control) or DM containing 200 nm TPA. The amount of Fos-B and Jun-D binding at the TH–AP-1 site was lower in the striatal neurons stimulated with TPA (lanes 11, 16) than in the control neurons (lanes 3, 8); c-Jun remained the same as control neurons (lanes 6, 14). Supershift complexes of c-Fos, Fra-1, Fra-2, and Jun-B were not detected in either control (lanes 2, 4, 5) or stimulated neurons (lanes 10, 12, 13).
Dopamine (DA)
Because DA serves as an important coactivator of aFGF in the induction of TH (Du and Iacovitti, 1995), we compared the supershift profile of striatal neurons grown in culture with DA to those kept in control media (Fig. 3B). Interestingly, only the binding of Jun-D (lane 16; compare with control in lane 8) was found to increase after DA treatment. In contrast, the binding of Fos-B and c-Jun to the TH–AP-1 site (lanes 11, 14) did not differ from control (lanes 3, 6). Moreover, DA-stimulated TH–AP-1 complexes did not contain either of the two c-Fos-like proteins (lane 10) nor Fra-1, Fra-2, or Jun-B (lanes 4, 5, 7, 12, 13, 15). These results suggest that the TH-inducing effects of DA at the TH–AP-1 site are limited to changes in the binding of Jun-D.
IBMX/forskolin
Although aFGF plus IBMX/forskolin induces the expression of TH in striatal neurons, IBMX/forskolin alone has no ability to do so (Du and Iacovitti, 1997b). After treatment with IBMX/forskolin only, we found the following changes in binding activity to the TH–AP-1 site (Fig. 3C): Fos-B was increased (lane 11; control,lane 3); Jun-D essentially was unchanged (lane 16; control, lane 8), as was c-Jun (lane 14; control, lane 6); c-Fos remained absent from the complexes (lane 10), as did Fra-1, Fra-2, and Jun-B (lanes 12, 13, 15). Note, however, that IBMX/forskolin treatment produced a preponderance of proteins in the high-affinity upper AP-1 complex, possibly reflecting increased Fos/Jun heterodimers or their increased phosphorylation.
Phorbol ester (TPA)
TPA and other activators of the PKC system can serve as potent coactivators of aFGF in the induction of TH expression in striatal neurons (Du and Iacovitti, 1997a). The results of gel shift studies, however, differed from other coactivators. Treatment with TPA resulted only in the decreased binding of Fos-B and Jun-D (Fig. 3D). Possibly, TPA exerted its effects at the TH–AP-1 site before the 1 hr time point when the cells were harvested and the nuclear extracts were analyzed. Indeed, the effects of TPA on the TH gene occur in a shorter time course than other coactivators; TH expression is induced within 6 hr of treatment with aFGF and TPA and has disappeared by 24 hr (Du and Iacovitti, 1997a).
Reduction of ATF-2 and CREM-1 binding to the TH–AP-1 site after stimulation
Somewhat surprisingly, we discovered in our preliminary competition experiments that TH–CRE oligonucleotides could partially deplete the TH–AP-1 complexes (data not shown). This raised the possibility that, besides Fos/Jun proteins, some members of the CREB/ATF transcription factor family also participated in the regulation of TH expression at the AP-1 site. In fact, the CRE consensus sequence (TGACGTCA) differs from the AP-1 consensus sequence (TGACTCA) by only one nucleotide (Pennypacker et al., 1995). Thus, it is possible for both CREB/ATF and Fos/Jun family to form heterodimers selectively by cross-family dimerization and bind to the AP-1 (Hai and Curran, 1991). To explore this possibility, we performed supershift experiments, using specific antibodies to CREB/ATF family members at the TH–AP-1 site (Fig.4A). The nuclear extracts were prepared from rat striatal neurons that were incubated either in control media or in media containing aFGF, DA, TPA, IBMX, and forskolin. Under basal conditions (control), we found that the TH–AP-1 complexes contained ATF-2 and CREM-1 transcription factors (lanes 5, 7). Despite the fact that stimulation of striatal neurons by aFGF and coactivators increased the amount of protein binding to the AP-1 site, ATF-2 and CREM-1 binding activity was reduced (lanes 13, 15) as compared with control. We found no detectable CREB-1, CREB-2, ATF-1, ATF-3, and CBP in the TH–AP-1 complexes under basal (lanes 2, 3, 4, 6, 8) or stimulated (lanes 10, 11, 12, 14, 16) conditions. Despite the reduction in binding at the AP-1 site, when they were examined by Western blot analysis (Fig. 4B), we found no change in ATF-2 or CREM-1 in stimulated cultures. These apparently discrepant findings suggest that the binding of these transcription factors may be increased elsewhere after stimulation, but at the AP-1 site on the TH gene, ATF-2 and CREM-1 exert their effects by a reduction in binding. Whether there also are important changes in the phosphorylative state of these transcription factors is not yet known.
Fig. 4.

ATF/CREB composition of TH–AP-1 binding protein complexes after TH induction in E14 striatal neurons. A, Autoradiogram of a representative supershift assay of TH–AP-1 binding in rat E14 striatal neurons fed DM (Control) or DM containing aFGF and the coactivators. The cultures were generated as described in the Figure 3 legend. At 1 hr before the addition of radiolabeled AP-1 probe, either no antibody (None) or 1 μl of antibody to the individual CREB family members (Santa Cruz Biotechnology) was added to 3 μg of nuclear extract proteins obtained from rat E14 striatal neurons treated with 10 ng/ml aFGF, 20 μm DA, 200 nm TPA, 0.25 mm IBMX, 50 μm forskolin, or control media at 4°C for 1 hr. The protein–DNA complexes were resolved as described in the Figure 3legend. B, Immunoblot analysis of control and induced striatal neurons, using the same antibodies as above. Molecular mass (kDa) is indicated by arrows. Despite the reduction in binding at the AP-1 site, there was no change in the expression of ATF-2 CREM-1 in both control and stimulated cultures.
In contrast to our supershift findings with aFGF and all of the coactivators, when each agent was analyzed for its individual effects, only IBMX/forskolin caused a similar decrease in ATF-2 and CREM-1 binding (Fig. 5B, lanes 13, 15 as compared with control lanes 5, 7). DA and TPA had no effect on CREB/ATF members (data not shown), and aFGF unexpectedly increased ATF-2 and CREM-1 binding (Fig. 5A,lanes 13, 15) as compared with controls (lanes 5, 7).
Fig. 5.

Supershift experiments at the TH–AP-1 site, using ATF/CREB family antibodies after treatment of E14 striatal neurons with aFGF alone or individual coactivators. A, Autoradiogram showing the results of a supershift experiment with striatal neurons treated with 10 ng/ml aFGF. The stimulation of striatal neurons by aFGF resulted in increased ATF-2 and CREM-1 binding (lanes 13, 15) as compared with controls (lanes 5, 7). Note that this is in contradistinction to the decrease in binding seen after treatment with aFGF and all of the coactivators. B, Autoradiogram showing the results of a supershift experiment with striatal neurons treated with 0.25 mm IBMX and 50 μm forskolin. Similar to the treatment with aFGF and the coactivators, IBMX/forskolin alone caused a decrease in ATF-2 and CREM-1 binding (lanes 13, 15) as compared with control (lanes 5, 7). DA and TPA had no effect on CREB/ATF members (data not shown).
MAPK: An essential pathway mediator of TH induction
Having established that treatment of striatal neurons with aFGF and the coactivators produces profound changes in the transcription factors binding the TH–AP-1 site, we next investigated signaling events occurring upstream of these changes. Several previous findings suggested that MAPK may be a significant player in this regard. Most importantly, all TH-inducing agents, including FGF (acting via its high-affinity receptor) PKA and PKC activators and DA (acting via the D1/D5 receptor), have resulted in the phosphorylation of MAPK (Ray and Sturgill, 1988; Ahn et al., 1990; Gomez et al., 1990; L’Allemain et al., 1991; Sutherland et al., 1993; Zhan et al., 1994) and ultimately in the activation of the AP-1 element in other systems (D’Arcangelo and Halegoua, 1993; Finkbeiner et al., 1997). In addition, extracellular signal-regulated protein kinases (ERKs), which are members of the MAPK family, are known to be important in the regulation of TH enzyme activity (Yamauchi and Fujisawa, 1979; Vulliet et al., 1980, 1984; Edelman et al., 1981; Albert et al., 1984; Campbell et al., 1986; Haycock et al., 1992; Sutherland et al., 1993; Halloran and Vulliet, 1994). Using commercial antibodies to phospho-specific MAPK, which cross-reacts with both ERK 1 (44 kDa) and ERK 2 (42 kDa) when phosphorylated at the Tyr 204 residue, we began our studies by testing whether aFGF and/or the coactivating substances increased the phosphorylation of MAPK. Cultures were fed defined media (DM) or DM containing 10 ng/ml aFGF and/or the coactivators (200 nmTPA + 20 μm DA + 0.25 mm IBMX/50 μm forskolin). At 30 min later the cultures were harvested, and the proteins were separated by SDS-PAGE and Western-blotted (antibodies diluted 1:1000). As depicted in Figure6A, in control cultures little, if any, phosphorylated ERK 1 (44 kDa) was detected despite the presence of modest levels of phosphorylated ERK 2 (42 kDa). In striking contrast, the addition of aFGF to the media resulted in the appearance of phosphorylated ERK 1 and increased levels of phosphorylated ERK 2. Surprisingly, stimulation by the coactivators in the absence of aFGF only slightly increased the phosphorylation of both ERKs. However, when the neurons were treated with all TH-inducing agents, the levels of both phosphorylated ERKs increased significantly.
Fig. 6.
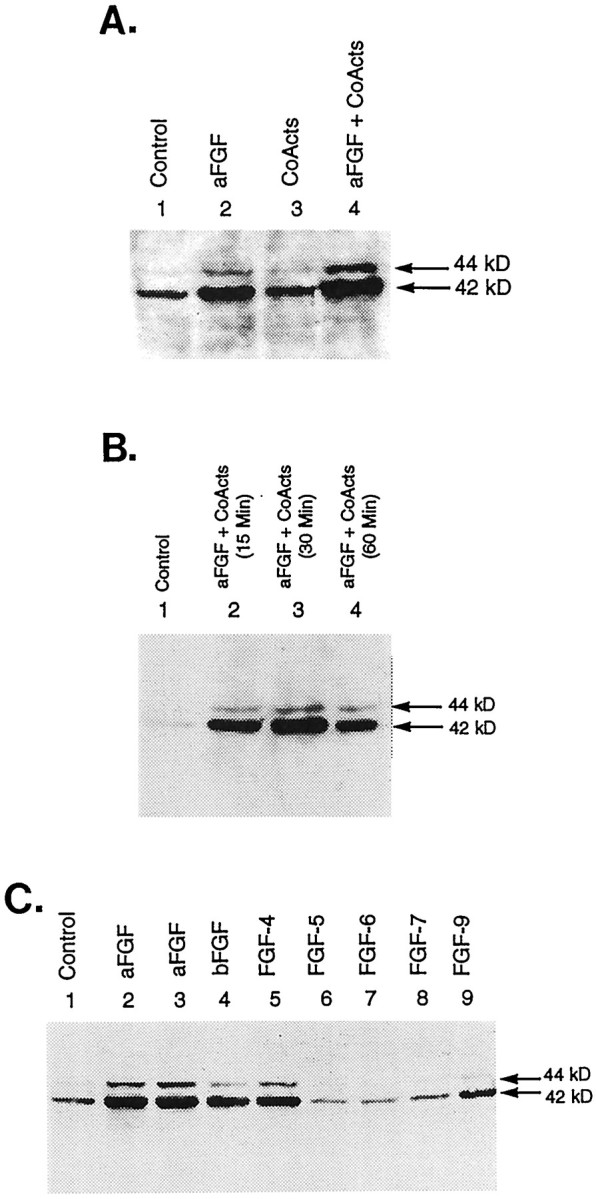
Effects of aFGF and its coactivators on the phosphorylation of MAPK. A, Immunoblot analysis of control and TH-induced E14 striatal neurons. Cultures were fed DM (Control, lane 1) or DM containing 10 ng/ml aFGF (lane 2), coactivators only (200 nm TPA + 20 μm DA + 0.25 mmIBMX/50 μm forskolin; lane 3), or aFGF plus coactivators (lane 4). The cultures were harvested 30 min later, and the proteins were separated by SDS-PAGE and Western-blotted, using commercial antibodies to phospho-specific MAPK, which cross-reacts with both ERK 1 (44 kDa) and ERK 2 (42 kDa) when phosphorylated at the Tyr 204 residue (antibodies diluted 1:1000).B, Time course of the phosphorylation of MAPK after treatment with aFGF and its coactivators. Cultures were fed DM (Control, lane 1) or DM containing 10 ng/ml aFGF + 200 nm TPA + 20 μm DA + 0.25 mm IBMX/50 μm forskolin. Cultures were harvested at various time intervals after treatment (15, 30, or 60 min) and analyzed as described in A. C, Phosphorylation of MAPK by the coactivators and various members of the FGF growth factor family. Cultures were fed DM (Control,lane 1) or DM containing 200 nm TPA + 20 μm DA + 0.25 mm IBMX/50 μmforskolin and 10 ng/ml aFGF (also known as FGF-1; lanes 2, 3), bFGF (also known as FGF-2; lane 4), FGF-4 (lane 5), FGF-5 (lane 6), FGF-6 (lane 7), FGF-7 (lane 8), or FGF-9 (lane 9). At 30 min after treatment the cultures were immunoblotted as described above.
Because of its profound effect on MAPK, time course experiments were initiated with aFGF. We found that phosphorylation of MAPK began as early as 15 min after treatment with aFGF, peaked at 30 min, and declined by 60 min (Fig. 6B). Thus, treatment with aFGF resulted in a rapid and time-dependent activation of the MAPK pathway.
To investigate further the role of MAPK in mediating TH induction, we compared the levels of MAPK phosphorylation after the incubation of neurons with the coactivators and various forms of FGF. Importantly, we found that only those FGFs that previously were shown to be competent to induce TH (Du and Iacovitti, 1995), that is, aFGF (also known as FGF-1), FGF-2, FGF-4, and FGF-9, were capable of phosphorylating MAPK (Fig. 6C). Moreover, the resultant levels of phosphorylated ERKs closely paralleled the TH-inducing potency of a particular FGF (i.e., FGF-1 = FGF-4 ≫ FGF-2 = FGF-9).
Finally, specific inhibitors of the MAPK signaling pathway were used to test whether they could prevent TH induction by aFGF and the coactivators. Cultures were preincubated for 1 hr with either apigenin, a specific inhibitor of MAPK, or PD98059, a specific inhibitor of MAPK and its upstream kinase, MAPK kinase (also known as MEK) before the addition of 10 ng/ml aFGF and/or 200 nm TPA, 20 μm DA, 0.25 mm IBMX/50 μmforskolin. Control cultures were treated with MAPK inhibitors alone, which had no inductive effect on TH expression (data not shown). However, 10–75 μm apigenin (Fig.7A) and 10–20 μm PD98059 (Fig. 7B) completely blocked TH induction by aFGF plus a single coactivator. Induction by aFGF plus multiple coactivators likewise was inhibited by concentrations of inhibitors >10 μm (data not shown).
Fig. 7.

Effects of MAPK inhibitors on TH induction by aFGF and its coactivators. A, Cultures of E14 striatal neurons were preincubated for 1 hr with various concentrations (1–75 μm) of apigenin, a specific inhibitor of MAPK, before the addition of 10 ng/ml aFGF plus individual or combined coactivators (200 nm TPA, 20 μm DA, and 0.25 mmIBMX/50 μm forskolin) or coactivators only. On the next day the cultures were fixed and stained immunocytochemically for the presence of TH. Then the percentage of TH-positive neurons was counted (at 10× magnification) in 50% of all microscopic fields.B, The same procedures were repeated as inA, using various concentrations (1–25 μm) of PD98059, a specific inhibitor of MAPK and its upstream kinase, MAPK kinase (also known as MEK).
Although these studies established that MAPK is an essential pathway mediator of TH induction and our gel shift studies established that, coincident with TH induction, there are striking changes in nuclear binding proteins at the TH–AP-1 site, they did not test the direct involvement of MAPK in producing these AP-1 binding changes. To do so, we examined cultures in which the MAPK pathway specifically was inhibited by PD 98059 1 hr before the incubation of cultures with DM (control) or DM plus aFGF and the coactivators. One hour later the nuclear proteins were harvested, and binding at the TH–AP-1 site was analyzed by supershift assay as described previously. We found that, after treatment with PD 98059, the amount of protein present in the AP-1 complexes was much lower and more variable, particularly in the high-affinity complex, in both control and treated cultures. Nonetheless, as shown in Figure 8, PD 98059 prevented all of the changes in nuclear binding to the TH–AP-1 site usually seen after treatment with aFGF and the coactivators (see Fig. 2A). Thus, there was no evidence of recruitment of either c-Fos protein, Fos-B, or Jun-D into the AP-1 complex. Interestingly, the usual binding of Fos-B, Jun-D, and c-Jun protein present under control conditions was also absent after treatment with PD 98059, suggesting that MAPK also may mediate their binding at the TH–AP-1 site. Taken together, these results suggest an essential role for MAPK in transmitting TH-inducing signals from outside the cell into the nucleus.
Fig. 8.

Fos/Jun composition of TH–AP-1 binding protein complexes after the inhibition of MAPK. Autoradiogram of a supershift assay of TH–AP-1 binding in rat E14 striatal neurons preincubated for 1 hr with 20 μm PD98059, a specific inhibitor of MAPK, before the addition of 10 ng/ml aFGF plus all of the coactivators (200 nm TPA, 20 μm DA, and 0.25 mmIBMX/50 μm forskolin). Cultures were harvested 1 hr later, and supershifts were performed with antibodies to the various members of the Fos/Jun family as described in the Figure 4 legend, with the addition of phosphorylated c-Jun (p-c-Jun).
DISCUSSION
Role of the AP-1 site in TH expression
With the use of deletion/mutation constructs, previous studies on PC12 cells have demonstrated convincingly the critical importance of the AP-1 site in regulating constitutive and enhanced expression of the TH gene (Gizang-Ginsberg and Ziff, 1990, 1994). This finding first prompted us to focus our studies on the role of the AP-1 site in signaling TH gene expression in striatal neurons. Using gel shift and supershift assays, we examined how the binding of the various transcription factors (Fos/Jun; ATF/CREB) to the AP-1 site changed when the TH gene was first expressed during transdifferentiation. Because the members of Fos and Jun families belong to a set of immediate early genes that are rapidly induced by various growth factors (Bravo et al., 1990; Herschman et al., 1991; Lau and Nathans, 1991), the swift recruitment of c-Fos into the protein complexes that specifically bound the TH–AP-1 element was anticipated. Unexpected, however, was the appearance of a second protein band of ∼84 kDa (band a), which cross-reacted with specific c-Fos antibodies. To the best of our knowledge, no such protein has been described previously. Although we do not yet know the identity or function of this molecule, our studies indicate that it is present in the AP-1 complex when TH is expressed in stimulated E14 neurons, and, conversely, it is absent in stimulated E18 neurons that have aged beyond the critical period and are therefore incapable of TH gene expression (our unpublished observations). Consequently, it may be important in signaling expression of the TH gene during early embryogenesis when neurons are first differentiating. In addition to the recruitment of both c-Fos proteins, supershift analysis further demonstrated an increased binding of Fos-B and Jun-D at the TH–AP-1 site, whereas c-jun binding remained at control levels.
Because ATF/CREB family members belong to the same bZIP class of DNA binding proteins as the Fos/Jun family, they also form dimers to bind to the AP-1 site (for review, see Kamei et al., 1996). Indeed, in our experiments we found constitutive binding of CREM-1 and ATF-2 to the TH–AP-1 site in extracts from untreated E14 striatal cells. However, unlike Fos/Jun, the binding of these factors decreased at the AP-1 site when the TH gene was expressed.
Because the Fos/Jun and ATF/CREB families of transcription factors can form homodimers or heterodimers or in some cases can cross-dimerize to bind the AP-1 site (Hai and Curran, 1991), their transactivational ability differs markedly, depending on their composition. On the basis of our supershift and competition assays, we speculate that, in unstimulated striatal neurons, the AP-1 complexes consist mainly of c-Jun/c-Jun and Jun-D/Jun-D homodimers or c-Jun/Jun-D heterodimers, all of which have low-to-medium DNA binding and transactivational activity, and possibly some heterodimers like Fos-B/c-Jun or Fos-B/Jun-D with high binding activity. In striking contrast, we postulate that, after treatment with aFGF and coactivators, TH–AP-1 protein complexes shift predominantly to the formation of c-Fos/Jun-D or Fos-B/Jun-D heterodimers that have the highest DNA binding and transactivational activity in vivo (Hai and Curran, 1991). Moreover, ATF-2/Jun-D heterodimers or CREM-1/CREM-1 homodimers that have low transactivational or even repressive activity at the AP-1 motif (Sassone-Corsi et al., 1988) are reduced in striatal neurons by the action of aFGF and the coactivators. In summary, we postulate that the components of TH–AP-1 complexes may switch from homodimers or heterodimers with low DNA binding and transactivational activity in control (non-TH-expressing) conditions to heterodimers with the highest DNA binding and transactivational activity after stimulation (TH-expressing conditions). We have generated a model to exemplify possible dimeric changes at the AP-1 site during TH induction (Fig.9).
Fig. 9.

A model of possible dimeric changes in transcription factors binding at the AP-1 site before and after TH induction by aFGF and the coactivators.
Although the supershift assay is a powerful analytical technique, there are a number of limitations that must be considered. First, the failure to see a supershifted band is not itself proof of the absence of a transcription factor, because antibodies may not have ready access to all proteins in a complex. Moreover, the post-translational modification of transcription factors, such as phosphorylation, greatly affect binding and transactivational activity. Because antibodies to phosphorylated forms of these proteins are not generally available, a supershift assay affords us only a partial picture of the events occurring at a given site. Despite these caveats, the supershift results reported here suggest that transcriptional changes at the AP-1 site occur concurrently with the initiation of TH gene expression in striatal neurons. Nonetheless, it remains to be demonstrated that these changes occur only in transdifferentiating neurons (80%) in the culture and that they are truly essential for TH expression. Final proof of the indispensability of the AP-1 element in TH expression will require additional experiments demonstrating a loss in the ability to express TH when striatal neurons are transfected with TH–AP-1-deleted constructs. It is indeed possible that other regulatory sites will prove to be more important in TH gene initiation and that changes at the AP-1 site merely regulate the level of that expression.
Balancing activation and repression of the TH gene
Activation of the TH gene
Although aFGF has no TH-inducing capacity of its own, all of our work, including the supershift studies presented here, indicate that it plays a role of central importance. When we examined the individual contributions of TH-inducing agents to transcriptional changes at the TH–AP-1 site, we found that only aFGF produced the same qualitative effects in Fos/Jun binding as those observed when the gene was expressed (i.e., after aFGF plus the coactivators).
However, despite the pivotal role of aFGF, the TH gene cannot be expressed from the quiescent state without synergy of at least one other coactivating molecule: DA, IBMX, forskolin, or TPA. How such diverse substances accomplish this crucial task remains a mystery. Clues gleaned from the present studies, however, suggest a number of possibilities. First, it is conceivable that aFGF-induced changes at the AP-1 site are needed to initiate TH gene expression, whereas coactivators serve to amplify that expression to a detectable level. Consistent with this notion is the previous demonstration that each of the coactivating substances can act as enhancers of transcription in TH-expressing cells (Lewis et al., 1983, 1987; McTigue et al., 1985;Lewis and Chikaraishi, 1987; Gizang-Ginsberg and Ziff, 1990, 1994;Icard-Liepkalns et al., 1992). Indeed, in our system the more coactivators present in the media, the greater is the expression of TH. One way this might be achieved is if each coactivator serves to amplify critically the activational effect of aFGF on the TH gene. At the AP-1 site the coactivators indeed produce binding changes similar to aFGF, although of a more limited scope. Thus, the effects of DA are confined to increases in Jun-D binding, whereas IBMX/forskolin increases the binding of Fos-B only. The net effect, however, is likely the same; that is, when combined with aFGF, each boosts the formation of high-affinity/high-transactivation heterodimers at the AP-1 site.
De-repression of the TH gene
Because neurons of the E14 striatum are GABergic (Max et al., 1996), expression of the TH gene normally is kept silenced in these cells. It stands to reason that initiating TH expression in striatal cells therefore might require not only transcriptional activation but also simultaneous de-repression of the gene. In this regard, changes in ATF-2 and CREM-1 are particularly interesting, because both have been associated with repressor activity at the AP-1 site (Foulkes et al., 1991; Masquilier and Sassone-Corsi, 1992; De Cesare et al., 1995). Counterintuitively, however, aFGF increased the binding of ATF-2 and CREM-1 repressors at the AP-1 site. One way in which coactivators may contribute to the TH-inducing process is by reversing these paradoxical effects of aFGF, which could be counterproductive to TH gene expression. Indeed, when coactivators are added to aFGF and the TH gene is expressed, there is a marked decline in the aFGF-induced transcription factors with potentially repressive activity.
Pathways involved in signaling TH expression
A question of vital interest that has been left open in these studies is how exogenously applied aFGF and its coactivating substances bring about these dramatic changes in nuclear transcription factors. Although conclusive proof is still lacking, it is believed that aFGF, acting via one or more of its high-affinity tyrosine kinase receptors, triggers intracellular kinase signaling cascades. In other systems (Morrison et al., 1988; Vainikka et al., 1992; Wang et al., 1994) these events lead to the activation of the kinases Raf, MEK, ERK, etc., before culminating in the activation of transcription factors at the AP-1 site (D’Arcangelo and Halegoua, 1993; Finkbeiner et al., 1997). Several lines of evidence suggest that aFGF follows a similar route to induce TH in striatal cells. First, our results demonstrate that phosphorylation of MAPK (ERK 1 and 2) occurs concomitantly with TH induction. The presence in the media of the appropriate FGF (1, 2, 4, 9), that is, one competent to induce TH, appears responsible for the major portion of this phosphorylative event. Moreover, when cells are pretreated with MEK/ERK inhibitors, there is total elimination of TH expression as well as the aFGF-associated changes in transcription factor binding at the TH–AP-1 site. Although the upstream signals (FGF receptors, grb2, Ras, and Raf) have not yet been worked out, MAPK appears to be an essential downstream mediator of aFGF stimulation. These studies suggest a direct link between TH gene expression and events occurring outside the cell (aFGF plus coactivators), pathways inside the cell (MAPK), and nuclear binding events (AP-1 changes).
Because coactivator treatment increases the phosphorylation of MAPK only slightly, its level of involvement is not yet entirely clear. Supershift experiments designed to test whether MAPK inhibitors prevent the AP-1 binding effects of individual coactivating substances will be necessary. Regardless of whether the coactivators partially mediate their effects via MAPK, the fact that their combined presence increases TH expression in an additive manner suggests that other pathways also are involved.
Summary and conclusions
We conclude that the initiation of TH expression requires the transmission of signals along pathways leading to the phosphorylation of MAPK and finally to sites on the TH gene. Ultimately, these events produce a complex choreography of changes at the AP-1 site (and probably many other sites on the TH gene as well) involving the crosstalk of multiple families of transcriptions factors (e.g., Fos/Jun and CREB/ATF). aFGF appears to play a central role, producing the necessary qualitative changes in the binding of Fos/Jun members at the AP-1 site. However, the growth factor is insufficient, requiring the cooperation of coactivating substances. Coactivators may participate in further amplifying the activating effects of aFGF on TH transcription or, conversely, in reversing the changes caused by aFGF that adversely affect TH expression. Because each agent (aFGF, DA, etc.) may have quite divergent effects at different sites on the gene, it is likely to be the summation of these opposing influences that tips the balance either in favor of expression or repression of the TH gene. How these effects relate to those observed in vivo when TH is first expressed in the developing brain is not known. Learning whether sonic hedgehog (Hynes et al., 1995a,b; Perrimon, 1995) or Nurr1 (Zetterstrom et al., 1997) gene products, both of which have been implicated in the differentiation of DA neurons in vivo, share pathways or transcriptional changes in common with those described here remains to be explored. Moreover, determining how cells become recalcitrant to these changes as they age beyond the critical period for their biochemical differentiation will be an important direction for the future.
Footnotes
This work was supported by National Institutes of Health Grants NS24204-09 and NS32519-03. We gratefully acknowledge the expert assistance of Ms. Natalie Stull in preparing primary cultures.
Correspondence should be addressed to Lorraine Iacovitti, Ph.D., Department of Neurobiology and Anatomy, Medical College of Pennsylvania and Hahnemann University, 3200 Henry Avenue, Philadelphia, PA 19129.
REFERENCES
- 1.Ahn NG, Weiel JE, Chan CP, Krebs EG. Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J Biol Chem. 1990;265:11487–11494. [PubMed] [Google Scholar]
- 2.Albert KA, Helmer-Matyjek NE, Nairn AC, Muller TH, Haycock JW, Greene LA, Goldstein M, Greengard P. Calcium/phospholipid-dependent protein kinase (protein kinase C) phosphorylates and activates tyrosine hydroxylase. Proc Natl Acad Sci USA. 1984;81:7713–7717. doi: 10.1073/pnas.81.24.7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Best JA, Chen Y, Piech KM, Tank AW. The response of the tyrosine hydroxylase gene to cyclic AMP is mediated by two cyclic AMP response elements. J Neurochem. 1995;65:1934–1943. doi: 10.1046/j.1471-4159.1995.65051934.x. [DOI] [PubMed] [Google Scholar]
- 4.Bravo R. Growth factor inducible genes in fibroblasts. In: Habenicht A, editor. Growth factors, differentiation factors, and cytokines. Springer; Berlin: 1990. pp. 324–343. [Google Scholar]
- 5.Cambi F, Fung B, Chikaraishi DM. 5′ Flanking sequences direct cell-specific expression of rat tyrosine hydroxylase. J Neurochem. 1989;53:1656–1659. doi: 10.1111/j.1471-4159.1989.tb08567.x. [DOI] [PubMed] [Google Scholar]
- 6.Campbell DG, Hardie DG, Vulliet PR. Identification of the four phosphorylation sites in the N-terminal region of tyrosine hydroxylase. J Biol Chem. 1986;261:10489–10492. [PubMed] [Google Scholar]
- 7.Carroll JM, Kim KS, Kim KT, Goodman HM, Joh TH. Effects of second messenger system activation of functional expression of tyrosine hydroxylase fusion gene constructs in neuronal and nonneuronal cells. J Mol Neurosci. 1991;3:65–74. doi: 10.1007/BF02885527. [DOI] [PubMed] [Google Scholar]
- 8.D’Arcangelo G, Halegoua S. A branched signaling pathway for nerve growth factor is revealed by Src-, Ras-, and Raf-mediated gene inductions. Mol Cell Biol. 1993;13:3146–3155. doi: 10.1128/mcb.13.6.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Cesare D, Vallone D, Caracciolo A, Sassone-Corsi P, Narlov C, Verde P. Heterodimerization of c-Jun with ATF-2 and c-Fos is required for positive and negative regulation of the human urokinase enhancer. Oncogene. 1995;11:365–376. [PubMed] [Google Scholar]
- 10.Du X, Iacovitti L. Synergy between growth factors and neurotransmitters required for catecholamine differentiation in brain neurons. J Neurosci. 1995;15:5420–5427. doi: 10.1523/JNEUROSCI.15-07-05420.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du X, Iacovitti L. Multiple signaling pathways direct the initiation of tyrosine hydroxylase gene expression in cultured brain neurons. Mol Brain Res. 1997a;50:1–8. doi: 10.1016/s0169-328x(97)00149-6. [DOI] [PubMed] [Google Scholar]
- 12.Du X, Iacovitti L. Protein kinase C activators work in synergy with specific growth factors to initiate tyrosine hydroxylase gene expression in striatal neurons in culture. J Neurochem. 1997b;68:564–569. doi: 10.1046/j.1471-4159.1997.68020564.x. [DOI] [PubMed] [Google Scholar]
- 13.Du X, Stull ND, Iacovitti L. Novel expression of the tyrosine hydroxylase gene requires both acidic fibroblast growth factor and an activator. J Neurosci. 1994;14:7688–7694. doi: 10.1523/JNEUROSCI.14-12-07688.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du X, Stull ND, Iacovitti L. Brain-derived neurotrophic factor works coordinately with partner molecules to initiate tyrosine hydroxylase expression in striatal neurons. Brain Res. 1995;680:229–233. doi: 10.1016/0006-8993(95)00215-c. [DOI] [PubMed] [Google Scholar]
- 15.Edelman AM, Raese JD, Lazar MA, Barchas JD. Tyrosine hydroxylase: studies of the phosphorylation of a purified preparation of the brain enzyme by cyclic AMP-dependent protein kinase. J Pharmacol Exp Ther. 1981;216:647–653. [PubMed] [Google Scholar]
- 16.Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- 17.Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell. 1991;64:739–749. doi: 10.1016/0092-8674(91)90503-q. [DOI] [PubMed] [Google Scholar]
- 18.Fu YM, Spirito P, Yu Z-X, Biro S, Sasse J, Lei J, Ferrans VJ, Epstein SE, Casscells W. Acidic fibroblast growth factor in the developing rat embryo. J Cell Biol. 1991;114:1261–1273. doi: 10.1083/jcb.114.6.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fung BP, Yoon SO, Chikaraishi DM. Sequences that direct rat tyrosine hydroxylase gene expression. J Neurochem. 1992;58:2044–2052. doi: 10.1111/j.1471-4159.1992.tb10945.x. [DOI] [PubMed] [Google Scholar]
- 20.Gizang-Ginsberg E, Ziff EB. Nerve growth factor regulates tyrosine hydroxylase gene transcription through a nucleoprotein complex that contains c-Fos. Genes Dev. 1990;4:447–491. doi: 10.1101/gad.4.4.477. [DOI] [PubMed] [Google Scholar]
- 21.Gizang-Ginsberg E, Ziff EB. Fos family members successively occupy the tyrosine hydroxylase gene AP-1 site after nerve growth factor or epidermal growth factor stimulation and can repress transcription. Mol Endocrinol. 1994;8:249–262. doi: 10.1210/mend.8.2.7909583. [DOI] [PubMed] [Google Scholar]
- 22.Gomez N, Tonks NK, Morrison C, Harmar T, Cohen P. Evidence for communication between nerve growth factor and protein tyrosine phosphorylation. FEBS Lett. 1990;271:119–122. doi: 10.1016/0014-5793(90)80386-w. [DOI] [PubMed] [Google Scholar]
- 23.Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci USA. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halloran SM, Vulliet PR. Microtubule-associated protein kinase-2 phosphorylates and activates tyrosine hydroxylase following depolarization of bovine adrenal chromaffin cells. J Biol Chem. 1994;269:17430–17440. [PubMed] [Google Scholar]
- 25.Haycock JW, Ahn NG, Cobb MH, Krebs EG. ERK1 and ERK2, two microtubule-associated protein kinases, mediate the phosphorylation of tyrosine hydroxylase at serine 31 in situ. Proc Natl Acad Sci USA. 1992;89:2365–2369. doi: 10.1073/pnas.89.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herschman HR. Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- 27.Hynes M, Porter JA, Chiang C, Chang D, Tessler-Lavigne M, Beachy PA, Rosenthal A. Induction of midbrain dopaminergic neurons by sonic hedgehog. Neuron. 1995a;15:35–44. doi: 10.1016/0896-6273(95)90062-4. [DOI] [PubMed] [Google Scholar]
- 28.Hynes M, Poulsen K, Tessler-Lavigne M, Rosenthal A. Control of neuronal diversity by the floor plate: contact-mediated induction of midbrain dopaminergic neurons. Cell. 1995b;80:95–101. doi: 10.1016/0092-8674(95)90454-9. [DOI] [PubMed] [Google Scholar]
- 29.Iacovitti L. Effects of a novel differentiation factor on the development of catecholamine traits in noncatecholamine neurons from various regions of the rat brain: studies in tissue culture. J Neurosci. 1991;11:2403–2409. doi: 10.1523/JNEUROSCI.11-08-02403.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iacovitti L, Stull ND. Induction of tyrosine hydroxylase in hNT neurons. NeuroReport. 1997;8:1471–1474. doi: 10.1097/00001756-199704140-00029. [DOI] [PubMed] [Google Scholar]
- 31.Iacovitti L, Evinger MJ, Joh TH, Reis DJ. A muscle-derived factor induces expression of a catecholamine phenotype in cultured rat cerebral cortex. J Neurosci. 1989;9:3529–3537. doi: 10.1523/JNEUROSCI.09-10-03529.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Icard-Liepkalns C, Biguet NF, Vyas S, Robert JJ, Sassone-Corsi P, Mallet J. AP-1 complex and c-fos transcription are involved in TPA-provoked and trans-synaptic inductions of the tyrosine hydroxylase gene: insights into long-term regulatory mechanisms. J Neurosci Res. 1992;32:290–298. doi: 10.1002/jnr.490320219. [DOI] [PubMed] [Google Scholar]
- 33.Kamei Y, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 34.Kim KS, Lee MG, Carroll J, Joh TJ. Both basal and inducible transcription of the tyrosine hydroxylase gene are dependent upon a cAMP response element. J Biol Chem. 1993a;268:15689–15695. [PubMed] [Google Scholar]
- 35.Kim KS, Park DH, Wessel TC, Song B, Wagner JA, Joh TJ. A dual role for the cAMP-dependent protein kinase in tyrosine hydroxylase gene expression. Proc Natl Acad Sci USA. 1993b;90:3471–3475. doi: 10.1073/pnas.90.8.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim KS, Tinti C, Song B, Cubells JF, Joh TJ. Cyclic AMP-dependent protein kinase regulates basal and cyclic AMP-stimulated but not phorbol ester-stimulated transcription of the tyrosine hydroxylase gene. J Neurochem. 1994;63:834–842. doi: 10.1046/j.1471-4159.1994.63030834.x. [DOI] [PubMed] [Google Scholar]
- 37.L’Allemain G, Sturgill TW, Weber MJ. Defective regulation of mitogen-activated protein kinase activity in a 3T3 cell variant mitogenically nonresponsive to tetradecanoyl phorbol acetate. Mol Cell Biol. 1991;11:1002–1008. doi: 10.1128/mcb.11.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lau LF, Nathans D. Genes induced by serum growth factors. In: Cohen P, Foulkes JG, editors. The hormonal control regulation of gene transcription. Elsevier; New York: 1991. pp. 165–201. [Google Scholar]
- 39.Lazaroff M, Patankar S, Yoon SO, Chikaraishi DM. The cyclic AMP response element directs tyrosine hydroxylase expression in catecholaminergic central and peripheral nervous system cell lines from transgenic mice. J Biol Chem. 1995;37:21579–21589. doi: 10.1074/jbc.270.37.21579. [DOI] [PubMed] [Google Scholar]
- 40.Lewis EJ, Chikaraishi DM. Regulated expression of the tyrosine hydroxylase gene by epidermal growth factor. Mol Cell Biol. 1987;7:3332–3336. doi: 10.1128/mcb.7.9.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis EJ, Tank AW, Weiner N, Chikaraishi DM. Regulation of tyrosine hydroxylase mRNA by glucocorticoid and cyclic AMP in a rat pheochromocytoma cell line: isolation of a cDNA clone tyrosine hydroxylase mRNA. J Biol Chem. 1983;258:14632–14637. [PubMed] [Google Scholar]
- 42.Lewis EJ, Harrington CA, Chikaraishi DM. Transcriptional regulation of the tyrosine hydroxylase gene by glucocorticoid and cyclic AMP. Proc Natl Acad Sci USA. 1987;84:3550–3554. doi: 10.1073/pnas.84.11.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masquilier D, Sassone-Corsi P. Transcriptional crosstalk: nuclear factors CREM and CREB bind to AP-1 sites and inhibit activation by Jun. J Biol Chem. 1992;267:22460–22466. [PubMed] [Google Scholar]
- 44.Max SR, Bossio A, Iacovitti L. Coexpression of tyrosine hydroxylase and glutamic acid decarboxylase in dopamine differentiation factor-treated striatal neurons in culture. Brain Res Dev Brain Res. 1996;91:140–142. doi: 10.1016/0165-3806(95)00163-8. [DOI] [PubMed] [Google Scholar]
- 45.McTigue M, Cremins J, Halegoua S. Nerve growth factor and other agents mediate phosphorylation and activation of tyrosine hydroxylase, a convergence of multiple kinase activities. J Biol Chem. 1985;260:9047–9056. [PubMed] [Google Scholar]
- 46.Morrison DK, Kaplan DR, Rapp U, Roberts TM. Signal transduction from membrane to cytoplasm: growth factors and membrane-bound oncogene products increase Raf-1 phosphorylation and associated protein kinase activity. Proc Natl Acad Sci USA. 1988;85:8855–8859. doi: 10.1073/pnas.85.23.8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nurcombe V, Ford MD, Wildschut JA, Barlett PF. Development regulation of neural response to FGF-1 and FGF-2 by heparan sulfate proteoglycan. Science. 1993;260:103–106. doi: 10.1126/science.7682010. [DOI] [PubMed] [Google Scholar]
- 48.Pennypacker KR, Hudson PM, Hong JS, McMillian MK. DNA binding activity of CREB transcription factors during ontogeny of the central nervous system. Dev Brain Res. 1995;86:242–249. doi: 10.1016/0165-3806(95)00033-a. [DOI] [PubMed] [Google Scholar]
- 49.Perrimon N. Hedgehog and beyond. Cell. 1995;80:517–520. doi: 10.1016/0092-8674(95)90503-0. [DOI] [PubMed] [Google Scholar]
- 50.Ray LB, Sturgill TW. Insulin-stimulated microtubule-associated protein kinase is phosphorylated on tyrosine and threonine in vivo. Proc Natl Acad Sci USA. 1988;85:3753–3757. doi: 10.1073/pnas.85.11.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sassone-Corsi P, Ransone LJ, Lamph WW, Verma IM. Direct interaction between fos and jun nuclear oncoproteins: role of the “leucine zipper” domain. Nature. 1988;336:692–695. doi: 10.1038/336692a0. [DOI] [PubMed] [Google Scholar]
- 52.Schnürch H, Risau W. Differentiating and mature neurons express the acidic fibroblast growth factor gene during chick neural development. Development. 1991;111:1143–1154. doi: 10.1242/dev.111.4.1143. [DOI] [PubMed] [Google Scholar]
- 53.Sutherland C, Alterio J, Campbell DG, LeBourdelles B, Mallet J, Haavik J, Cohen P. Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur J Biochem. 1993;217:715–722. doi: 10.1111/j.1432-1033.1993.tb18297.x. [DOI] [PubMed] [Google Scholar]
- 54.Vainikka S, Partanen J, Bellosa P, Coulier F, Basilio C, Jaye M, Alitalo C. Fibroblast growth factor receptor-4 shows novel features in genomic structure, ligand binding, and signal transduction. EMBO J. 1992;11:4273–4280. doi: 10.1002/j.1460-2075.1992.tb05526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vulliet PR, Langan TA, Weiner N. Tyrosine hydroxylase: a substrate of cyclic AMP-dependent protein kinase. Proc Natl Acad Sci USA. 1980;77:92–96. doi: 10.1073/pnas.77.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vulliet PR, Woodgett JR, Cohen P. Phosphorylation of tyrosine hydroxylase by calmodulin-dependent multiprotein kinase. J Biol Chem. 1984;259:13680–13683. [PubMed] [Google Scholar]
- 57.Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilcox B, Unnerstall J. Expression of acidic fibroblast growth factor mRNA in the developing and adult rat brain. Neuron. 1991;6:397–409. doi: 10.1016/0896-6273(91)90248-x. [DOI] [PubMed] [Google Scholar]
- 59.Yamauchi T, Fujisawa Regulation of bovine adrenal tyrosine-3-monooxygenase by phosphorylation–dephosphorylation reaction, catalyzed by adenosine 3′,5′-monophosphate-dependent protein kinase and phosphoprotein phosphatase. J Biol Chem. 1979;254:6408–6413. [PubMed] [Google Scholar]
- 60.Yoon SO, Chikaraishi DM. Tissue-specific transcription of the rat tyrosine hydroxylase gene requires synergy between and AP-1 motif and an overlapping E box-containing dyad. Neuron. 1992;9:55–67. doi: 10.1016/0896-6273(92)90220-8. [DOI] [PubMed] [Google Scholar]
- 61.Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 62.Zhan X, Plourde C, Hu X, Friesel R, Maciag T. Association of fibroblast growth factor receptor-1 with c-Src correlates with association between c-Src and cortactin. J Biol Chem. 1994;269:20221–20224. [PubMed] [Google Scholar]