

Abstract
Amyloid β-protein, Aβ, is normally produced in brain and is cleared by unknown mechanisms. In Alzheimer’s disease (AD), Aβ accumulates in plaque-like deposits and is implicated genetically in neurodegeneration. Here we investigate mechanisms for Aβ degradation and Aβ toxicity in vivo, focusing on the effects of Aβ40, which is the peptide that accumulates in apolipoprotein E4-associated AD. Chronic intraventricular infusion of Aβ40 into rat brain resulted in limited deposition and toxicity. Coinfusion of Aβ40 with the cysteine protease inhibitor leupeptin resulted in increased extracellular and intracellular Aβ immunoreactivity. Analysis of gliosis and TUNEL in neuron layers of the frontal and entorhinal cortex suggested that leupeptin exacerbated Aβ40 toxicity. This was supported further by the neuronal staining of cathepsin B in endosomes or lysosomes, colocalizing with intracellular Aβ immunoreactivity in pyknotic cells. Leupeptin plus Aβ40 caused limited but significant neuronal phospho-tau immunostaining in the entorhinal cortex. Intriguingly, Aβ40 plus leupeptin induced intracellular accumulation of the more toxic Aβ, Aβ42, in a small group of septal neurons. Leupeptin infusion previously has been reported to interfere with lysosomal proteolysis and to result in the accumulation of lipofuscin, dystrophic neurites, tau- and ubiquitin-positive inclusions, and structures resembling paired helical filaments. Coinfusion of Aβ40 with the serine protease inhibitor aprotinin also increased diffuse extracellular deposition but reduced astrocytosis and TUNEL and was not associated with intracellular Aβ staining. Collectively, these data suggest that an age or Alzheimer’s-related defect in lysosomal/endosomal function could promote Aβ deposition and DNA fragmentation in neurons and glia similar to that found in Alzheimer’s disease.
Keywords: Alzheimer’s disease, Aβ, lysosome, cathepsins, leupeptin, aprotinin, neurotoxicity, in vivo, rat
Alzheimer’s disease (AD) is characterized by the age-related accumulation of deposits of a 40 or 42 amino acid peptide, amyloid β-protein (Aβ). This peptide is produced and secreted normally by cultured cells (Haass et al., 1992;Shoji et al., 1992) and is found in normal CSF (Seubert et al., 1992). Familial AD caused by genetic mutations is associated either with increased Aβ production of the more amyloidogenic peptide, Aβ1–42 (Scheuner et al., 1996; Selkoe, 1997), or, as with apolipoprotein E4 (ApoE4), with increased accumulation of Aβ40 (Ishii et al., 1997; Mann et al., 1997). However, the majority of AD cases is not early onset and does not appear to be linked to mutations that directly increase Aβ. An alternative mechanism for the late-onset cases could be an age-related decline in the removal or degradation rate for Aβ. These studies sought to investigate this mechanism in vivo.
There are two obvious pathways for the degradation of secreted Aβ: degradation by extracellular proteases or reuptake into the endosomal/lysosomal system, followed by intracellular degradation. Others have shown that extracellular proteases, including trypsin or trypsin-like serine proteases and metalloproteases, readily degrade Aβ secreted by cultured cell lines (Naidu et al., 1995; Backstrom et al., 1996; Qiu et al., 1997). Further, on the basis of N-terminal modifications in deposited peptides, aminopeptidases also may initiate Aβ degradation (Kuda et al., 1997).
Aβ degradation also can occur intracellularly in endosomes and lysosomes of cultured fibroblasts (Palmert et al., 1989; Knauer et al., 1992), microglia (Shaffer et al., 1995; Ard et al., 1996), and a human neuroblastoma cell line (Ida et al., 1996). Aβ aggregates, notably from Aβ1–42, can accumulate intracellularly in the endosomal/lysosomal system and mediate toxicity (Yang et al., 1995;Burdick et al., 1997). Most in vitro Aβ toxicity data focus on the toxic effects of fibrils that generally are believed to act at the cell surface. Lipoproteins may be responsible for the delivery of Aβ to the endosomal/lysosomal system for degradation (Narita et al., 1997). A sizable pool of Aβ rides on carrier proteins, notably lipoproteins containing ApoE, a known mediator of AD risk, and ApoJ (Ghiso et al., 1993; Biere et al., 1996; Koudinov et al., 1996). Rat Aβ infusion paradigms suggest that microglia or related monocytes actively take up and accumulate Aβ in rodent brain (Frautschy et al., 1992, 1996). When microglia cultures are treated with soluble Aβ and leupeptin, which inhibits its degradation, the Aβ readily accumulates in lysosomes period (Ard et al., 1996). Collectively, these data suggest that compromising lysosomal functionin vivo would lead to a significant accumulation of Aβ.
Although both extracellular and endosomal/lysosomal Aβ degradation pathways are active in various in vitro systems and could be active in the brain, we sought to test whether specific degradation pathways were active in the brain by using a rat Aβ infusion paradigm. Aβ40 was chosen because ApoE4 dosage, a major genetic risk factor for AD, appears to correlate positively with Aβ40 accumulation (Ishii et al., 1997; Mann et al., 1997). In recognition of the possibility that the inhibition of Aβ degradation might promote toxicity, we also evaluated toxicity and glial responses.
MATERIALS AND METHODS
Surgeries and Aβ infusion. In this study 4-month-old Sprague Dawley female rats (200–250 gm body weight) were used. Surgical and animal care procedures were performed with strict adherence to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication number 86-23, Bethesda, MD). Rats were quarantined for 1 week after arrival at the housing facility to avoid specific contagious pathogens. The rats were kept on a 12 hr light/dark cycle and given food and water ad libitum.
Surgery. After quarantine, rats were allowed 1 additional week to adapt to their new environment. After anesthesia (1.875 mg/kg acepromazine, 37.5 mg/kg ketamine, and 1.9 mg/kg xylazine, i.m.), stainless steel brain catheters (Alza, Palo Alto, CA) were implanted with the use of a David Kopf stereotaxic instrument [at coordinates −0.8 mm anteroposterior (AP), −1.4 mm mediolateral (ML) to bregma, and −3.5 mm dorsoventral (DV) to cranium] and were attached with polyethylene tubing to subcutaneous miniosmotic pumps (Alzet 2004, Alza) under the dorsal neck, as described previously (Frautschy et al., 1996). The pump contents, released over 4 weeks, contained one of five treatments: (1) vehicle (4 mm HEPES, pH 8.0), (2) Aβ40, (3) aprotinin (Sigma, St. Louis, MO), (4) leupeptin (Sigma), (5) Aβ40 coinfused with aprotinin, or (6) Aβ40 coinfused with leupeptin. Over the 1 month period each rat was infused with a total of 20 μg (5 nmol) of Aβ40 and/or 2 mg of protease inhibitors (31 μmol of aprotinin and 4.2 μmol of leupeptin).
Immunocytochemistry. After infusion the rats were anesthetized and perfused with 4% paraformaldehyde, as previously described (Frautschy et al., 1996). Brains were removed and cut rostrally into blocks that included the region of the cannula and 3 mm posterior. Blocks were paraffin-embedded, sectioned at 8 μm, mounted, and processed for immunohistochemistry, using standard techniques (Mak et al., 1994). For Aβ antibodies, a 10 min period of 70% formic acid pretreatment was used (Yang et al., 1994). Sections stained for phosphotyrosine (PT; Sigma) (Korematsu et al., 1994) or glial fibrillary acidic protein (GFAP; Sigma) were pretreated with a citrate buffer (antigen unveiling system; Vector Laboratories, Burlingame, CA) in a pressure cooker for 1 min after boiling, which facilitated consistent and homogenous labeling throughout the sections. Cathepsin B (graciously supplied by Dr. R. A. Nixon, Nathan Kline Institute, New York University Medical Center, Orangeburg, NY) was diluted at 1:200. Immunostaining for cathepsin B was improved greatly by the pretreatment of sections with Tris-buffered saline (TBS) in the microwave until the buffer boiled for 2 sec, followed by 10 min of incubation or until the buffer temperature decreased from 90 to 70°C. After hydrogen peroxide quenching, blocking with 5% normal serum dissolved in PBS plus 0.3% Triton X-100, and incubation with primary antibodies overnight at 4°C, the sections were labeled with secondary biotinylated antibodies and treated with the Vector ELITE ABC kit or ABC-AP kit. We used the peroxidase substrate metal-enhanced diaminobenzidine (DAB; Pierce, Rockford, IL) (Yang et al., 1994) and the alkaline phosphatase substrate Vector blue for double staining. Some sections were counterstained with contrast green (Vector Laboratories) to locate nuclei. Congo red and thioflavin S staining was performed as previously described (Hsiao et al., 1996). Adjacent sections from each treatment were stained with antibody preabsorbed with Aβ42 peptide to determine the specificity of the immunostaining (Frautschy et al., 1996). 10G4 monoclonal to the 1–13 region of native human Aβ40 (Yang et al., 1994) was used for the quantitative analysis of deposition. Additional sections were labeled with 4G8 monoclonal to Aβ17–24 (Senetek, Maryland Heights, MO) and Saido42, an end-specific antibody against Aβ42 (Saido et al., 1995).
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL). The “Apoptag” kit (Oncor, Gaithersburg, MD) was used for in situ end labeling of the DNA fragments as per the manufacturer’s instructions, with 30 min of proteinase K room temperature pretreatment and DAB as the peroxidase substrate. Then some TUNEL-stained sections were stained for choline acetyltransferase with ChAT goat IgG antibody (Chemicon, Temecula, CA) diluted 1:100 and were incubated for 1 hr at 37°C. Biotinylated anti-goat secondary antibody (1:1000; Vector Laboratories) was added, followed by ABC-alkaline phosphatase (1:100) and Vector blue alkaline phosphatase substrate.
Image and statistical analysis. All histological and immunohistochemical images were acquired from an Olympus Vanox-T (model AHBT) microscope with an Optronix Engineering LX-450A charge-coupled device video camera system. Then the video signal was routed into a Power Center 120 Macintosh-compatible microcomputer via a Scion Corporation AG-5 averaging frame grabber. Once digitized, the images were analyzed with National Institutes of Health Image public domain software (developed at National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/). Custom Pascal macro subroutines were written and used to calculate area, particle size, and density of microglial, astrocytic, or TUNEL-positive cells. Each capture was exposed to the same computer subroutine and density slice thresholding. The same slides were analyzed single-blind by two different microscopists to ensure that trends were independent of individual bias. The “cell density analysis” macro was run at 200× optical magnification for the accurate resolution of cells. The parameters measured were percentage area immunostained, number of cells per square millimeter, and average size of cell. Layers II–III of the frontal cortex and layer II of the piriform cortex were analyzed as an average of four fields per rat. The “plaque analysis macro”, run at 100× optical magnification, allowed for high resolution of plaques and the analysis of multiple plaques in one field. Results from this analysis revealed the average diameter of the plaque, number of plaques, location of plaques, and total immunoreactive Aβ area per plaque region (hippocampus, thalamus, or cortex). Plaque diameter was defined as the projected diameter of the plaque if the total area of the plaque was converted to a circle with the same total area: (2 · square root [total area/3.1426]). Data from the image analysis macros were exported to an EXCEL file for appropriate formatting before being exported to SuperANOVA (1.11) or StatView (version 4.5, Abacus, Berkeley, CA) on a Power Macintosh for statistical analysis.
RESULTS
Selection of protease inhibitor candidates
In preliminary experiments (data not shown) we screened a battery of protease inhibitors for their ability to inhibit the degradation of soluble Aβ40 by 10× concentrated serum-free media conditioned by human fetal astrocyte cultures. Aβ degradation was monitored by the time-dependent disappearance of Aβ-immunoreactivity (Aβ-ir) on Western blots labeled with 10G4 antibody. In these experiments the serine protease inhibitor aprotinin was an effective inhibitor of the loss of 10G4 Aβ-ir incubated with astrocyte-conditioned media. Metalloprotease inhibitors like EDTA and 1,10-phenanthroline also showed significant inhibition, but their marked toxicity ruled out their use in our long-term infusion experiments. Because we previously had found that leupeptin was an effective inhibitor of Aβ degradation by microglia and others had shown that the degradation of Aβ-containing peptides in cultured cells is reduced by leupeptin (Cole et al., 1992; Golde et al., 1992), this agent also was selected.
As in previous Aβ40 infusion experiments (Frautschy et al., 1996), plaque-like Aβ-ir was limited to a few scattered, small, diffuse deposits in animals infused with Aβ40 alone. In contrast, the inclusion of protease inhibitors, notably leupeptin, resulted in numerous plaque-like deposits in cortex, hippocampus, and thalamus. Figure 1 illustrates Aβ-ir plaque-like deposits in the hippocampus and cortex. Figure 1C shows the intracellular Aβ-ir accumulation observed in the leupeptin plus Aβ-treated rats, including punctate labeling of processes extending from the Aβ-containing cells (Fig. 1C, arrows), which is a higher magnification of the field shown in Figure6F2. Figure 1, D and E, shows how the addition of leupeptin to Aβ-infused rats resulted in more Aβ-ir cell staining. Similar deposits were seen with 4G8, which appeared to detect 75% of the plaques stained for 10G4 (data not shown). To gain insight into the peptide species being stained by 10G4 and 4G8, we used confocal microscopy staining both for an end-specific antibody to Aβ42 (FITC) and in conjunction with 10G4 (rhodamine). Although 10G4-ir and Aβ42-ir colocalized in some regions, notably in selected neurons in the septum (Fig.2A, arrowhead) and vessels, extracellular diffuse plaques did not stain for Aβ42 (Fig.2A, inset). Other cells contained two compartments of Aβ-ir: intracellular staining of 10G4-ir alone (Fig. 2A, arrows), surrounded by periplasmic staining of both antigens. Figure 2, B and C, demonstrates the individual rhodamine and FITC channels of Figure 2A. Attempts to demonstrate the presence of intracellular Aβ40 by using end-specific antibodies produced weak and ambiguous labeling (data not shown), possibly because lysosomal carboxypeptidases may have destroyed the majority of free Aβ40 epitope.
Fig. 1.

Aβ immunoreactivity (Aβ-ir) with 10G4 monoclonal to Aβ in hippocampus (A) and frontal cortex (B) of rats coinfused intraventricularly with leupeptin plus Aβ40 for 1 month. The inset shows a higher magnification of a cortical plaque centered on a darkly stained cell, possibly a neuron. C, Illustrated is punctate intracellular 10G4 Aβ immunoreactivity in the entorhinal cortex, which was widespread in leupeptin plus Aβ-treated rats. Fragmented cell processes emanated from Aβ-containing cells (thick arrows). Aβ-ir cells were prevalent in leupeptin plus Aβ-treated rats (leu+Aβ; D) but were rare in rats infused with leupeptin alone (leu; E, thin arrows). Scale bars: A–E, 100 μm;inset in B, 25 μm.
Fig. 6.
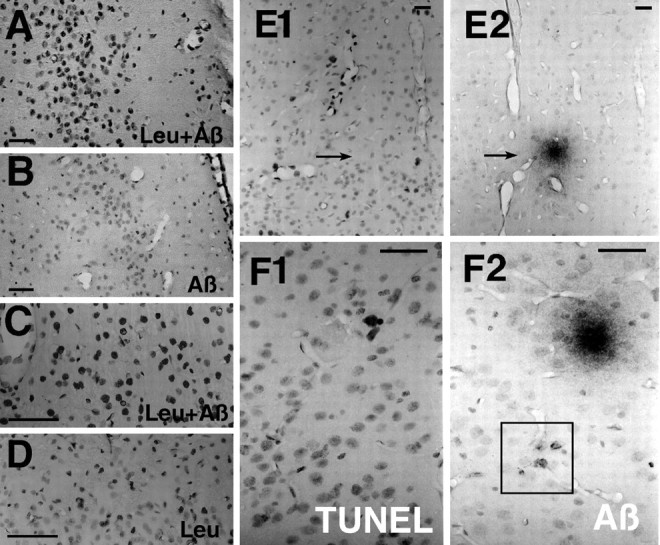
An example of TUNEL labeling in the entorhinal cortex of leupeptin (Leu) plus Aβ-treated rats (A) versus Aβ40 alone (B). Higher-power micrographs demonstrate the differential TUNEL nuclear labeling affected by Leu plus Aβ40 (C) versus Leualone (D). The irregular morphology of the densely labeled nuclei is consistent with nuclear damage.E, F, TUNEL and Aβ labeling in adjacent sections. Most commonly, the Aβ deposits (arrows) are not coincident with, but are central to, the TUNEL-stained cells in deposits that are hypocellular, as in E1 andE2. Occasionally, TUNEL-positive cells are found in the area of Aβ deposits, as shown here in the entorhinal cortex (F1, F2). Note the boxed area that shows the intracellular Aβ-immunostained cells depicted at higher power in Figure 1C. Scale bars:A–D, 200 μm; E, F, 50 μm.
Fig. 2.
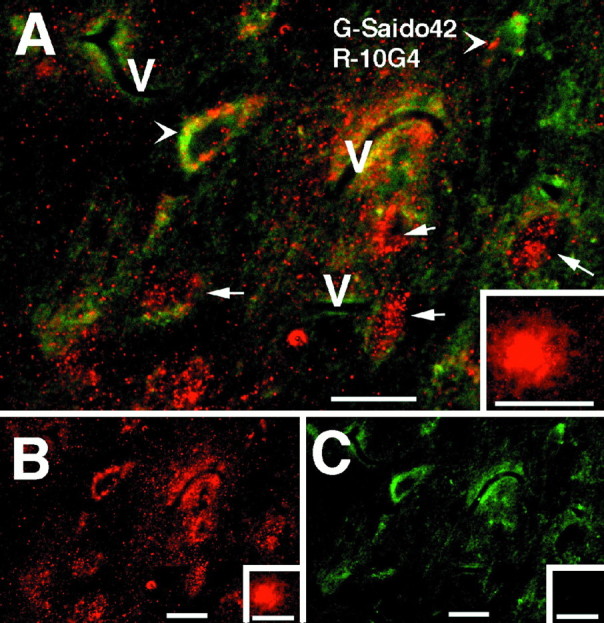
Confocal microscopy of Aβ and end-specific Saido42 staining in the septum of a rat treated with leupeptin plus Aβ40. A, The arrowhead indicates colocalization of 10G4 and Saido42 immunoreactivity (yellow) in a large cell of neuron size.Arrows indicate those cell compartments in which 10G4 staining is apparent (rhodamine) without Saido42 staining (FITC). Theinset shows an extracellular plaque in the same region that stains with 10G4, but not with Saido42. B, Shown is the same field as A with the rhodamine (10G4) channel alone. C, Demonstrated is the same field asA with the FITC channel alone (Saido42).V, Vessels. Scale bar, 50 μm.
Quantitative analysis of plaques showed that both aprotinin and leupeptin increased Aβ40-induced Aβ deposition and that leupeptin had the more robust effect (Fig. 3). For example, compared with Aβ40 alone, aprotinin plus Aβ40 resulted in both a statistically significant increase in total Aβ-ir area (Fig.3B) per whole-brain section and an increase in the number of deposits in the thalamus (Fig. 3A). Leupeptin plus Aβ40 induced the largest increase in total Aβ-ir area and the number of deposits in thalamus (Fig. 3A) as well as in the hippocampus and cortex (data not shown). Unlike aprotinin, leupeptin increased plaque diameter in Aβ40-treated rats (Fig. 3C). These results demonstrate that protease inhibitors can increase Aβ deposition in this model.
Fig. 3.

Quantitative image analysis of 10G4 Aβ-ir deposits in brain sections of rats infused with Aβ40 alone or Aβ40 plus leupeptin (leu) or Aβ40 plus aprotinin (apro). Compared with Aβ40 alone, coinfusion with either protease inhibitor resulted in statistically significant increases in the number of plaques per section in the thalamus (A) and the total Aβ-ir per whole-brain section (B), whereas only leupeptin plus Aβ40 resulted in an increased mean plaque diameter per whole-brain section (C). Lower case letters above thecolumns indicate values that are significantly different from a (Aβ alone), b (leupeptin + Aβ), or c (aprotinin + Aβ). Error bars represent SD.
Activated microglia and reactive astrocytes were observed with PT (Fig.4A) and GFAP (data not shown), respectively, and were assessed quantitatively. Figure4A depicts PT-labeled microglia in and around the cannula site in animals treated with Aβ plus leupeptin. Three ameboid microglia are apparent within the cannula tract as well as an enlarged microglia cell within the larger Aβ-ir plaque, in addition to many ramified microglia distal to the plaque and cannula tract. Reactive gliosis (GFAP and PT) were quantitated as indices of the tissue response to treatments. Anti-cathepsin B (brown) was used to label endosomes/lysosomes, followed by anti-Aβ labeling (blue). Although most neurons showed intense cathepsin B labeling (Fig.4B) in the leupeptin plus Aβ-treated rats, occasional cells also showed significant and overlapping punctate blue-purple Aβ-ir without morphological evidence of degeneration. Other cells (Fig. 4C–E) had markedly enlarged cathepsin B-positive granules with some apparent diffuse labeling and intense Aβ-ir granules clustered in one region, suggestive of a loss of organization and degeneration.
Fig. 4.

A, Phosphotyrosine (PT;brown) double labeling with 10G4 Aβ-ir (blue) was used to determine the degree of Aβ-associated microgliosis. The area shown here is adjacent to the cannula track where three amoeboid PT-positive cells can be seen in a rat infused with Aβ40 alone. Double-labeling sections from a leupeptin plus Aβ-treated rat for cathepsin B (brown) and Aβ (10G4; blue) revealed various degrees of overlap of Aβ (B) with cathepsin B, containing neurons ranging from apparently healthy cells showing only cathepsin B to occasional cells containing both cathepsin B and Aβ-ir (purple). C–E, Cells with grossly enlarged cathepsin B-positive granules and more diffuse cathepsin labeling as well as polarized clusters of intensely Aβ-ir-enlarged granules. F, G, Choline acetyltransferase-ir (blue) and TUNEL labeling of nuclei (brown) in the septum. The arrowheadshows a ChAT-ir neuron with TUNEL nuclei blebbing.G, Inset, Higher magnification. Scale bars: A–E, 25 μm; F, G, 50 μm.
Figure 5 shows the quantitative analysis of immunohistochemistry for GFAP-ir and PT-ir. As shown in Figure5A, the experimental infusions resulted in a similar level of astrogliosis that was decreased in the aprotinin-alone animals and markedly increased in the leupeptin plus Aβ group. An Aβ-induced increase in GFAP-labeled astrocyte cell size, suggesting the hypertrophy of cells, was attenuated by aprotinin (Fig. 5B). In contrast, aprotinin plus Aβ increased the microgliosis relative to injury or Aβ or aprotinin alone, consistent with a microglial response to extracellular Aβ. Both leupeptin and leupeptin plus Aβ treatments resulted in similar levels of microgliosis, which was more than in all other groups except for aprotinin plus Aβ (Fig.5C). The increased microgliosis with leupeptin alone is consistent with a toxic effect.
Fig. 5.
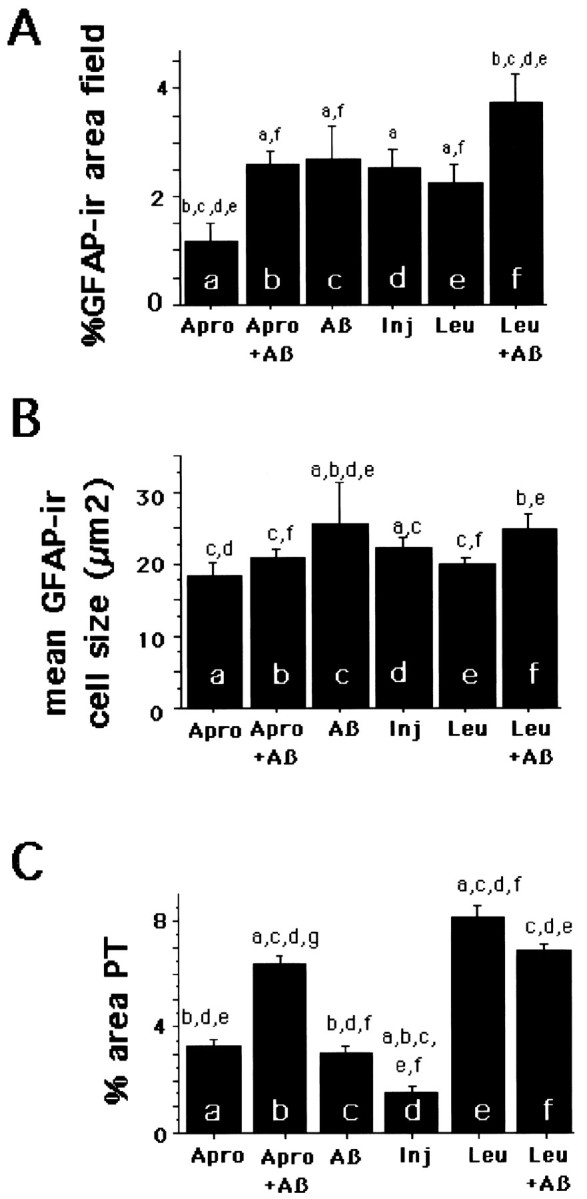
Results of quantitative image analysis of sections taken 0.8 mm posterior to the cannula site, where the maximum number of deposits was induced. Injury represents results from vehicle-infused animals. A, Shown is the analysis for GFAP as the percentage of GFAP-ir area per field. Leupeptin plus Aβ40 increased and aprotinin alone decreased the percentage of GFAP as compared with all other groups. The mean area of GFAP-ir cells (B) was decreased by aprotinin relative to Aβ alone but was not altered by leupeptin. C, Shown is the analysis for microgliosis as the percentage of PT-ir area per field. Aprotinin plus Aβ, leupeptin, and leupeptin plus Aβ increased microgliosis, as compared with Aβ alone. Error bars represent SE.
To evaluate the persistent toxic effects of treatments in sections taken after 1 month of infusion, we chose the TUNEL method as an index of DNA damage. Although increased TUNEL labeling can be indicative of apoptosis, we use it here as a quantitative index of damage resulting from DNA fragmentation. TUNEL labeling revealed increased staining in patches and neuronal layers in cortex and hippocampus of Aβ-infused animals, which clearly was increased by leupeptin coinfusion. Figure6, A and B, depicts examples of this labeling in the entorhinal cortex. Figure 6,C and D, illustrates a higher magnification of TUNEL-labeled nuclei in the frontal cortex. Many of the densely labeled nuclei were irregularly shaped and appeared to be degenerating. The TUNEL-positive cell population of leupeptin plus Aβ appeared larger than that of the leupeptin-alone group, suggesting that Aβ was influencing a large cell population. TUNEL-labeled patches (or layers) of cells were sometimes (Fig. 6E1,E2), but not typically (Fig. 6F1,F2), coincident with Aβ deposits in this model.
Quantitation by image analysis of TUNEL-positive nuclei was performed in neuronal layers of the piriform and frontal cortex (Fig.7). The density of TUNEL-labeled nuclei in both regions was increased by Aβ infusion and reduced by the inclusion of aprotinin, which was neuroprotective. In contrast, leupeptin increased the TUNEL labeling associated with Aβ infusion in both the frontal cortex and, even more dramatically, in the piriform cortex. Analysis showed that the TUNEL area stained per nucleus was 30% of the total nuclear area. The Aβ plus leupeptin group and Aβ-treated groups were associated with larger nuclear area stained (Fig. 7C), presumably because larger nuclei such as neurons were being stained.
Fig. 7.

Results of image analysis of TUNEL-labeled nuclei density in neuron layers of the piriform layer II (A) and frontal cortex layers II–III (B). Leupeptin exacerbated and aprotinin protected against Aβ-associated toxicity in both regions.C, Nuclear area per cell labeled with TUNEL in layers II–III of the frontal cortex demonstrates that, in the Aβ40 and Aβ40 plus leupeptin groups, TUNEL labeled a larger area per nucleus. [Counterstaining with hematoxylin shows that TUNEL is labeling an average of 30% of the nuclear area.] Error bars represent SE.
Finally, as an additional index of neurodegenerative changes in treated animals, sections were labeled with AT8, an antibody to phosphorylated tau protein (serine 202) found in neurofibrillary tangles and dystrophic neurites. In the leupeptin plus Aβ-treated rats, but not in other treatment groups, AT8-ir cells were identified in the hippocampus and entorhinal cortex as being associated with large diffuse plaques (Fig. 8).
Fig. 8.
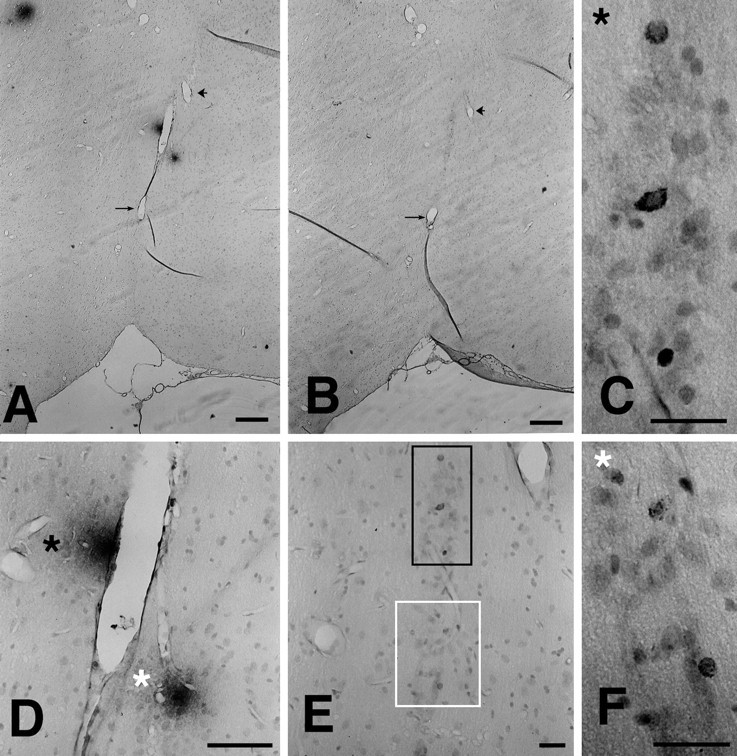
AT8 phospho-tau and Aβ immunostaining in the entorhinal cortex and adjacent thalamus of a leupeptin plus Aβ-treated rat. A, B, Depicted are two adjacent sections immunostained for 10G4 and AT8 phospho-tau, respectively. These low-magnification sections are depicted at higher magnification in D (10G4) and E (AT8). The black-lined box in E, shown at higher magnification in C, corresponds to the plaque labeled by the black asterisk in D. Thewhite-lined box in E, shown at higher magnification in F, corresponds to the plaque labeled by the white asterisk in D. Similar AT8/Aβ colocalization was found in the hippocampus. Scale bar, 100 μm.
DISCUSSION
Unlike leupeptin, aprotinin does not enter cells readily, yet both aprotinin and leupeptin increased the plaque-like deposition of intraventricularly infused Aβ. Only leupeptin plus Aβ caused the intracellular accumulation of Aβ-ir and increased toxicity, raising the possibility that the rate of intracellular degradation may limit toxicity.
Aβ immunoreactivity
Because plaque Aβ-ir with various Aβ antibodies was increased by the infusion of Aβ peptide, was blocked by preabsorption with Aβ42, and was not stained by amyloid precursor protein (APP) antibodies, the Aβ40 infusion-dependent increases in Aβ-ir likely represent, at least in part, Aβ. However, it cannot be ruled out that some of the staining represents an accumulation of Aβ plus leupeptin infusion-dependent C-terminal APP fragments that contain 10G4 and 4G8 epitopes. This supports data from culture experiments showing that these protease inhibitors reduce Aβ degradation (data not shown).
Leupeptin effects on Aβ-ir
Leupeptin inhibits lysosomal degradation of Aβ or Aβ-containing peptides, and coinfusion of leupeptin increases Aβ deposition. We also have found that the coinjection of leupeptin into rodent brain reduces the loss of Aβ detected by sandwich ELISA (our unpublished observations). Leupeptin is well known to inhibit the thiol cathepsins B, H, and L, calpain, and some serine proteases, like trypsin. A likely explanation is the inhibition of cathepsin B, a protease that has been shown to degrade Aβ and that has high carboxypeptidase activity at acidic pH (MacKay et al., 1997). Because the leupeptin effects correlate with the endosome labeling of Aβ, we believe the primary effects that were observed are lysosomal. Because calpain may increase Aβ42 in vitro (Yamazaki et al., 1997) and can cause tau staining (Rebeck et al., 1995), we cannot eliminate that calpain inhibition is involved in our observed effects, although it would be difficult to explain how cytosolic calpain could have access to Aβ. Leupeptin does not inhibit cathepsin D, a lysosomal aspartyl protease that cleaves Aβ between residues L34–M35 and F19–F20 and F20–F21 (Hamazaki, 1996; McDermott and Gibson, 1996) that is responsible for the major Aβ-degrading activity in extracts of human brain (Hamazaki, 1996; McDermott and Gibson, 1996).
Intracellular Aβ, leupeptin, and toxicity
Intense Aβ labeling of enlarged cathepsin B-positive granules is consistent with a degenerative process in which affected cells build up Aβ-ir. Individual cells with gross accumulations of Aβ-ir and degenerative morphology illustrate a pathological progression (see Fig.4B–E). Because only some enlarged granules (see Fig.4C–E) are intensely Aβ-ir, the colocalization could represent the massive accumulation of Aβ peptide fragments within individual or fused endosomes or lysosomes in which seeding has occurred. In AD brain 4G8 labels secondary lysosomes and lipofuscin (Bancher et al., 1989), and the intracellular accumulation of ApoE correlates with the intracellular accumulation of Aβ and cell death (LaFerla et al., 1997). These data suggest that leupeptin reduces intracellular Aβ degradation, which may promote Aβ toxicityin vivo.
Aprotinin effects on Aβ-ir
APP contains a serine protease inhibitor domain (Cole et al., 1991). α-1 antichymotrypsin, α-1 trypsin, and other serpins are in plaques (Gollin et al., 1992), raising the possibility that the inhibition of serine proteases could promote Aβ deposition. Serine proteases prepared from human brain (Matsumoto et al., 1996), as well as trypsin-like serine proteases (Yang et al., 1995; Burdick et al., 1997), effectively degrade Aβ. Aprotinin, a broad spectrum serine protease inhibitor, can be used to examine extracellular Aβ degradation because it does not enter cells readily. Aprotinin-induced increase in Aβ deposition is consistent with the involvement of extracellular serine proteases in Aβ degradation. Our in vivo data for the neuroprotective effects of aprotinin on Aβ-induced TUNEL support previously reported neuroprotective effects of serpins in Aβ toxicity (Schubert, 1997). Unlike leupeptin plus Aβ, aprotinin plus Aβ does not cause the intracellular accumulation of Aβ. Although both protease inhibitors increase deposition, their differential effects on toxicity suggest the significance of intracellular Aβ accumulation in Aβ toxicity.
Other Aβ toxicity studies
Intraventricular Aβ infusion allows for the evaluation of persistent effects of Aβ far from an injection site and after the initial response to injection trauma has subsided. We chose TUNEL as an index of ongoing toxicity that could detect strand breaks caused by Aβ-induced oxidative damage. TUNEL staining allows for the detection and quantification of nuclei with excessive DNA fragmentation. The TUNEL-positive cells evaluated in the piriform cortex are predominantly neurons, because this is a dense neuron layer. TUNEL-positive neurons in the septum were identified by double labeling (see Fig.4F,G). The Aβ-induced increase in TUNEL-positive nuclei in the frontal cortex was visualized in cells with larger nuclei than those affected by leupeptin alone (presumably neurons; see Figs.6B,C, 7C). Although we cannot eliminate the possibility that Aβ affects TUNEL in glia, this evidence suggests that Aβ can increase TUNEL staining in neurons. Coinfusion of leupeptin exacerbates and coinfusion of aprotinin alleviates Aβ40 toxicity.
As an indirect measurement of toxicity, we evaluated GFAP-ir astrocytes and found that leupeptin plus Aβ40 increased total GFAP-ir cortical area as compared with Aβ40, whereas aprotinin plus Aβ40 reduced astrocyte cell size. This strengthens the results observed with TUNEL, suggesting a possible glial response to neuron (DNA fragmentation) damage. We also examined PT-labeled microglia, which become hypertrophied in plaques of APPsw mice (Frautschy et al., 1998). In our model we observed a less dramatic but significant hypertrophy of microglia, especially in the large diffuse Aβ deposits (see Fig. 4A). Both leupeptin and aprotinin increased the microglial response to Aβ despite the apparent neuroprotective effects of aprotinin. This may reflect effects of the protease inhibitors and Aβ on microglia that are independent of neuron damage. Nevertheless, the observed Aβ-dependent increases in TUNEL and persistently increased astrogliosis support ongoing Aβ-associated toxicity.
There is now extensive literature showing that high levels of aggregating or fibrillar Aβ can be toxic to isolated neurons or cell lines in culture (Yankner et al., 1990; Yankner, 1996; Blanc et al., 1997). When high levels of Aβ are focally injected into rats with the use of toxic solvents, acute toxicity occurs at the injection site (Kowall et al., 1991); injection of fibrillar Aβ can lead to a selected loss of some neurons (Weldon et al., 1998), but the relevance of these models is unclear (Waite et al., 1992). In vivoresults, using lower doses and nontoxic solvents, demonstrate negative or equivocal toxicity (Kosik and Coleman, 1992; Winkler et al., 1994). APP transgenic mice, which accumulate high levels of deposited Aβ, have not shown quantifiable neuron loss (Irizarry et al., 1997). It is unclear whether this relates to reduced toxicity in rodents or other factors such as the overexpression of APP, which may exert neurotrophic effects.
Our results suggest an alternative explanation for limited Aβ toxicity in young rodent models. Infused soluble Aβ is removed or degraded readily, which may explain why few persistent deposits are found in the absence of additional cofactors (Snow et al., 1994;Frautschy et al., 1996). Rodent Aβ aggregates as readily as human Aβ (Hilbich et al., 1991); therefore, with aging, the persistence of effective Aβ clearance mechanisms also may be a major reason why rodents normally do not develop age-related Aβ deposits.
Other effects of protease inhibitor infusions
Chronic intraventricular infusion of leupeptin, but not aprotinin, induces the formation of lysosome-derived lipofuscin (Ivy et al., 1989a). In contrast to saline, leupeptin infusion also causes the accumulation of tau and ubiquitin (Ivy et al., 1989b), APP C-terminal fragments (Hajimohammadreza et al., 1994), and intraneuronal Aβ-ir (Mielke et al., 1997) and reduced ChAT, somatostatin, and neuropeptide Y (Kuki et al., 1996). Ultrastructural analysis demonstrated the appearance of dense-body packed dystrophic neurites (Kuki et al., 1996) and paired helical filament (PHF)-like structures (Takauchi and Miyoshi, 1995). Why we failed to see increased APP C-terminal immunostaining with leupeptin treatment remains unclear but could reflect methodological differences in tissue preparation or time course because of our longer infusion. Acute uptake of Aβ1–42 aggregates by endosomes or lysosomes can induce C-terminal fragments (Yang et al., 1995). Because of the possible resemblance of these results to changes found during normal aging and AD, it is possible that leupeptin-induced changes in lysosomes may overlap with these phenomena.
Because the formation of lipofuscin from lysosomes is a common and well confirmed structural age change in postmitotic cells, studies of age-related changes in lysosomal enzymes and lysosomes have looked for and found defects in aged rodent brains (Nakamura et al., 1989). Widespread alterations in neuronal endosomes and lysosomes in AD occur at early stages, far in excess of changes in other neurodegenerative conditions (Cataldo et al., 1994, 1996, 1997). Despite the activation and proliferation of the endosomal and lysosomal system in AD, it is unable to prevent the accumulation of Aβ deposits. Aβ aggregates accumulate in the lysosomes of cultured cells and may seed further aggregation of Aβ at low pH (Burdick et al., 1997), whereas neurotoxicity associated with Aβ42 treatment upregulates cathepsins and causes leakage of lysosomes (Yang et al., 1997). Aβ42 is particularly resistant to degradation (Yamazaki et al., 1997), especially in its fibrillar form (Nordstedt et al., 1994).
Conclusions
These in vivo coinfusion studies demonstrate that protease inhibitors can increase Aβ infusion-induced plaque-like Aβ-ir deposits. Tests are underway that use more specific inhibitors of putative Aβ-degrading enzymes in brain. Our results show evidence of widespread Aβ toxicity in the infusion model, contrasting neuroprotective and neurotoxic effects of aprotinin and leupeptin, both of which increase Aβ deposition. Finally, our toxicity data are consistent with a role for the uptake of intracellular Aβ into the endosomal/lysosomal system in vivo, which may be attributable, in part, to the induction of endosomal accumulation of toxic Aβ42.
Footnotes
This work was supported by grants from Veterans Administration Merit (to S.A.F. and G.M.C.) and by National Institute on Aging Grants AG1125 (to G.M.C.) and AG10685 (to S.A.F.). We are grateful to Elizabeth and Thomas Plott and their family for their continued support (to G.M.C.). We thank Ping Ping Chen and Leticia Calderón for technical assistance.
Correspondence should be addressed to Dr. Sally A. Frautschy, Sepulveda Veterans Association Medical Center, GRECC11E, 16111 Plummer Street, Sepulveda, CA 91343.
REFERENCES
- 1.Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer’s amyloid β-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Backstrom JR, Lim GP, Cullen MJ, Tokes ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40). J Neurosci. 1996;16:7910–7919. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bancher C, Grundke-Iqbal K, Iqbal K, Koo KS, Wisniewski HM. Immunoreactivity of neuronal lipofuscin with monoclonal antibodies to the amyloid β-protein. Neurobiol Aging. 1989;10:125–132. doi: 10.1016/0197-4580(89)90021-3. [DOI] [PubMed] [Google Scholar]
- 4.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 5.Blanc EM, Toborek M, Mark RJ, Hennig B, Mattson MP. Amyloid β-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells. J Neurochem. 1997;68:1870–1881. doi: 10.1046/j.1471-4159.1997.68051870.x. [DOI] [PubMed] [Google Scholar]
- 6.Burdick D, Kosmoski J, Knauer MF, Glabe CG. Preferential adsorption, internalization, and resistance to degradation of the major isoform of the Alzheimer’s amyloid peptide, Aβ1–42, in differentiated PC12 cells. Brain Res. 1997;746:275–284. doi: 10.1016/s0006-8993(96)01262-0. [DOI] [PubMed] [Google Scholar]
- 7.Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994;640:68–80. doi: 10.1016/0006-8993(94)91858-9. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal–lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole GM, Masliah E, Shelton ER, Chan HW, Terry RD, Saitoh T. Accumulation of amyloid precursor protein fragment in Alzheimer plaques. Neurobiol Aging. 1991;12:85–91. doi: 10.1016/0197-4580(91)90046-m. [DOI] [PubMed] [Google Scholar]
- 11.Cole GM, Bell L, Truong QB, Saitoh T. An endosomal lysosomal pathway for degradation of amyloid precursor protein. Ann NY Acad Sci. 1992;674:103–117. doi: 10.1111/j.1749-6632.1992.tb27480.x. [DOI] [PubMed] [Google Scholar]
- 12.Frautschy S, Cole GM, Baird A. Phagocytosis and deposition of vascular β-amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 13.Frautschy SA, Yang F, Calderón L, Cole GM. Rodent models of Alzheimer’s disease: rat Aβ infusion approaches to amyloid deposits. Neurobiol Aging. 1996;17:311–321. doi: 10.1016/0197-4580(95)02073-x. [DOI] [PubMed] [Google Scholar]
- 14.Frautschy SA, Yang F, Irizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 15.Ghiso J, Matsubara E, Koudinov A, Choi-Miura NH, Tomita M, Wisniewski T, Frangione B. The cerebrospinal fluid-soluble form of Alzheimer’s amyloid-β is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane–attack complex. Biochem J. 1993;293:27–30. doi: 10.1042/bj2930027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 17.Gollin PA, Kalaria RN, Eikelenboom P, Rozemuller A, Perry G. α1-Antitrypsin and α1-antichymotrypsin are in the lesions of Alzheimer’s disease. NeuroReport. 1992;3:201–203. doi: 10.1097/00001756-199202000-00020. [DOI] [PubMed] [Google Scholar]
- 18.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 19.Hajimohammadreza I, Anderson VER, Cavanagh JB, Seville MP, Nolan CC, Anderton BH, Leigh PN. β-Amyloid precursor protein fragments and lysosomal dense bodies are found in rat brain neurons after ventricular infusion of leupeptin. Brain Res. 1994;640:25–32. doi: 10.1016/0006-8993(94)91853-8. [DOI] [PubMed] [Google Scholar]
- 20.Hamazaki H. Cathepsin D is involved in the clearance of Alzheimer’s β-amyloid protein. FEBS Lett. 1996;396:139–142. doi: 10.1016/0014-5793(96)01087-3. [DOI] [PubMed] [Google Scholar]
- 21.Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Human and rodent sequence analogs of Alzheimer’s amyloid-β A4 share similar properties and can be solubilized in buffers of pH 7.4. Eur J Biochem. 1991;201:61–69. doi: 10.1111/j.1432-1033.1991.tb16256.x. [DOI] [PubMed] [Google Scholar]
- 22.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 23.Ida N, Masters CL, Beyreuther K. Rapid cellular uptake of Alzheimer amyloid-β A4 peptide by cultured human neuroblastoma cells. FEBS Lett. 1996;394:174–178. doi: 10.1016/0014-5793(96)00948-9. [DOI] [PubMed] [Google Scholar]
- 24.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPsw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Fraser PE, Takahashi H, Tsuji S, Gearing M, Mizutani T, Yamada S, Kato M, St. George-Hyslop PH, Mirra SS, Mori H. Aβ1–40 but not Aβ1–42 levels in cortex correlate with apolipoprotein E E4 allele dosage in sporadic Alzheimer’s disease. Brain Res. 1997;748:250–252. doi: 10.1016/s0006-8993(96)01363-7. [DOI] [PubMed] [Google Scholar]
- 26.Ivy GO, Kanai S, Ohta M, Smith G, Sato Y, Kobayashi M, Kitani K. Lipofuscin-like substances accumulate rapidly in brain, retina, and internal organs with cysteine protease inhibition. Adv Exp Med Biol. 1989a;266:31–45. doi: 10.1007/978-1-4899-5339-1_3. [DOI] [PubMed] [Google Scholar]
- 27.Ivy GO, Kitani K, Ihara Y. Anomalous accumulation of tau and ubiquitin immunoreactivities in rat brain caused by protease inhibition and by normal aging: a clue to PHF pathogenesis? Brain Res. 1989b;498:360–365. doi: 10.1016/0006-8993(89)91117-7. [DOI] [PubMed] [Google Scholar]
- 28.Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/β protein. Proc Natl Acad Sci USA. 1992;89:7437–7441. doi: 10.1073/pnas.89.16.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korematsu K, Goto S, Nagahiro S, Ushio Y. Microglial response to transient focal cerebral ischemia: an immunocytochemical study on the rat cerebral cortex using anti-phosphotyrosine antibody. J Cereb Blood Flow Metab. 1994;14:825–830. doi: 10.1038/jcbfm.1994.103. [DOI] [PubMed] [Google Scholar]
- 30.Kosik KS, Coleman PD. Special issue: is β-amyloid neurotoxic? Neurobiol Aging. 1992;13:535–625. [PubMed] [Google Scholar]
- 31.Koudinov AR, Koudinova NV, Kumar A, Beavis RC, Ghiso J. Biochemical characterization of Alzheimer’s soluble amyloid β-protein in human cerebrospinal fluid: association with high density lipoproteins. Biochem Biophys Res Commun. 1996;223:592–597. doi: 10.1006/bbrc.1996.0940. [DOI] [PubMed] [Google Scholar]
- 32.Kowall NW, Beal MF, Busciglio J, Duffy LK, Yankner BA. An in vivo model for the neurodegenerative effects of β-amyloid and protection by substance P. Proc Natl Acad Sci USA. 1991;88:7247–7251. doi: 10.1073/pnas.88.16.7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuda T, Shoji M, Arai H, Kawashima S, Saido TC. Reduction of plasma glutamyl aminopeptidase activity in sporadic Alzheimer’s disease. Biochem Biophys Res Commun. 1997;231:526–530. doi: 10.1006/bbrc.1996.5920. [DOI] [PubMed] [Google Scholar]
- 34.Kuki K, Maeka K, Takauchi S, Kakigi T, Maeda S, Tanaka C. Neurochemical and pathological alterations following infusion of leupeptin, a protease inhibitor, into the rat brain. Dementia. 1996;7:233–238. doi: 10.1159/000106885. [DOI] [PubMed] [Google Scholar]
- 35.LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G. Neuronal cell death in Alzheimer’s disease correlates with ApoE uptake and intracellular Aβ stabilization. J Clin Invest. 1997;100:310–320. doi: 10.1172/JCI119536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKay EA, Ehrhard A, Moniatte M, Guenet C, Tardif C, Tarnus C, Sorokine O, Heintzelmann B, Nay C, Remy JM. A possible role for cathepsins D, E, and B in the processing of β-amyloid precursor protein in Alzheimer’s disease. Eur J Biochem. 1997;244:414–425. doi: 10.1111/j.1432-1033.1997.00414.x. [DOI] [PubMed] [Google Scholar]
- 37.Mak K, Yang F, Vinters HV, Frautschy SA, Cole GM. Polyclonals to β-amyloid (1–42) identify most plaque and vascular deposits in Alzheimer cortex, but not striatum. Brain Res. 1994;667:138–142. doi: 10.1016/0006-8993(94)91725-6. [DOI] [PubMed] [Google Scholar]
- 38.Mann DMA, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH. Preferential deposition of amyloid β-protein (Aβ) in the form of Aβ40 in Alzheimer’s disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci Lett. 1997;221:81–84. doi: 10.1016/s0304-3940(96)13294-8. [DOI] [PubMed] [Google Scholar]
- 39.Matsumoto A, Enomoto T, Fujiwara Y, Baba H, Matsumoto R. Enhanced aggregation of β-amyloid-containing peptides by extracellular matrix and their degradation by the 68 kDa serine protease prepared from human brain. Neurosci Lett. 1996;220:159–162. doi: 10.1016/s0304-3940(96)13235-3. [DOI] [PubMed] [Google Scholar]
- 40.McDermott JR, Gibson AM. Degradation of Alzheimer’s β-amyloid protein by human cathepsin D. NeuroReport. 1996;7:2163–2166. doi: 10.1097/00001756-199609020-00021. [DOI] [PubMed] [Google Scholar]
- 41.Mielke JG, Maritz J, Bengualid KM, Ivy GO. Chloroquine administration in mice increases β-amyloid immunoreactivity and attenuates kainate-induced blood–brain barrier dysfunction. Neurosci Lett. 1997;227:169–172. doi: 10.1016/s0304-3940(97)00340-6. [DOI] [PubMed] [Google Scholar]
- 42.Naidu A, Quon D, Cordell B. β-Amyloid peptide produced in vitro is degraded by proteinases released by cultured cells. J Biol Chem. 1995;270:1369–1374. doi: 10.1074/jbc.270.3.1369. [DOI] [PubMed] [Google Scholar]
- 43.Nakamura Y, Takeda M, Suzuki H, Morita H, Tada K, Hariguchi S, Nishimura T. Lysosome instability in aged rat brain. Neurosci Lett. 1989;97:215–220. doi: 10.1016/0304-3940(89)90166-3. [DOI] [PubMed] [Google Scholar]
- 44.Narita M, Holtzman DM, Schwartz AL, Bu GJ. α2-Macroglobulin complexes with and mediates the endocytosis of β-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem. 1997;69:1904–1911. doi: 10.1046/j.1471-4159.1997.69051904.x. [DOI] [PubMed] [Google Scholar]
- 45.Nordstedt C, Naslund J, Tjernberg LO, Karlstrom AR, Thyberg J, Terenius L. The Alzheimer Aβ peptide develops protease resistance in association with its polymerization into fibrils. J Biol Chem. 1994;269:30773–30776. [PubMed] [Google Scholar]
- 46.Palmert MR, Podlisny MB, Witker DS, Oltersdorf T, Younkin LH, Selkoe DS, Younkin SG. The β-amyloid protein precursor of Alzheimer disease has soluble derivatives in human brain and cerebrospinal fluid. Proc Natl Acad Sci USA. 1989;86:6338–6342. doi: 10.1073/pnas.86.16.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid β-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem. 1997;272:6641–6646. doi: 10.1074/jbc.272.10.6641. [DOI] [PubMed] [Google Scholar]
- 48.Rebeck GW, Harr SD, Strickland DK, Hyman BT. Multiple, diverse senile plaque-associated proteins are ligands of an apolipoprotein E receptor, the α2-macroglobulin. Ann Neurol. 1995;37:211–217. doi: 10.1002/ana.410370212. [DOI] [PubMed] [Google Scholar]
- 49.Saido TC, Iwatsubo T, Mann DMA, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(PE) in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 50.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Selkoe D, Younkin S. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 51.Schubert D. Serpins inhibit the toxicity of amyloid peptides. Eur J Neurosci. 1997;9:770–777. doi: 10.1111/j.1460-9568.1997.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 52.Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–631. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- 53.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantitation of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 54.Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR. Amyloid β-protein removal by neuroglial cells in culture. Neurobiol Aging. 1995;16:737–745. doi: 10.1016/0197-4580(95)00055-j. [DOI] [PubMed] [Google Scholar]
- 55.Shoji M, Golde TE, Cheung TT, Ghiso J, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG. Production of the Alzheimer amyloid β-protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 56.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG. An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar Aβ-amyloid in rat brain. Neuron. 1994;12:219–134. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 57.Takauchi S, Miyoshi K. Cytoskeletal changes in rat cortical neurons induced by long-term intraventricular infusion of leupeptin. Acta Neuropathol (Berl) 1995;89:8–16. doi: 10.1007/BF00294253. [DOI] [PubMed] [Google Scholar]
- 58.Waite J, Cole GM, Frautschy SA, Connor DJ, Thal LJ. Solvent effects on β-protein toxicity in vivo. Neurobiol Aging. 1992;13:595–599. doi: 10.1016/0197-4580(92)90062-3. [DOI] [PubMed] [Google Scholar]
- 59.Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, Mantyh PW. Fibrillar β-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci. 1998;18:2161–2173. doi: 10.1523/JNEUROSCI.18-06-02161.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winkler J, Connor DJ, Frautschy SA, Behl C, Waite JJ, Cole GM, Thal LJ. Lack of long-term effects after β-amyloid protein injections in rat brain. Neurobiol Aging. 1994;15:601–607. doi: 10.1016/0197-4580(94)00054-9. [DOI] [PubMed] [Google Scholar]
- 61.Yamazaki T, Haass C, Saido TC, Omura S, Ihara Y. Specific increase in amyloid β-protein 42 secretion ratio by calpain inhibition. Biochemistry. 1997;36:8377–8383. doi: 10.1021/bi970209y. [DOI] [PubMed] [Google Scholar]
- 62.Yang AJ, Knauer M, Burdick DA, Glabe C. Intracellular Aβ1–42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J Biol Chem. 1995;270:14786–14792. doi: 10.1074/jbc.270.24.14786. [DOI] [PubMed] [Google Scholar]
- 63.Yang AJ, Chandswangbuvana D, Margol L, Glabe CG. Abnormal sequestration of lysosomal hydrolases in Aβ-mediated neurotoxicity. Soc Neurosci Abstr. 1997;23:1663. [Google Scholar]
- 64.Yang F, Mak K, Vinters HV, Frautschy SA, Cole GM. Monoclonal antibody to the C-terminus of β-amyloid. NeuroReport. 1994;15:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- 65.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 66.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid β-protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]