

Abstract
Exogenous application of acetylcholine elicits inward currents in hippocampal interneurons that are mediated via α-bungarotoxin-sensitive nicotinic acetylcholine receptors, but synaptic responses mediated via such receptors have never been reported in mammalian brain. In the present study, EPSCs were evoked in hippocampal interneurons in rat brain slices by electrical stimulation and were recorded by using whole-cell voltage-clamp techniques. Nicotinic EPSCs were isolated pharmacologically, using antagonists to block other known types of ligand-gated ion channels, and then were tested with either α-bungarotoxin or methyllycaconitine, which are selective antagonists for nicotinic acetylcholine receptors that contain the α7 receptor subunit. Each antagonist proved highly effective at reducing the remaining synaptic current. Evoked α7-mediated nicotinic EPSCs also were desensitized by superfusion with 1 μm nicotine, had extrapolated reversal potentials near 0 mV, and showed strong inward rectification at positive potentials. In several interneurons, methyllycaconitine-sensitive spontaneous EPSCs also were observed that exhibited a biphasic decay rate very similar to that of the α7-mediated evoked response. These studies provide the first demonstration of a functional cholinergic synapse in the mammalian brain, in which the primary postsynaptic receptors are α-bungarotoxin-sensitive nicotinic acetylcholine receptors.
Keywords: nicotine, hippocampus, interneuron, rat, acetylcholine, electrophysiology
Nicotinic acetylcholine receptors (nAChRs) that are highly sensitive to blockade by the snake toxin α-bungarotoxin (αBTx) play a primary role in the generation of end plate potentials at neuromuscular synapses (Lee, 1972; Magazanik and Vyskocil, 1972). In mammalian brain, high-affinity binding sites for [125I]-αBTx were discovered >20 years ago (Eterovic and Bennett, 1974; Polz-Tejera et al., 1975; Moore and Brady, 1976), but the inability to demonstrate functional responses that could be blocked by αBTx led to speculation that central [125I]-αBTx binding sites did not represent functional nAChRs (Quik and Geertsen, 1988). However, the cloning and functional expression of the α7 nAChR subunit (Couturier et al., 1990; Séguéla et al., 1993), which binds [125I]-αBTx with high affinity, and the demonstration that α7 mRNA is expressed in brain strongly suggested that the central [125I]-αBTx binding sites were functional nAChRs. Recent physiological studies have demonstrated that αBTx-sensitive nAChRs in brain act presynaptically to modulate synaptic transmission, rather than postsynaptically to mediate fast synaptic transmission (McGehee et al., 1995; Alkondon et al., 1996;Gray et al., 1996). Where nicotinic responses have been found in the CNS that appear to be mediated by postsynaptic receptors, such as in the nucleus ambiguous (Zhang et al., 1993), dorsal motor nucleus of the vagus (Ito et al., 1989), medial vestibular nucleus (Phelan and Gallagher, 1992), and at the motor neuron→Renshaw cell synapse in spinal cord (Curtis and Ryall, 1966; Duggan et al., 1976), synaptic transmission is mediated by nAChRs that have pharmacological characteristics inconsistent with those of α7-containing nAChRs. On the basis of such evidence, it has been proposed that a major role of α-BTx-sensitive receptors in the brain may be to modulate synaptic transmission via actions at presynaptic sites (McGehee and Role, 1996;Role and Berg, 1996).
Nevertheless, in avian ciliary ganglion, postsynaptic responses are mediated partially by αBTx-sensitive nAChRs, although the αBTx-sensitive component of the synaptic current is mediated by extrasynaptic receptors (Zhang et al., 1996; Ullian et al., 1997). In addition, αBTx-sensitive nicotinic currents can be elicited from hippocampal neurons in culture (Alkondon and Albuquerque, 1991;Alkondon et al., 1992; Zorumski et al., 1992; Komourian and Quik, 1996), suggesting a possible postsynaptic role, and cultured hippocampal neurons from mice with a null mutation affecting the α7 subunit lack fast nicotinic currents (Orr-Urtreger et al., 1997). The hippocampus expresses high levels of α7 mRNA (Séguéla et al., 1993) and is heavily innervated by cholinergic afferents arising from the medial septum. Moreover, it has been demonstrated in our laboratory (Frazier et al., 1996, 1998) as well as others (Alkondon et al., 1997; Jones and Yakel, 1997) that responses evoked by the local application of acetylcholine onto hippocampal interneurons are readily blocked by αBTx as well as by another highly selective antagonist of α7-containing nAChRs, methyllycaconitine (MLA). In the present study we demonstrate evoked synaptic responses in stratum radiatum interneurons that are mediated via αBTx- and MLA-sensitive nAChRs. This constitutes the first report of a functioning synapse in the mammalian brain in which α7-containing nAChRs mediate the postsynaptic response.
MATERIALS AND METHODS
Whole-cell recording. Young (14- to 27-d-old) male Sprague Dawley rats were decapitated, and a Vibratome (Pelco, Ted Pella, Redding, CA) was used to prepare 300-μm-thick coronal slices of hippocampus. During incubation the slices were submerged at room temperature in artificial CSF containing (in mm): 124 NaCl, 3.3 KCl, 2.4 MgCl2, 10d-glucose, 2.5 CaCl2, 1.2 KH2PO4, and 25.9 NaHCO3, saturated with 95% O2/5% CO2. All experiments were performed at room temperature while the tissue was superfused with buffer at a rate of 2 ml/min. Whole-cell patch-clamp recordings were made with glass pipettes pulled on a Flaming/Brown electrode puller (Sutter Instrument, Novato, CA). The resistance of the pipettes was 6–10 MΩ when they were filled with a potassium gluconate-based internal solution that consisted of (in mm): 130 K-gluconate, 1 EGTA, 2 MgCl2, 0.1–0.5 CaCl2, 2.54 ATP (di Na+), and 10 HEPES (free acid), adjusted to pH 7.25 with additional KOH. To generateI–V plots of evoked synaptic potentials, we used a cesium gluconate-based internal solution in which cesium gluconate replaced the K-gluconate, and we used QX-314 (5 mm) to block sodium-dependent action potentials. Individual hippocampal interneurons were visualized with an upright microscope equipped with differential interference contrast (Nomarski) optics. EPSCs were recorded in voltage-clamped cells with an AxoClamp 2A amplifier (Axon Instruments, Foster City, CA) operating in continuous clamp mode. Interneurons were identified initially on the basis of their location, but they also were identified electrophysiologically on the basis of their resting membrane potential, ability to sustain a high firing rate in response to a depolarizing pulse, and a short interspike interval (Frazier et al., 1998). Evoked EPSCs were generated by twisted bipolar stimulating electrodes constructed from 0.0026 inch Formvar-coated nichrome wire that were placed under visual guidance in either stratum radiatum or stratum oriens. To achieve maximal stability of the evoked responses, we delivered stimulation at an interpulse interval of 2–3 min; all responses were recorded on a microcomputer with NeuroPro software (RC Electronics) and were analyzed in Microsoft Excel with a custom-built add-in. Spontaneous EPSCs (sEPSCs) were filtered at 1.25 KHz, stored on tape with a Racal FM tape recorder, and digitized off-line at a frequency of 5–10 KHz. sEPSCs were detected by an automated event discriminator (Strathclyde CDR V3.6; courtesy of Dr. John Dempster, Department of Physiology, University of Strathclyde), and then all events were inspected visually to avoid the inclusion of noise that spuriously met our event detection criteria. Housing and treatment of all animals were in accordance with institutional guidelines.
Drugs used in the present experiments were obtained from Research Biochemicals (Natick, MA), including MLA, 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX),dl-(−)-2-amino-5-phosphonovaleric acid (APV), bicuculline methiodide (BMI), 1αH, 3α, 5αH-tropan-3-yl-3, 5-dichlorobenzoate (MDL 72222), and suramin. Nicotine, mecamylamine, atropine, and αBTx were obtained from Sigma (St. Louis, MO). QX-314-Cl was purchased from Alomone Labs (Jerusalem, Israel). All drugs that were superfused were added directly to the superfusion system from concentrated stock solutions via calibrated syringe pumps (Razel, Stamford, CT). In initial experiments it was noted that not all of the antagonists were required in every instance to isolate α7-mediated synaptic responses. DNQX and APV usually were required and thus were applied to every cell that was examined during the course of these experiments; in most cases MDL 72222, mecamylamine, and BMI were used also. In later experiments all of the antagonists were added simultaneously, often before whole-cell recording, to simplify the experimental protocol and to maximize the time during which the α7-mediated synaptic events could be recorded.
Kinetic analyses. Kinetic analysis of spontaneous and evoked EPSCs was performed with SlideWrite 4.0 (Advanced Graphics Software, Carlsbad, CA). Either individual or averaged responses (excluding the stimulus artifact) were fit by using an iterative nonlinear-curve-fitting algorithm (Levenberg-Marquardt) to each of the equations listed below:
| Equation 1 |
| Equation 2 |
| Equation 3 |
where i represents synaptic current, trepresents time, k0 andk1 are constants that govern the amplitude of the response, T1 and T2are time constants that determine the rate of rise of the response, τ1 and τ2 are time constants that determine the rate of decay of the response, and a and bare exponents that help to determine the rate of rise of the response.
Equation 1 corresponded to the simplest model incorporating exponentially rising and decaying phases for the response. Equation 2was based on the empirical observation that nearly all of the evoked EPSCs clearly showed two components to the decay of the evoked response and on the hypothesis that the α7-containing nAChR requires the binding of two acetylcholine molecules for the channel to open. Equation 3 was tested to see if a better fit could be obtained on the basis of the hypothesis that there were two independent components with unknown exponents. Equation 1 provided an adequate fit (r2 > 0.90) to evoked EPSCs in only 3 of 11 cases and did not provide the best fit to any of the responses. However, it provided a good fit (r2 > 0.94 in three cells) to the spontaneous events and the best fit in one of three of the cases examined. Equation 3 provided an equally good fit (r2 = 0.97 ± 0.02,n = 11) to the evoked EPSCs when it was compared with Equation 2 (r2 = 0.95 ± 0.02;p < 0.004, paired Student’s t test). However, in all but one case Equation 3 converged on solutions with what appeared to be unreasonable parameters (exponents >10, extremely long time constants, or responses that were the summation of simultaneously decaying inward and outward currents). Given the fact that both equations fit the data quite well, Equation 2 was used for the subsequent determination of the kinetic parameters of evoked EPSCs.
RESULTS
Isolation of nicotinic EPSCs
Interneurons in stratum radiatum of rat hippocampus were identified visually (Fig.1A), and recordings were made under voltage clamp, using whole-cell patch electrodes. Synaptic responses were elicited in area CA1 stratum radiatum interneurons by low-frequency electrical stimulation of the afferent fibers in either stratum radiatum or stratum oriens (Fig.1B). To isolate synaptic potentials mediated by nAChRs, we blocked glutamatergic synaptic responses with a combination of the ionotropic glutamate receptor antagonists DNQX (10–20 μm) and APV (40 μm). Under those conditions fast inward currents were often still observed in stratum radiatum interneurons that were voltage-clamped at −65 to −70 mV. To eliminate the contribution of other types of non-nicotinic ligand-gated ion channels, we also superfused slices with the GABAA receptor antagonist BMI (20–30 μm), the selective 5HT3 antagonist MDL 72222 (500 nm), or the ATP receptor antagonist suramin (100 μm). Although suramin occasionally reduced the amplitude of the DNQX- and APV-insensitive EPSC, MDL 72222 and BMI typically had no effect. The ineffectiveness of MDL 72222 suggested that under our conditions 5HT3receptors usually were not activated; the BMI data may indicate that GABAA receptors were not activated, but it is more likely that GABAergic currents were not readily apparent because the cells were clamped near the chloride reversal potential. Currents mediated via GABAB receptors were not blocked specifically in these experiments, but they have a much slower time course than the EPSCs that were recorded. Finally, to reduce any portion of the remaining synaptic current mediated by nAChRs that did not contain the α7 subunit, we applied mecamylamine (2 μm; a nonselective nAChR antagonist) in most experiments. At that concentration the mecamylamine only rarely affected the amplitude of the synaptic potentials that were observed in the present study. That finding is consistent with our previous report indicating that 2 μmmecamylamine has only a minimal effect on the responses mediated by α7-containing nAChRs expressed by radiatum interneurons (Frazier et al., 1998). The glycine antagonist strychnine was not tested because of its effectiveness as an α7 antagonist (Séguéla et al., 1993; Zhang et al., 1996), but glycine would be unlikely to contribute to inward currents at the holding potentials that were tested. In most of the subsequent experiments the slices were superfused simultaneously with the full complement of antagonists described above (DNQX/APV/MDL 72222/BMI/mecamylamine/suramin; referred to as the “inhibitor cocktail”), because it became apparent that such treatment afforded the highest probability of isolation of putative nicotinic EPSCs, although in some cases not all of those antagonists were required.
Fig. 1.

Localization of stimulation and recording electrodes and identification of CA1 interneurons. A, A high-power view of a single stratum radiatum interneuron during recording with a patch electrode. The pyramidal cell layer is visible at the top of the view. Stratum radiatum interneurons were identified by their clear displacement from the pyramidal cell layer as well as by their characteristic patterns of electrophysiological activity (see Materials and Methods). Scale bar, 10 μm. B, A low-power view of the hippocampal slice preparation showing the location of the bipolar stimulation electrode (top left), which was placed most frequently in stratum oriens (S. Oriens), as well as the patch recording electrode located in stratum radiatum (S. Rad) of the CA1 region. Scale bar, 100 μm.
Pharmacology of nicotinic EPSCs
The synaptically evoked responses that remained in the presence of the inhibitor cocktail were tested with low concentrations of MLA or αBTx (Fig. 2), both of which are selective antagonists of nAChRs that contain the α7 subunit. In nine cells either MLA (50–75 nm) or αBTx (100 nm) significantly reduced the EPSC amplitude; the average degree of block for those antagonists was 77 ± 4.6% (Fig. 2E). In individual instances (e.g., Figs. 2C, 3A, 4) the block of the EPSC was essentially complete. To ensure that adequate concentrations of antagonists had been tested, we superfused four additional cells with higher concentrations of MLA (100–150 nm). Although there was a slightly greater inhibition of the EPSC in those experiments (Fig. 2E), the difference was not statistically significant (p= 0.49, unpaired one-tailed Student’s t test). Atropine (5 μm) was without effect on these fast EPSCs (data not shown). The antagonism of the EPSC by MLA could be quite rapid (Fig.3A), whereas the block seen with αBTx was slow to develop, usually taking 15–20 min to approach the maximal level of inhibition. The αBTx-induced blockade proved to be irreversible, and recovery from MLA was very slow, with most experiments ending before complete recovery could be achieved. Overall, the characteristics of the MLA and αBTx blockade of the synaptic potentials recorded in the radiatum interneurons that were observed in the present study were similar to those we have reported previously concerning the blockade of responses to exogenously applied acetylcholine (Frazier et al., 1998). Given the pharmacological selectivity of these antagonists, it is apparent that the majority of the synaptic current observed in the presence of the inhibitor cocktail was mediated via the activation of α7-containing nAChRs, presumably by synaptically released acetylcholine.
Fig. 2.

Pharmacology of nicotinic responses in interneurons. Averaged evoked responses are illustrated before and during superfusion with nicotinic agonists and antagonists. Slices were stimulated electrically (arrows), and three to five successive responses were averaged before and during drug superfusion.A and B illustrate the effects of two different concentrations of MLA, whereas C shows the effect of αBTx. Some washout from the effects of MLA were seen, but the effects of αBTx were essentially irreversible for the duration of our experiments. D, A functional antagonism induced by superfusion with nicotine. E, The mean ± SEM response in the presence of each of these agents is presented as a percentage of the mean EPSC amplitude during the period before drug superfusion. The numbers superimposed on thebars are the numbers of cells tested with the drugs at the indicated concentrations (in nm) (<100 represents the combined results from experiments with 50 and 75 nm MLA; >100 includes data from experiments with 100 and 150 nmMLA). F, An I–V plot for a synaptically evoked potential. For the I–V determination the cell was clamped with a Cs-gluconate/QX-314-filled electrode at the indicated voltages for 10 sec before the synaptic stimulus to inactivate most voltage-dependent conductances. The reversal potential extrapolated from a linear fit to the first four points of the curve (straight line) was 15 mV. Similar inwardly rectifying responses were obtained from four of four interneurons in which the synaptic current was inhibited by superfusion with MLA. A few cells showed noninwardly rectifying responses, but none of them was affected by MLA. Stimulus artifacts were clipped in most records. Calibration: 25 msec/200 pA in A; 25 msec/50 pA inB–D.
Fig. 3.

Time course of inhibition of EPSCs by MLA and nicotine. The time course of inhibition of synaptically evoked responses is illustrated for 50 nm MLA (A) and for 1 μm nicotine (B). Individual responses (insets) are shown before, at the time of peak drug effect, and after (nicotine only) drug superfusion. The response to MLA in A was unusually rapid and complete as compared with the average drug effect, possibly because this neuron was quite close to the surface of the slice. The inhibition of the synaptic response (▵, B) by nicotine was not accompanied by any change in the holding current (○, B), input resistance, or access resistance, all of which were monitored throughout the experiment. Stimulus artifacts were clipped in most records, and the time of stimulation is indicated byarrows. Calibration: 10 msec/50 pA in A; 25 msec/25 pA in B.
Fig. 4.

Effect of MLA on spontaneous and evoked EPSCs. In this experiment the effect of MLA superfusion (150 nm) was determined simultaneously on responses evoked by electrical stimulation and on spontaneously occurring events. A, Time course in the decline of the evoked synaptic potential (E-EPSC) during superfusion with MLA (the beginning of superfusion was at 1 min). Traces displayed at the top are averages of each successive pair of points. B, Amplitude and time of occurrence of each of the spontaneous events recorded in the same experiment; the records at the top are averages of all of the single events obtained during the same epochs as the evoked responses in A (the last record is a comparable average of baseline noise; no events could be detected).
Because many types of nAChRs are susceptible to desensitization by nicotine, the sensitivity of the evoked EPSCs to the bath application of nicotine also was used to determine whether the responses were mediated via nAChRs. In four cells, superfusion with 1 μmnicotine reversibly reduced the amplitude of the MLA-sensitive EPSC by 74 ± 7.3% (see Fig. 2D,E) without producing detectable changes in the holding current (Fig. 3B). We previously have shown that α7 receptor-mediated responses elicited by exogenous application of acetylcholine to stratum radiatum interneurons are blocked in a nearly identical manner by nicotine superfusion (Frazier et al., 1998), presumably by agonist-induced receptor desensitization. Thus, from a functional standpoint the slow application of low concentrations of nicotine has an antagonistic rather than agonistic effect on transmission at these synapses.
Although the currents observed in the presence of the inhibitor cocktail were often MLA- or αBTx-sensitive, that was not always the case. More than 125 cells were tested to observe 17 that reliably exhibited α7-mediated currents. Nearly one-half of all cells failed to show an appreciable current in the presence of the inhibitor cocktail, and some of those that did were not stable enough to allow for a complete pharmacological characterization. Further, some cells showed EPSCs that could not be blocked by any of the antagonists that we tested and showed other properties (e.g., outward rectification) that clearly distinguished them from nicotinic responses. The relatively low proportion of cells tested that exhibited α7-mediated currents is likely to be a reflection of the difficulty involved in stimulating the appropriate afferents or of a relative lack of intact afferent projections in the coronal slice preparation. We do not believe it is likely to indicate a paucity of cholinergic afferentation in the intact brain, because we have shown previously that a strong majority (70 of 76) of stratum radiatum interneurons respond to local acetylcholine application with α7-mediated currents (Frazier et al., 1998). In the future other preparations (e.g., septohippocampal slices) may provide a greater rate of success when attempts are made to record α7-mediated synaptic potentials. Nevertheless, the results presented in the current work include only those from cells in which the majority of the inward current (in the presence of the inhibitor cocktail) could be blocked by either MLA or αBTx. Even of those cells, 76% continued to exhibit a residual current that clearly could be distinguished from the noise (i.e., >6 pA) after treatment with an α7-selective nAChR antagonist. On average, the amplitude of those residual currents was 26 ± 3.2% of the control amplitude. Although the kinetics of the residual currents were usually very similar to the MLA-sensitive current, suggesting that they were mediated via the same receptors, it is possible that a component of the MLA-insensitive current may have been mediated via an unidentified transmitter that was insensitive to MLA and αBTx.
Spontaneous synaptic events
To provide further evidence of a functional synaptic input to hippocampal interneurons mediated by postsynaptic α7-containing nAChRs, we examined spontaneous synaptic events that persisted in the presence of the inhibitor cocktail and compared them with evoked synaptic responses in the same cells. In 3 of ∼15 cells with low and stable access resistance, spontaneous synaptic events were observed and subsequently were challenged with α7-selective nicotinic antagonists. The events occurred with a low frequency (0.25–0.75 Hz) and had average amplitudes between 10 and 20 pA in cells clamped at −70 mV. During superfusion with 150 nm MLA, these spontaneous events became undetectable. The time of their disappearance coincided with the loss of the synaptically evoked response (Fig.4), further suggesting that the evoked and the spontaneous MLA-sensitive currents were mediated by receptors with similar pharmacological properties.
Physiological characteristics of evoked and spontaneous α7-mediated nicotinic currents
Like the α7-mediated inward currents elicited by local application of acetylcholine to hippocampal interneurons (Frazier et al., 1998), those evoked by electrical stimulation had extrapolated reversal potentials near 0 mV and showed strong inward rectification at potentials positive to reversal (see Fig. 2F). The refractory period of the MLA- and αBTx-sensitive responses was fairly brief in that paired stimulation with interpulse intervals of 100–200 msec showed neither significant facilitation or inhibition.
To characterize the kinetics of EPSCs, we fit evoked responses with functions consisting of an exponentially rising phase and an exponentially decaying phase with either one or two time constants (see Materials and Methods). Although the majority of the synaptic responses had relatively smooth rising and decaying phases and could be fit well by these functions (r2 > 0.90; 10 of 14 cells), a number of the EPSCs showed multiple peaks in the responses and could not be fit readily by simple exponential functions (see, for example, Fig. 2C,D). These multiple peaks appeared at the same time points in successive responses and disappeared in parallel during antagonist superfusion. For the responses with simple kinetics the average time constant for the rising phase of the evoked EPSCs was 3.6 ± 0.9 msec (n = 10). The decay phase was fit better by the sum of two exponentially decaying components rather than by a monoexponential decay (r2 = 0.88 for single and 0.96 for the sum of two exponentials; t = 3.26; p < 0.008, paired Student’s t test). The primary component of the decay reflected, on average, 91% of the peak synaptic current and decayed with a τ of 5.1 ± 1.0 msec. The secondary component (9% of the current) decayed with a τ of 49 ± 15 msec. The MLA/αBTx-sensitive component of the response (i.e., the control current minus the current that remained in the presence of MLA or αBTx) clearly had two decay components as well; in six of six cases the antagonist-sensitive current was fit better by the sum of two exponential functions than by a monoexponential decay (p < 0.002, paired Student’s ttest).
MLA-sensitive spontaneous events recorded in the presence of the inhibitor cocktail were analyzed in a similar manner. As would be expected, when they were compared with the synaptically evoked responses, the spontaneous events had somewhat faster rise times and similar decay kinetics (Fig.5C). The τ values for the rise times ranged from 0.50 to 0.67 msec (compared with an average of 3.6 msec for evoked responses), and τ values for the decay of spontaneous events ranged from 4.9 to 11.4 msec (the average for the fast component of the evoked responses was 5.1 msec). There was not a clearly distinguishable slow component to the decay of the sEPSCs, which may be a reflection of the difficulty involved in separating the low-amplitude slow component from the noise.
Fig. 5.
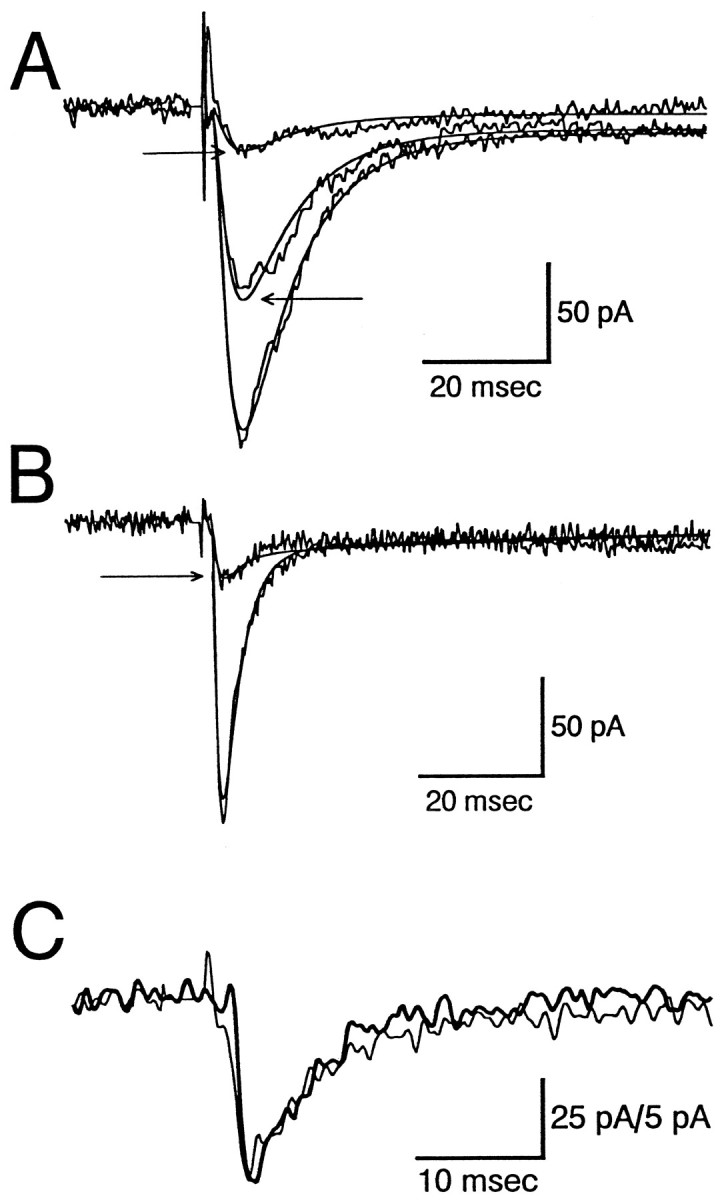
Effects of MLA on the kinetics of nicotinic EPSCs.A, Responses from a neuron recorded under control conditions (large response) and two responses recorded during the initial stages of superfusion with 150 nm MLA (denoted with arrows). The τ values for the best fit (r2 = 0.993) to the control response were 2.1 msec for the rise time and 8.7 and 205 msec for the decay; the fast-decaying component of the response accounted for 95% of the amplitude of the response. The smooth line corresponds to a function with these parameters. The fits to the MLA responses were obtained by assuming that the time constants were unaltered but that the magnitude of the fast and slowly decaying currents were reduced independently. For the smaller of the two responses the fast component was inhibited by 88% and the slow component by 68%. B, Another cell tested as in A, although in this case it was superfused with 50 nm MLA. The τ values for the best fit (r2 = 0.968) to the control response were 0.83 msec for the rise time and 3.0 and 105 msec for the decay; the fast-decaying component of the response accounted for 93% of the amplitude of the response. In this cell MLA reduced the fast-decaying component of the response by 88%, and the slow component was increased by 12%. C, Time course of averaged spontaneous synaptic events (heavy line) is compared with the evoked response (light line). In this cell, spontaneous events were recorded that had kinetics quite similar to those of electrically evoked EPSCs but that had approximately fivefold lower amplitudes (calibration: 25 pA for evoked and 5 pA for spontaneous responses). The time constants for the spontaneous and evoked responses, respectively, were 0.58 and 0.84 msec for the rise time, 5.0 and 4.3 msec for the fast decay, and 29 msec for the slow component of the decay of the evoked EPSC, which comprised 15% of the response.
DISCUSSION
The present experiments demonstrate that the stimulation of cholinergic afferents in the hippocampal slice preparation can elicit excitatory postsynaptic responses in interneurons that are mediated via nAChRs that contain the α7 subunit. The identity of these EPSCs was confirmed by their lack of sensitivity to antagonists of other known fast ligand-gated ion channels (AMPA, NMDA, 5-HT3, ATP, and GABAA receptors), by their sensitivity to two selective antagonists of α7-containing nAChRs (MLA and αBTx), and by the ability of nicotine to desensitize the response. The MLA-sensitive EPSCs also had extrapolated reversal potentials near 0 mV and showed strong inward rectification at positive potentials, both characteristic of α7-mediated currents.
Several aspects of the kinetics of the α-BTx-sensitive nicotinic EPSCs deserve further discussion. First, there was some variability in the rise and decay times for the evoked responses between different cells (although within a cell the kinetics of individual responses were quite consistent). Second, the decay of nearly all of the responses were fit better by the sum of two exponential functions than by a monoexponential function. Although those observations might suggest that different populations of receptors mediate the kinetically distinct components of the response, there was little evidence to that effect. For example, there was no significant relationship between the overall time course of the responses and their sensitivity to MLA or α-BTx, nor was there evidence that the fast and slow components of the decay of the EPSCs were differentially sensitive to blockade. These results distinguish the present system from the situation in the ciliary ganglion (Ullian et al., 1997) in which two distinct populations of receptors mediate the synaptic response. An unexplored possibility is that, as has been suggested by others (Sargent, 1993), there may be multiple populations of α7-containing receptors (but seeChen and Patrick, 1997), each exhibiting similar sensitivity to MLA and α-BTx but having different kinetics that arise as a result of the incorporation of other receptor subunits. Alkondon and colleagues (1997) have suggested that there also may be non-α7-containing nAChRs on interneurons. Although we have not examined that possibility in detail, the presence of mecamylamine in most experiments and the relatively uniform sensitivity of all components of the EPSCs to MLA suggest that non-α7-containing nAChRs are unlikely to account for the biexponential decay rate of the EPSCs observed in the present study. A final explanation for the differences in the rise and decay times from cell to cell is based on anatomical considerations, i.e., the EPSCs demonstrating slower kinetics might correspond to the activation of more distal synapses. However, the lack of a significant correlation between the rise and decay time within individual cells suggests that this explanation is unlikely.
It was noted further that the α7-mediated spontaneous events had rise times that were significantly faster than the majority of the evoked responses. Similar observations have been made in other transmitter systems and could reflect either asynchrony in the evoked release process (i.e., nonsynchronous release of individual quanta), which would slow the rate of rise of evoked EPSCs, or a predominance of spontaneous events originating at proximal synapses, where they could be detected more easily than events occurring at distal synapses. The relatively low frequency of the sEPSCs is expected because presumably they occur in an action potential-independent manner, i.e., via the terminals of the severed axons of cholinergic cell bodies located in the medial septum. Alternatively, it is possible that the spontaneous events arise from the action potential-dependent activity of the sparse cholinergic neurons in hippocampus (Matthews et al., 1987). In that event the low level of internal cholinergic circuitry also might explain the low frequency with which the spontaneous events were observed.
The present experiments have established clearly that cholinergic afferents, presumably originating in the septal nuclei, elicit fast EPSCs mediated by α7-containing nAChRs in hippocampal CA1 interneurons. Although there are intrinsic hippocampal cholinergic neurons (Matthews et al., 1987), they provide only a very small part of the normal cholinergic innervation of the hippocampus and are unlikely to account for the responses observed here. Previous attempts to elicit cholinergic responses in hippocampal neurons in vitro have demonstrated that trains of stimulation can activate a slow EPSP mediated by muscarinic receptors in CA1 pyramidal neurons (Cole and Nicoll, 1983; Madison et al., 1987) but did not detect a nicotinic component to the responses. Together with the findings of the present study, those results suggest that, when septal cholinergic neurons fire at relatively low rates, the primary effect will be a nicotinic activation of hippocampal interneurons. Because of the extensive projections of interneurons to pyramidal neurons (Freund and Buzsáki, 1996), the activation of interneurons by cholinergic afferents from the medial septum is likely to result in the GABAergic inhibition of pyramidal cells. At higher rates of activity this relatively transient inhibitory effect would be opposed by the slow cholinergic EPSP in the pyramidal cells. The combination of a slow excitatory potential (Cole and Nicoll, 1983), a decrease in the Ca2+-dependent afterhyperpolarization (Madison et al., 1987), and synchronized transient IPSPs might provide a mechanism that could underlie the phasic bursts of pyramidal neuron activity that occur during theta rhythm activity. Such patterns of firing have been hypothesized to be an important component of the endogenous patterns of activity that can induce LTP at hippocampal CA1 synapses (Rose and Dunwiddie, 1986). The additional effect of the inhibition of hippocampal interneurons by GABAergic septal neurons (Toth et al., 1997) adds another layer of complexity, and further studies in a more intact system will be required to assess the net effect of activation of both cholinergic and GABAergic septal afferents.
Another aspect of the synaptically evoked α7-mediated nicotinic EPSCs is that they essentially were blocked by bath superfusion with nicotine, although nicotine superfusion elicited no detectable inward currents. The present experiments did not distinguish between a possible presynaptic effect of nicotine (inhibition of release) versus a postsynaptic effect (e.g., desensitization of postsynaptic receptors). However, our previous studies have demonstrated that α7-containing nAChRs on interneurons can be desensitized completely by nanomolar concentrations of nicotine (Frazier et al., 1998) and that this occurs under conditions in which the possibility of a presynaptic action of nicotine (i.e., release of acetylcholine or other neurotransmitters) is blocked. That result strongly suggests that the effect of nicotine in the present study also was mediated postsynaptically. In that event the disruption of fast cholinergic transmission by low concentrations of nicotine (such as those achieved during the use of tobacco products) must now be considered as a likely mechanism contributing to the overall effect of nicotine on hippocampal function. In combination with the presynaptic effects of α7-containing nAChRs that have been described in hippocampus (Gray et al., 1996), it is apparent that the net effect of nicotine on hippocampal activity is likely to be quite complex.
Given that it has been >25 years since the demonstration of high-affinity binding of [125I]-αBTx to sites in the CNS, it is surprising that there have not been previous reports of αBTx-sensitive synaptic potentials in the brain. However, there are several aspects of this response that have made it difficult to isolate. First, hippocampal pyramidal neurons in situ appear to lack αBTx-sensitive receptors (Jones and Yakel, 1997; Frazier et al., 1998), and, until the development of visualized slice recording techniques, it was not possible to record consistently from GABAergic inhibitory neurons in slices. Furthermore, the disynaptic GABAergic response that might be evoked in pyramidal neurons as a result of driving interneurons via synaptic activation of nAChRs would be impossible to dissociate from the monosynaptic GABAergic responses that would result from such stimulation. Second, electrical stimulation can elicit a variety of EPSCs mediated via fast ligand-gated ion channels; in earlier experiments the pharmacological tools necessary to isolate α7-mediated EPSCs were not available. Finally, as has been suggested by Sargent (1993), the extremely rapid desensitization of the α7-containing nAChR has made it considerably more difficult to study. Indeed, we have shown that the α7-containing nAChRs located on stratum radiatum interneurons cannot be activated by bath superfusion with agonists and can be desensitized very easily by the leakage of small amounts of agonist from drug pipettes; successful activation requires techniques that permit both the rapid application and rapid removal of agonist (Frazier et al., 1998). With these factors in mind, it would appear likely that similar approaches to those used in the present study may demonstrate the existence of fast synaptic responses mediated by α7-containing nAChRs in other brain regions in which the receptor is expressed.
Note added in proof. The present observations regarding synaptic responses mediated by α7-containing receptors in hippocampal interneurons have recently been confirmed by another laboratory (M. Alkondon, E. F. R. Pereira, and E. X. Albuquerque, Brain Res, in press).
Footnotes
This work was supported by Grant MH44212 from the National Institute of Mental Health and the Veterans Administration Medical Research Service.
Correspondence should be addressed to Dr. Thomas Dunwiddie, Department of Pharmacology C236, University of Colorado Health Sciences Center, 4200 East 9th Avenue, Denver, CO 80262.
Dr. Frazier’s present address: Department of Physiology and Biophysics, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106-4970.
REFERENCES
- 1.Alkondon M, Albuquerque EX. Initial characterization of the nicotinic acetylcholine receptors in rat hippocampal neurons. J Recept Res. 1991;11:1001–1021. doi: 10.3109/10799899109064693. [DOI] [PubMed] [Google Scholar]
- 2.Alkondon M, Pereira EF, Wonnacott S, Albuquerque EX. Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol. 1992;41:802–808. [PubMed] [Google Scholar]
- 3.Alkondon M, Rocha ES, Maelicke A, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat brain. V. α-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther. 1996;278:1460–1471. [PubMed] [Google Scholar]
- 4.Alkondon M, Pereira EFR, Barbosa CTF, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates γ-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther. 1997;283:1396–1411. [PubMed] [Google Scholar]
- 5.Chen DN, Patrick JW. The α-bungarotoxin-binding nicotinic acetylcholine receptor from rat brain contains only the α7 subunit. J Biol Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- 6.Cole AE, Nicoll RA. Acetylcholine mediates a slow synaptic potential in hippocampal pyramidal cells. Science. 1983;221:1299–1301. doi: 10.1126/science.6612345. [DOI] [PubMed] [Google Scholar]
- 7.Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M. A neuronal nicotinic acetylcholine receptor subunit (α7) is developmentally regulated and forms a homo-oligomeric channel blocked by α-BTx. Neuron. 1990;5:847–856. doi: 10.1016/0896-6273(90)90344-f. [DOI] [PubMed] [Google Scholar]
- 8.Curtis DR, Ryall RW. The synaptic excitation of Renshaw cells. Exp Brain Res. 1966;2:81–96. doi: 10.1007/BF00234362. [DOI] [PubMed] [Google Scholar]
- 9.Duggan AW, Hall JG, Lee CY. α-Bungarotoxin, cobra neurotoxin, and excitation of Renshaw cells by acetylcholine. Brain Res. 1976;107:166–170. doi: 10.1016/0006-8993(76)90106-2. [DOI] [PubMed] [Google Scholar]
- 10.Eterovic VA, Bennett EL. Nicotinic cholinergic receptor in brain detected by binding of α-(3H) bungarotoxin. Biochim Biophys Acta. 1974;362:346–355. doi: 10.1016/0304-4165(74)90227-x. [DOI] [PubMed] [Google Scholar]
- 11.Frazier CJ, Proctor WR, Rose GM, Dunwiddie TV. Identification of a novel current mediated by α-7 nicotinic receptors in rat hippocampal stratum radiatum interneurons, but not in CA1 pyramidal cells. Soc Neurosci Abstr. 1996;22:334. [Google Scholar]
- 12.Frazier CJ, Rollins YD, Breese CR, Leonard S, Freedman R, Dunwiddie TV. Acetylcholine activates an α-bungarotoxin-sensitive nicotinic current in rat hippocampal interneurons, but not pyramidal cells. J Neurosci. 1998;18:1187–1195. doi: 10.1523/JNEUROSCI.18-04-01187.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 14.Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- 15.Ito C, Fukuda A, Nabekura J, Oomura Y. Acetylcholine causes nicotinic depolarization in rat dorsal motor nucleus of the vagus in vitro. Brain Res. 1989;503:44–48. doi: 10.1016/0006-8993(89)91701-0. [DOI] [PubMed] [Google Scholar]
- 16.Jones S, Yakel JL. Functional nicotinic ACh receptors on interneurones in the rat hippocampus. J Physiol (Lond) 1997;504:603–610. doi: 10.1111/j.1469-7793.1997.603bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komourian J, Quik M. Characterization of nicotinic receptors in immortalized hippocampal neurons. Brain Res. 1996;718:37–45. doi: 10.1016/0006-8993(96)00024-8. [DOI] [PubMed] [Google Scholar]
- 18.Lee CY. Chemistry and pharmacology of polypeptide toxins in snake venoms. Annu Rev Pharmacol Toxicol. 1972;12:265–286. doi: 10.1146/annurev.pa.12.040172.001405. [DOI] [PubMed] [Google Scholar]
- 19.Madison DV, Lancaster B, Nicoll RA. Voltage-clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magazanik LG, Vyskocil F. The loci of α-bungarotoxin action on the muscle postjunctional membrane. Brain Res. 1972;48:420–423. doi: 10.1016/0006-8993(72)90203-x. [DOI] [PubMed] [Google Scholar]
- 21.Matthews DA, Salvaterra PM, Crawford GD, Houser CR, Vaughn JE. An immunocytochemical study of choline acetyltransferase-containing neurons and axon terminals in normal and partially deafferented hippocampal formation. Brain Res. 1987;402:30–43. doi: 10.1016/0006-8993(87)91044-4. [DOI] [PubMed] [Google Scholar]
- 22.McGehee DS, Role LW. Presynaptic ionotropic receptors. Curr Opin Neurobiol. 1996;6:342–349. doi: 10.1016/s0959-4388(96)80118-8. [DOI] [PubMed] [Google Scholar]
- 23.McGehee DS, Heath MJS, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- 24.Moore WM, Brady RN. Studies of nicotinic acetylcholine receptor protein from rat brain. Biochim Biophys Acta. 1976;444:252–260. doi: 10.1016/0304-4165(76)90242-7. [DOI] [PubMed] [Google Scholar]
- 25.Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack α-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phelan KD, Gallagher JP. Direct muscarinic and nicotinic receptor-mediated excitation of rat medial vestibular nucleus neurons in vitro. Synapse. 1992;10:349–358. doi: 10.1002/syn.890100410. [DOI] [PubMed] [Google Scholar]
- 27.Polz-Tejera G, Schmidt J, Karten HJ. Autoradiographic localisation of α-bungarotoxin-binding sites in the central nervous system. Nature. 1975;258:349–351. doi: 10.1038/258349a0. [DOI] [PubMed] [Google Scholar]
- 28.Quik M, Geertsen S. Neuronal nicotinic α-bungarotoxin sites. Can J Physiol Pharmacol. 1988;66:971–979. doi: 10.1139/y88-160. [DOI] [PubMed] [Google Scholar]
- 29.Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 30.Rose GM, Dunwiddie TV. Induction of hippocampal long-term potentiation using physiologically patterned stimulation. Neurosci Lett. 1986;69:244–248. doi: 10.1016/0304-3940(86)90487-8. [DOI] [PubMed] [Google Scholar]
- 31.Sargent PB. The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- 32.Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toth K, Freund TF, Miles R. Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. J Physiol (Lond) 1997;500:463–474. doi: 10.1113/jphysiol.1997.sp022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ullian EM, McIntosh JM, Sargent PB. Rapid synaptic transmission in the avian ciliary ganglion is mediated by two distinct classes of nicotinic receptors. J Neurosci. 1997;17:7210–7219. doi: 10.1523/JNEUROSCI.17-19-07210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M, Wang YT, Vyas DM, Neuman RS, Bieger D. Nicotinic cholinoceptor-mediated excitatory postsynaptic potentials in rat nucleus ambiguus. Exp Brain Res. 1993;96:83–88. doi: 10.1007/BF00230441. [DOI] [PubMed] [Google Scholar]
- 36.Zhang ZW, Coggan JS, Berg DK. Synaptic currents generated by neuronal acetylcholine receptors sensitive to α-bungarotoxin. Neuron. 1996;17:1231–1240. doi: 10.1016/s0896-6273(00)80253-6. [DOI] [PubMed] [Google Scholar]
- 37.Zorumski CF, Thio LL, Isenberg KE, Clifford DB. Nicotinic acetylcholine currents in cultured postnatal rat hippocampal neurons. Mol Pharmacol. 1992;41:931–936. [PubMed] [Google Scholar]