

Abstract
Nitric oxide (NO) plays a complex role in the pathophysiology of cerebral ischemia. In this study, mutant mice with disrupted type I (neuronal) NO synthase (nNOS) were compared with wild-type littermates after permanent focal ischemia. Cerebral blood flow in the central and peripheral zones of the ischemic distribution were measured with laser doppler flowmetry. Simultaneously, microdialysis electrodes were used to measure extracellular amino acid concentrations and DC potential in these same locations. Blood flow was reduced to <25 and 60% of baseline levels in the central and peripheral zones, respectively; there were no differences in nNOS mutants versus wild-type mice. Within the central ischemic zone, DC potentials rapidly shifted to −20 mV in all mice. In the ischemic periphery, spreading depression (SD)-like waves of depolarization were observed. SD-like events were significantly fewer in the nNOS mutant mice. Concurrent with these hemodynamic and electrophysiological perturbations, extracellular elevations in amino acids occurred after ischemia. There were no detectable differences between wild-type and mutant mice in the ischemic periphery. However, in the central zone of ischemia, elevations in glutamate and GABA were significantly lower in the nNOS mutants. Twenty-four hour infarct volumes in the nNOS mutant mice were significantly smaller than in their wild-type littermates. Overall, the number of SD-like depolarizations and the integrated efflux of glutamate were significantly correlated with infarct size. These results suggest that NO derived from the nNOS isoform contributes to tissue damage after focal ischemia by amplifying excitotoxic amino acid release in the core and deleterious waves of SD-like depolarizations in the periphery.
Keywords: ischemia, excitotoxicity, knock-out mice, microdialysis, NO, spreading depression
Nitric oxide (NO) is a highly diffusible gas, and biomathematical models have suggested that it can act at distances far from the original sites of release (Gally et al., 1990; Wood and Garthwaite, 1994). Recently, it has been shown that NO can diffuse away from the original synapse where it is released and can increase neurotransmitter efflux in adjacent synapses (Schuman and Madison, 1994). In synaptosomal preparations, NMDA-stimulated glutamate release was significantly attenuated by inhibition of NO synthase (NOS) (Montague et al., 1994). In hippocampal slices, NO donors increased and NOS inhibitors decreased NMDA-stimulated release of glutamate, respectively (Jones et al., 1995). Similar results were obtained in cerebellar slices, in which NO potentiated NMDA-stimulated aspartate release (Dickie et al., 1992). Recently, we used microdialysis to show that NMDA-stimulated glutamate release was attenuated in mutant mice with a disrupted type I (neuronal) NOS (nNOS) gene compared with wild-type mice (Kano et al., 1998). These results suggest that NO can act as an amplifier of neurotransmitter release in vivo. Under conditions of cerebral ischemia in which NO release is elevated (Malinski et al., 1993), NO might serve to potentiate the efflux of glutamate, thus acting as a molecular amplifier of acute excitotoxicity.
In focal ischemia, the center of the lesion comprises tissue with dense ischemia (core), whereas the peripheral zones comprise tissue with mild to moderate reductions in blood flow (penumbra) (Astrup et al., 1981). Maturation of the ischemic infarct is related to expansion of the core and collapse of the penumbra. It has been suggested that penumbral collapse is mediated by waves of spreading depression (SD)-like depolarizations (Hossman, 1996). Because these SD-like events involve elevations in extracellular glutamate (Fabricius et al., 1993; Marranes et al., 1988), it is reasonable to hypothesize that after focal ischemia, NO amplifies the excitotoxic release of glutamate and mediates the occurrence of SD-like events in the ischemic periphery.
In the present study, we investigate this hypothesis by comparing the ischemic profiles of SD-like events and glutamate release in wild-type mice versus mutant mice with disrupted nNOS genes. The central and peripheral zones of focal ischemia are examined separately, and these measurements are correlated with final infarct size.
MATERIALS AND METHODS
Animal model. Male and female mutant mice with disruptions in the gene encoding for type I or nNOS were used together with their wild-type littermates (Huang et al., 1993). Body weights were in the 20–27 gm range. Some of these mice were genotyped to confirm disruption of the gene in the mutants.
Mice were anesthetized with α-chloralose (40 mg · kg−1 · hr−1, i.v.). A catheter was inserted into the femoral artery for continuous measurement of mean arterial blood pressure (Maclab/8S, AD Instruments), and the trachea was cannulated. Thereafter, the mice were artificially ventilated with a mechanical ventilator (SAR-830/P, CWE) using a mixture of 70% N2O and 30% O2. Ventilation parameters were set at optimal rates that have been previously determined: 0.15–0.2 sec inspiration time, 110/min respiratory rate, and 250–350 ml/min inspiratory flow. Once the animals were stabilized, oxygen concentrations in the flow mix were adjusted to obtain arterial PO2 levels of ∼140–160 mmHg. End-tidal CO2 was continuously monitored with a microcapnometer (Columbus Instruments) and maintained at 4.8–5.5%, which corresponds to PCO2 levels of ∼30–40 mmHg. Arterial blood gases and pH were measured at the end of each experiment. Rectal core temperatures were maintained at 36.9–37.1°C using a thermostatically controlled heating pad.
After obtaining baseline recordings of hemodynamic, microdialysis, and electrical status (Fig. 1) for at least 30 min, focal cerebral ischemia was induced by permanent occlusion of the middle cerebral artery (Chiamulera et al., 1992). Briefly, a 2 mm craniotomy was drilled in the inferior portion of the temporal bone to expose the right middle cerebral artery, which was then cauterized at a level just superior to the inferior cortical vein. In this model, the lenticulostriate arteries were left intact; thus only cortical ischemia should be obtained. This approach was adapted from that used to obtain focal cortical ischemia in rats (Brint et al., 1988). Hemodynamic, microdialysis, and electrical data were collected for 90 min after ischemia. After that, all catheters were removed, and mice were allowed to return to their cages for a 24 hr recovery period before being killed to quantify infarct volumes (see below).
Fig. 1.

Schematic showing the location of microdialysis probes and electrodes and laser doppler flow probes (LDF) in relation to the zones of focal cortical ischemia induced via occlusion of the distal middle cerebral artery in mice.
In vivo microdialysis. Microdialysis probes (0.25 mm outer diameter, 1 mm membrane length; Applied Neuroscience, London, UK) were stereotactically inserted into the cortex in locations that corresponded to what had been previously determined in pilot studies to be either central or core areas of the ischemic focus (from bregma: 3.5 mm lateral, 1.5 mm caudal, 1 mm depth), or peripheral zones of the ischemic distribution (from bregma: 1.5 mm lateral, 1.5 mm caudal, 1 mm depth). The correct location of these probes was confirmed in all experiments by observing the levels of blood flow reduction that were measured via laser doppler flowmetry (Fig. 1; also see below). The probes were perfused with artificial CSF at a rate of 2 μl/min. Samples were collected at 10 min fractions resulting in 20 μl sample volumes. Microdialysis samples were then analyzed with reversed phase HPLC to quantify concentrations of various amino acid neurotransmitters and neuromodulators, as previously described (Lo et al., 1998a,b).l-Glu and GABA were measured because they represent the major excitatory and inhibitory transmitters in mammalian brain (Hertz, 1979; Tossman et al., 1986). d-Ser and Gly were measured because they are the primary neuromodulators at the NMDA receptor site (Wroblenski et al., 1989; Kemp and Leeson, 1993), and NMDA-mediated toxicity is a major pathological mechanism after cerebral ischemia (Choi, 1992). Tau was assessed because there are data suggesting that it may be a neuroprotective inhibitory neuromodulator (Lehmann et al., 1984; Schurr et al., 1987). Phosphoethanolamine (PEA) was assessed because it is a lipid component that may reflect membrane damage and recycling (Hagberg et al., 1985). l-Ser was examined as a “control” enantiomer for d-Ser. Finally,l-Ala was examined because it is considered a non-neuroactive amino acid (Hagberg et al., 1985).
Surface electrical recordings. Steady-state DC potentials and electrocorticograms were recorded with the electrode that was built into the microdialysis probe (see above). An Ag/AgCl reference electrode was placed subcutaneously in the scalp. Electrical signals were captured (Axoprobe 1A, Axon Instruments) and analyzed with the Maclab data system (Maclab/8S, AD Instruments). The characteristics of these combination microdialysis probes and electrodes have been previously described (Obrenovitch et al., 1993).
Laser doppler flowmetry. Regional cerebral blood flow was assessed at the microdialysis site by a laser doppler flowmeter (Periflux PF2B, Perimed). The flow probe (0.6 mm diameter) was placed onto the cortex adjacent to the microdialysis probe and away from large surface vessels (Fig. 1). Steady-state baseline values were recorded before middle cerebral artery occlusion, and blood flow data were then expressed as percentage of the preocclusion baseline.
Determination of infarct volume. Mice were killed to quantify infarct volumes at 24 hr after focal ischemia. Brains were removed and frozen in isopentane (−35°C). Coronal brain sections (20 μm thick) were cut with a cryostat, collected at 1 mm intervals, and stained with hematoxylin–eosin. Infarct areas were analyzed with a digital image analyzer, and infarct volumes were calculated by integrating along five equally spaced coronal sections. Two methods of calculating infarct volume were used. In the first method, infarcted areas were directly integrated. In the second method, effects of edema and brain swelling were normalized with a standard formula whereby noninfarcted areas were subtracted from contralateral hemispheric areas (Lin et al., 1992). This approach yields an “indirect” measurement of infarct volume.
Data analysis. Data are reported as mean ± SEM. Continuous measurements over time (blood pressure, laser doppler flow, and microdialysis) were examined with two-way repeated measures ANOVA for comparisons between nNOS mutants and their wild-type littermates. Additionally, post hoc Fisher’s PLSD analyses were performed at ischemic time points to compare wild-type versus nNOS mutant mice. Microdialysis measurements of extracellular amino acid concentrations were also analyzed in terms of total integrated efflux over the course of ischemia as well as maximal absolute release during that time. Electrical recordings were analyzed in terms of the degree of DC depolarization and the number of transient cortical SD-like events after ischemia. Differences in these parameters between mutants and wild-type mice were analyzed by two-tailed Student’s ttests. Comparisons of infarct sizes between wild-type and mutant mice were also performed with two-tailed Student’s t tests. Linear regressions were performed to assess the correlations between infarct sizes and the electrophysiological and microdialysis measurements. p < 0.05 was considered significant.
RESULTS
Physiological parameters and cerebral blood flow
Arterial pH and blood gases were within the normal range for all mice (Table 1). Mean arterial blood pressure remained at ∼80–90 mmHg throughout all experimental periods, and there were no differences between the nNOS mutants and their wild-type littermates (Fig. 2). Laser doppler flowmetry showed that occlusion of the middle cerebral artery reduced blood flow in the central zone of ischemia <25% of baseline levels (Fig. 3A). In the ischemic periphery, blood flow was reduced to ∼50–60% of baseline (Fig. 3B). There were no differences in the levels of ischemic blood flow reduction in wild-type mice versus nNOS mutant mice.
Table 1.
Blood pH and gas parameters
| pH (mmHg) | PCO2(mmHg) | PO2 (°C) | |
|---|---|---|---|
| Wild-type mice | 7.282 ± 0.04 | 34.5 ± 1.9 | 157 ± 8 |
| nNOS mutants | 7.289 ± 0.04 | 34.3 ± 1.7 | 145 ± 11 |
Fig. 2.

Mean arterial blood pressures remained between 80 and 90 mmHg throughout the experimental measurement period. There were no differences between the nNOS mutants and their wild-type littermates.
Fig. 3.
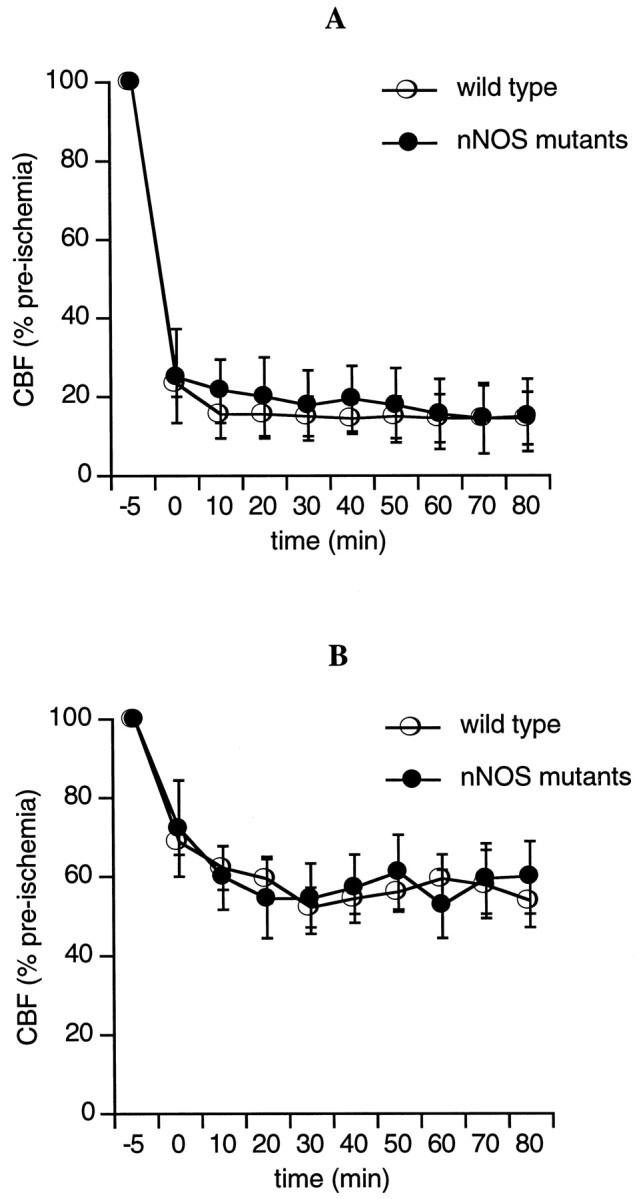
Laser doppler flowmeter probes showed that focal ischemia resulted in severe blood flow deficits within the central zones of the ischemic distribution (A) and mild to moderate blood flow deficits in the ischemic periphery (B). There were no differences in the degree of blood flow reductions between wild-type and nNOS mutant mice.
Neurotransmitter and neuromodulator release
Before cerebral ischemia, there were no differences in baseline concentrations of amino acid neurotransmitters and neuromodulators between wild-type and nNOS mutant mice. After onset of focal ischemia, large and rapid elevations in neurotransmitters (l-Glu and GABA) occurred in the central zones of ischemia in wild-type mice (Fig.4). However, in the NOS mutants, ischemic efflux of l-Glu and GABA were significantly reduced (Fig.4, Table 2). For other amino acids, including the NMDA receptor site modulators d-Ser and Gly, the inhibitory modulator Tau, and the lipid component PEA, focal ischemia induced statistically comparable levels of release in wild-type and mutant mice (Fig. 4). Similarly, the non-neuroactive amino acids l-Ser and l-Ala showed slow progressive accumulations after ischemia in all mice (Fig. 4).
Fig. 4.

Microdialysis measurements of extracellular amino acid concentrations in the center of the ischemic zone showed large elevations in almost all amino acids. Two-way repeated measures ANOVA showed that elevations in l-Glu (F = 5.645; p = 0.04) and GABA (F = 7.422; p = 0.03) were significantly lower in nNOS mutant mice (•) compared with wild-type littermates (○). See Table2 for quantitative efflux parameters. *p < 0.05 via post hoc Fisher’s PLSD analyses.
Table 2.
Efflux parameters in central ischemic zone
| Maximum efflux (fold increase over baseline) | Integrated efflux (μm min) | |||
|---|---|---|---|---|
| WT | nNOS mutants | WT | nNOS mutants | |
| l-Glu | 71 ± 30 | 19 ± 11* | 791 ± 261 | 227 ± 94* |
| GABA | 50 ± 14 | 11 ± 4* | 187 ± 67 | 40 ± 19* |
*p < 0.05 between wild-type (WT) and mutant mice.
In the ischemic periphery, amino acid efflux was considerably lower compared with that found in the central ischemic zone. Significant efflux over baseline was only detected for l-Glu, GABA, Tau, and PEA (Fig. 5). No elevations ind-Ser, Gly, l-Ser, and l-Ala were observed. There were no differences in efflux between nNOS mutants and their wild-type littermates.
Fig. 5.

Microdialysis measurements of extracellular amino acid concentrations in the peripheral zones of the ischemic distribution showed small elevations in l-Glu, GABA, Tau, and PEA. No changes in d-Ser, Gly, l-Ser, andl-Ala were observed after ischemic onset. There were no significant differences in response between wild-type (○) and mutant (•) mice.
Ischemic depolarizations
In the central zone of ischemia, a rapid negative shift in DC potential was observed, consistent with anoxic depolarization (Fig.6A). There were no differences between the magnitude of these depolarizations in wild-type mice and nNOS mutant mice (approximately −20 mV; Table3). In contrast, electrical recordings in the ischemic periphery revealed the presence of multiple SD-like depolarizations (Fig. 6B). These waves of depolarization typically involved transient negative DC shifts of ∼6–8 mV. There were no differences in the magnitude of these depolarizations in wild-type versus nNOS mutant mice. However, the number of SD-like depolarizations was significantly reduced in the nNOS mutants compared with their wild-type littermates (Table 3).
Fig. 6.

Representative examples of electrophysiological DC potential measurements after onset of focal ischemia. A, In the central zones of ischemia, rapid negative shifts in DC potential occurred. B, In contrast, transient SD-like events were present in the ischemic periphery. The data shown here are from a wild-type mouse. Vertical lines at the beginning of the traces indicate start and completion of the electrocoagulation procedure for middle cerebral artery occlusion.
Table 3.
Electrophysiological parameters after ischemia
| Central zone | Peripheral zone | |
|---|---|---|
| DC shifts (mV) | ||
| WT | −21.4 ± 3.4 | NA |
| nNOS mutants | −21.5 ± 2.3 | NA |
| No. of SD-like events | ||
| WT | 0 | 5.7 ± 0.6 |
| nNOS mutants | 0 | 3.0 ± 0.5* |
| Magnitude of SD-like events (mV) | ||
| WT | NA | −6.8 ± 3.4 |
| nNOS mutants | NA | −8.3 ± 2.5 |
NA, Not applicable.
*p < 0.05 between wild-type (WT) and mutant mice.
Infarct size and correlations with amino acid efflux and SD-like events
Permanent occlusion of the distal middle cerebral artery resulted in focal infarction at 24 hr involving the cortex without any detectable striatal damage in all mice. Infarct volumes in the nNOS mutant mice were significantly smaller compared with their wild-type littermates (Fig. 7A,B). The sizes of the infarcts were linearly correlated with the number of SD-like events measured in the ischemic periphery (r = 0.704; p < 0.01). Infarct sizes were also linearly correlated with the cumulative efflux of l-Glu (r = 0.786; p < 0.05) and GABA (r = 0.637; p < 0.05) within the central zones of focal ischemia. However, there were no significant correlations between infarct size and efflux of l-Glu or GABA within the ischemic periphery.
Fig. 7.
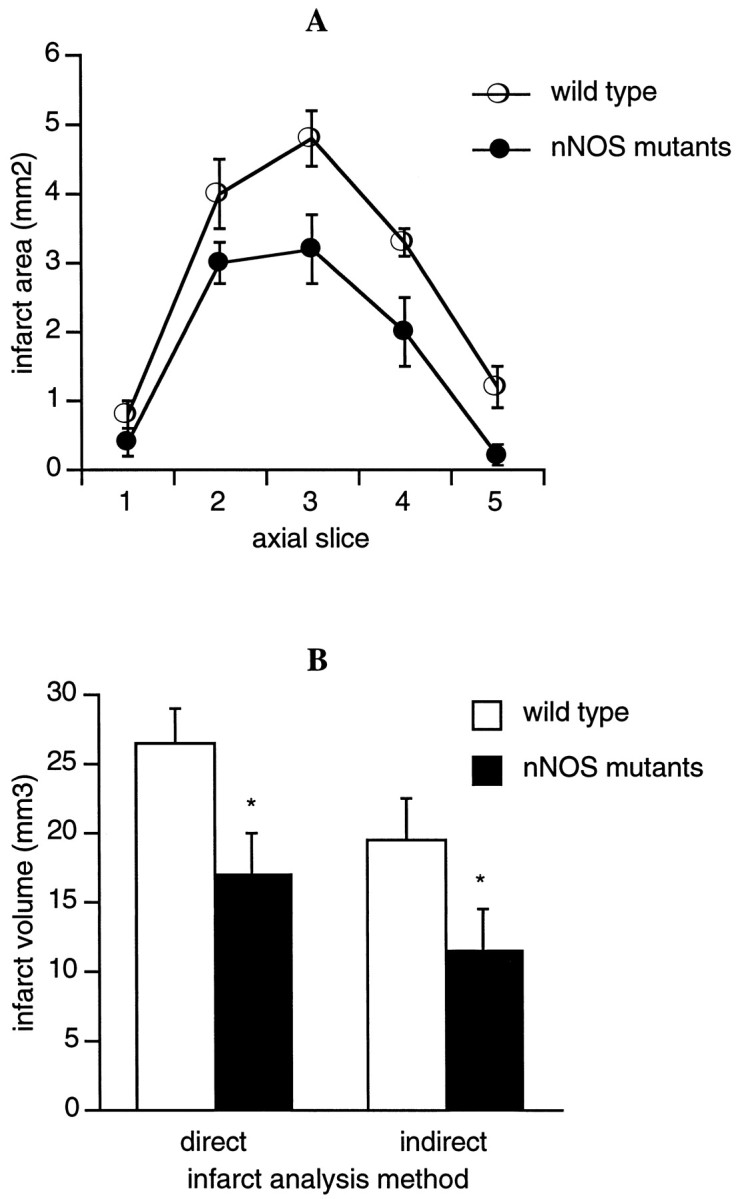
Cortical infarct size after permanent focal cerebral ischemia measured with hematoxylin–eosin staining 24 hr after occlusion. A, Direct infarct areas measured on representative axial slices. B, Infarct volumes were calculated with both “direct” and “indirect” approaches (see Materials and Methods). Infarct volumes in nNOS mutant mice were significantly smaller compared with their wild-type littermates. *p < 0.05 between wild-type and mutant mice.
DISCUSSION
It has been previously shown that mutant mice with disruptions in the genes encoding for nNOS were significantly protected against cerebral injury after focal (Huang et al., 1994) and global (Panahian et al., 1996) ischemia. In the present study, we showed that reduced cerebral infarction after permanent focal ischemia in these nNOS mutant mice was significantly correlated with (1) decreased efflux of glutamate within the center of the ischemic zone and (2) reduced numbers of SD-like depolarizations in the ischemic periphery.
NO plays a complex role in the pathophysiology of cerebral ischemia (Dawson, 1994; Iadecola et al., 1994; Iadecola, 1997; Huang and Lo, 1998). Depending on the redox milieu, it can either manifest itself as the nitrosonium ion NO+ or the free radical NO⋅ (Lipton et al., 1993). The former molecular species can act in a beneficial manner after cerebral ischemia by downregulating deleterious NMDA currents (Lei et al., 1992), whereas the latter species can contribute to free radical damage to cellular membranes, proteins, and DNA (Beckman, 1994). Another reason for the multiplicity of action of NO involves the many potential sources of NO in brain. Three isoforms of NOS have been described: type I or nNOS, type II or inducible NOS (iNOS), and type III or endothelial NOS (eNOS). NO derived from nNOS and iNOS isoforms can play detrimental roles in cerebral ischemia (Iadecola, 1997). Indeed, mutant mice with disrupted nNOS or iNOS genes suffer from smaller infarcts after focal ischemia compared with wild-type mice (Huang et al., 1994; Iadecola et al., 1997). In contrast, mutant mice with disrupted eNOS genes have larger infarcts after focal ischemia (Huang et al., 1996). The protective role for eNOS may involve hemodynamic mechanisms of compensatory vasodilation and collateral recruitment that sustain peripheral zones of the ischemic distribution (Lo et al., 1996). Additionally, NO can also have effects on platelet response and the inflammatory cascade (Moncada et al., 1991). Based on these multiple actions of NO, it is not surprising that somewhat variable outcomes have been obtained by pharmacological inhibition of NO synthesis in various experimental models of focal cerebral ischemia (Buisson et al., 1992; Dawson et al., 1992; Yamamoto et al., 1992; Yoshida et al., 1994).
Our working hypothesis here is that, in addition to these actions, NO also serves as an amplifier of acute excitotoxicity after cerebral ischemia. This hypothesis is based on a wide spectrum of data showing that NO can potentiate neurotransmitter release in locally defined neuronal networks (Gally et al., 1990; Dickie et al., 1992; Montague et al., 1994; Schuman and Madison, 1994; Jones et al., 1995; Kano et al., 1998). In the present study, excitotoxic efflux of glutamate after focal cerebral ischemia was significantly reduced in mutant mice with disrupted nNOS genes. However, these effects were only present in the central zones of ischemia, and no differences between mutant and wild-type mice were seen in the ischemic periphery. The most likely reason for these results is that, although nNOS is considered a constitutive enzyme, it can be significantly upregulated after cerebral ischemia (Huang and Lo, 1998). In a mouse model of focal ischemia, upregulation of nNOS takes place primarily in the ischemic core (Hara et al., 1997). Direct measurements of NO in a cat model of focal ischemia showed that large elevations in NO concentration only occurred in the ischemic core and not in the ischemic periphery (Ohta et al., 1996). Therefore, it is conceivable that after focal ischemia, nNOS is upregulated in the ischemic core, and the subsequent surge of NO then serves to amplify the efflux of excitotoxic glutamate in these regions. These effects of NO were specific for neurotransmitters only (l-Glu and GABA); no differences in ischemic efflux were seen for the neuromodulators (d-Ser, Gly, Tau, and PEA).
The scenario described above leaves unanswered the question of why there were reduced numbers of SD-like depolarizations but no differences in glutamate efflux in the ischemic periphery of the nNOS mutants compared with wild-type mice. If ischemic SD-like events are indeed mediated by glutamate (Marranes et al., 1988; Fabricius et al., 1993), and ischemic glutamate efflux is amplified by NO, then we should expect to see reduced glutamate release in the peripheral zones of focal ischemia as well. One possible explanation might be that the reduced SD-like depolarizations simply reflected the reduction in overall ischemic injury in the nNOS mutants (Ijima et al., 1992) and merely represented an epiphenomenon not directly related to the presence or absence of nNOS at all. Alternatively, it is possible that amplification of gluatamte efflux within the ischemic periphery may have occurred at the synaptic level without massive spillover into extracellular space. Consistent with this possibility, the release of gluatamte was 10-fold lower in the ischemic periphery compared with the ischemic center. In this case, subtle differences within the penumbra between nNOS mutant mice and their wild-type littermates would not be detectable with the microdialysis technique (Benveniste and Hansen, 1991).
There were significant differences in the profiles of neurotransmitter and neuromodulator efflux in the ischemic center versus ischemic periphery. In the center, massive efflux of almost all amino acids occurred. In the ischemic periphery, however, only the neurotransmitters (l-Glu and GABA) and the neuromodulators (Tau and PEA) were released. No changes in the non-neuroactive amino acids (l-Ser and l-Ala) were detected. Elevations in the extracellular concentrations of non-neuroactive amino acids may reflect decreased extracellular space attributable to astrocytic swelling (Kimelberg, 1995). These data suggest that severe tissue injury with massive astrocytic swelling occurs only in the ischemic core and not the ischemic periphery. The NMDA receptor site modulators d-Ser and Gly also showed similar profiles, being significantly released only in the ischemic center and not the ischemic periphery. Recently, it has been suggested thatd-Ser is primarily localized within astrocytes and may be released after stimulation of AMPA receptors that are present on astrocytic cell membranes (Schell et al., 1997). One might speculate that release of d-Ser in the center of the ischemic zone occurs via astrocytic swelling and/or AMPA receptor activation. In the latter scenario, the lack of differences in d-Ser release between wild-type mice and nNOS mutants may be explained by the fact that nNOS activation is closely linked to the neuronal NMDA receptor rather than the AMPA receptor. In any case, d-Ser and Gly significantly potentiate NMDA currents and can therefore contribute to further excitotoxic injury after cerebral ischemia.
Although reductions in l-Glu efflux in the nNOS mutants would be consistent with decreased excitotoxicity after ischemia, it is important to note that GABA efflux is also significantly reduced. The similarity between ischemic efflux profiles of l-Glu and GABA underscore the fact that both amino acids are considered classical neurotransmitters that are primarily released via calcium-dependent vesicular systems (Hertz, 1979; Tossman et al., 1986). The interpretation of the changes in GABA efflux may be more difficult. GABA, via its inhibitory actions, has been proposed to be potentially neuroprotective after cerebral ischemia (Sternau et al., 1989; Lyden and Hedges, 1992). However, it is known that high concentrations of GABA can be depolarizing (Alger and Nicoll, 1982; Staley et al., 1995;van den Pol et al., 1996), and large doses of GABA have been shown to be exacerbate hypoxic injury in neuronal cell culture models (Muir et al., 1996).
There are a few limitations associated with the present study. First, our measurements were restricted to the acute phase of ischemia only. Therefore, we were unable to determine whether differences in excitotoxic glutamate efflux in the nNOS mutants persisted as the infarct matured over time. However, it has been shown in larger animal models of focal ischemia that sustained elevations in glutamate accompany the propagation of cerebral injury; i.e., excitotoxic mechanisms are likely to contribute to the collapse of the ischemic penumbra over time (Matsumoto et al., 1993). Therefore, it is possible that NO can continue to amplify excitotoxic glutamate efflux even during prolonged ischemia. Second, we were only able to obtain measurements at single locations, whereas the spatial profile of the ischemic penumbra is clearly three-dimensional. Nevertheless, the appropriateness of our selected locations was confirmed with blood flow and electrophysiological measurements. Therefore, it is likely that our data were representative of the overall events in the central and peripheral zones of ischemia. Finally, our correlations between decreased infarct size and reductions in l-Glu efflux or decreased SD-like events in the ischemic periphery cannot unequivocally prove causality. Further experiments at the synaptic or vesicular level may be required.
In conclusion, we showed that reduced infarctions after permanent focal ischemia in nNOS mutant mice were significantly correlated with (1) attenuated efflux of glutamate in the center of the ischemic distribution and (2) reduced numbers of SD-like depolarizations in the ischemic periphery. These results are consistent with the idea that in addition to its other multiple actions after cerebral ischemia, NO can act as an amplifier of acute excitotoxicity.
Footnotes
This work was supported in part by National Institutes of Health Grants R29 NS32806 and R01 NS37074 (to E.H.L.), R01 NS33335 (to P.L.H.), and P50 NS10828 (to M.A.M.) and an Established Investigatorship from the American Heart Association (to P.L.H.).
Correspondence should be addressed to Dr. Eng H. Lo, Neuroprotection Research Laboratory, Departments of Neurology and Radiology, Harvard Medical School, Massachusetts General Hospital East Building 149, Room 2322, Charlestown, MA 02129.
REFERENCES
- 1.Alger BE, Nicoll RA. Pharmacological evidence for two kinds of GABA receptor on rat hippocampal pyramidal cells studied in vitro. J Physiol (Lond) 1982;328:125–141. doi: 10.1113/jphysiol.1982.sp014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia: the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- 3.Beckman JS. Peroxynitrite vs hydroxyl radical: the role of NO in superoxide-mediated cerebral injury. Ann NY Acad Sci. 1994;738:69–75. doi: 10.1111/j.1749-6632.1994.tb21791.x. [DOI] [PubMed] [Google Scholar]
- 4.Benveniste H, Hansen AJ. Practical aspects of using microdialysis for determination of brain interstitial concentrations. In: Robinson TE, Justice JB, editors. Microdialysis in the neurosciences, Chap 4. Elsevier; Amsterdam: 1991. [Google Scholar]
- 5.Brint S, Jacewicz M, Keissling M, Tanabe J, Pulsinelli W. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral artery and ipsilateral common carotid arteries. J Cereb Blod Flow Metab. 1988;8:474–485. doi: 10.1038/jcbfm.1988.88. [DOI] [PubMed] [Google Scholar]
- 6.Buisson A, Plotkine M, Boulu RG. The neuroprotective effect of a NOS inhibitor in rat focal cerebral ischemia. Br J Pharmacol. 1992;106:766–767. doi: 10.1111/j.1476-5381.1992.tb14410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiamulera C, Terron A, Reggiani A, Cristofori P. Qualitative and quantitative analysis of the progressive cerebral damage after middle cerebral artery occlusion in mice. Brain Res. 1992;606:251–258. doi: 10.1016/0006-8993(93)90992-v. [DOI] [PubMed] [Google Scholar]
- 8.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 9.Dawson DA. Nitric oxide and focal cerebral ischemia: multiplicity of actions and diverse outcomes. Cerebrovasc Brain Metab Rev. 1994;6:299–324. [PubMed] [Google Scholar]
- 10.Dawson DA, Kusumoto K, Graham DI, McCulloch J, Macrae IM. Inhibition of NO synthesis does not reduce infarct volume in rat focal cerebral ischemia. Neurosci Lett. 1992;142:151–154. doi: 10.1016/0304-3940(92)90361-a. [DOI] [PubMed] [Google Scholar]
- 11.Dickie BGM, Lewis MJ, Davies JA. NMDA-induced release of nitric oxide potentiates aspartate overflow from cerebellar slices. Neurosci Lett. 1992;138:145–148. doi: 10.1016/0304-3940(92)90492-p. [DOI] [PubMed] [Google Scholar]
- 12.Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 1993;612:61–69. doi: 10.1016/0006-8993(93)91644-8. [DOI] [PubMed] [Google Scholar]
- 13.Gally JA, Montague PR, Reeke GN, Edelman GM. The NO hypothesis: possible effects of a short-lived rapidly diffusible signal in the development and function of the nervous system. Proc Natl Acad Sci USA. 1990;87:3547–3551. doi: 10.1073/pnas.87.9.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagberg H, Lehmann A, Sandberg M, Nystrom B, Jacobsen I, Hamberger A. Ischemic-induced shift of inhibitory and excitatory amino acids from intra to extracellular compartments. J Cereb Blood Flow Metab. 1985;5:413–419. doi: 10.1038/jcbfm.1985.56. [DOI] [PubMed] [Google Scholar]
- 15.Hara H, Ayata C, Huang PL, Waeber C, Ayata G, Fuji M, Moskowitz MA. 3H-l-Ng-nitroarginine binding after transient focal ischemia and NMDA induced excitotoxicity in type I and type III NOS null mice. J Cereb Blood Flow Metab. 1997;17:515–526. doi: 10.1097/00004647-199705000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Hertz L. Functional interactions between neurons and astrocytes. Prog Neurobiol. 1979;13:277–323. doi: 10.1016/0301-0082(79)90018-2. [DOI] [PubMed] [Google Scholar]
- 17.Hossman KA. Peri-infarct depolarizations. Cerebrovasc Brain Metab Rev. 1996;8:195–208. [PubMed] [Google Scholar]
- 18.Huang PL, Lo EH (1998) Genetic analysis of NOS isoforms using nNOS and eNOS knockout animals. Prog Brain Res, in press. [DOI] [PubMed]
- 19.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 21.Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, Moskowitz MA. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–987. doi: 10.1097/00004647-199609000-00023. [DOI] [PubMed] [Google Scholar]
- 22.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997;20:132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 23.Iadecola C, Pelligrino DA, Moskowitz MA, Lassen NA. Nitric oxide synthase inhibition and cerebrovascular regulation. J Cereb Blood Flow Metab. 1994;14:175–192. doi: 10.1038/jcbfm.1994.25. [DOI] [PubMed] [Google Scholar]
- 24.Iadecola C, Zhang F, Casey R, Nagayama M, Ross E. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ijima T, Mies G, Hossman KA. Repeated negative DC deflections in rat cortex following MCA occlusions are abolished by MK801. J Cereb Blood Flow Metab. 1992;12:727–733. doi: 10.1038/jcbfm.1992.103. [DOI] [PubMed] [Google Scholar]
- 26.Jones NM, Loiacono RE, Beart PM. Roles for nitric oxide as an intra and interneuronal messenger at NMDA release-regulating receptors: evidence from studies of the NMDA evoked release of 3H-noradrenaline and d-3H-aspartate from rat hippocampal slices. J Neurochem. 1995;64:2057–2063. doi: 10.1046/j.1471-4159.1995.64052057.x. [DOI] [PubMed] [Google Scholar]
- 27.Kano T, Shimizu-Sasamata M, Huang PL, Moskowitz MA, Lo EH. Effects of nitric oxide synthase gene knockout on neurotransmitter release in vivo. Neuroscience. 1998;86:695–699. doi: 10.1016/s0306-4522(98)00179-1. [DOI] [PubMed] [Google Scholar]
- 28.Kemp JA, Leeson PD. The glycine site of the NMDA receptor. Trends Pharmacol Sci. 1993;14:20–25. doi: 10.1016/0165-6147(93)90108-v. [DOI] [PubMed] [Google Scholar]
- 29.Kimelberg HK. Current concepts of brain edema: review of laboratory investigations. J Neurosurg. 1995;83:1051–1059. doi: 10.3171/jns.1995.83.6.1051. [DOI] [PubMed] [Google Scholar]
- 30.Lehmann A, Hagberg H, Hamberger A. A role for taurine in the maintenance of homeostasis in the central nervous system during hyperexcitation? Neurosci Lett. 1984;52:341–343. doi: 10.1016/0304-3940(84)90185-x. [DOI] [PubMed] [Google Scholar]
- 31.Lei SZ, Pan Z, Aggarwal SK, Chen H, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 32.Lin TN, Yong YH, Wa G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal ischemia model in rats. Stroke. 1992;23:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 33.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of NO and related nitroso compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 34.Lo EH, Hara H, Rogowska J, Trocha M, Pierce AR, Huang PL, Fishman MC, Wolf GL, Moskowitz MA. Temporal correlation mapping analysis of the hemodynamic penumbra in mutant mice deficient in endothelial nitric oxide synthase gene expression. Stroke. 1996;27:1381–1386. doi: 10.1161/01.str.27.8.1381. [DOI] [PubMed] [Google Scholar]
- 35.Lo EH, Bosque-Hamilton P, Meng W. Inhibition of PARP: reduction of ischemic injury and attenuation of NMDA-induced neurotransmitter dysregulation. Stroke. 1998a;29:830–836. doi: 10.1161/01.str.29.4.830. [DOI] [PubMed] [Google Scholar]
- 36.Lo EH, Pierce AR, Matsumoto K, Kano T, Evans CJ, Newcomb R. Alterations in K+ evoked profiles of neurotransmitter and neuromodulator amino acids after focal ischemia-reperfusion. Neuroscience. 1998b;83:449–458. doi: 10.1016/s0306-4522(97)00434-x. [DOI] [PubMed] [Google Scholar]
- 37.Lyden PD, Hedges B. Protective effect of synaptic inhibition during cerebral ischemia in rats and rabbits. Stroke. 1992;23:1463–1469. doi: 10.1161/01.str.23.10.1463. [DOI] [PubMed] [Google Scholar]
- 38.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 39.Marranes R, Willems R, Prins ED, Wauquier A. Evidence for a role of the NMDA receptor in cortical spreading depression in the rat. Brain Res. 1988;457:226–240. doi: 10.1016/0006-8993(88)90690-7. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto K, Graf R, Rosner G, Taguchi J, Heiss WD. Elevation of neuroactive substances in the cortex of cats during prolonged focal ischemia. J Cereb Blood Flow Metab. 1993;13:586–594. doi: 10.1038/jcbfm.1993.76. [DOI] [PubMed] [Google Scholar]
- 41.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 42.Montague PR, Gancayco CD, Winn MJ, Marchase RB, Friedlander MJ. Role of NO production in NMDA receptor mediated neurotransmitter release in cerebral cortex. Science. 1994;263:973–977. doi: 10.1126/science.7508638. [DOI] [PubMed] [Google Scholar]
- 43.Muir KK, Lobner D, Monyer H, Choi DW. GABA-A receptor activation attenuates excitotoxicity but exacerbates oxygen-glucose deprivation induced neuronal injury in vitro. J Cereb Blood Flow Metab. 1996;16:1211–1218. doi: 10.1097/00004647-199611000-00015. [DOI] [PubMed] [Google Scholar]
- 44.Obrenovitch TP, Richards DA, Sarna G, Symon L. Combined intracerebral microdialysis and electrophysiological recordings: methods and applications. J Neurosci Methods. 1993;47:139–145. doi: 10.1016/0165-0270(93)90030-u. [DOI] [PubMed] [Google Scholar]
- 45.Ohta K, Graf R, Rosner G, Kumura E, Heiss WD. Early nitric oxide increase in depolarized tissue of cat focal cerebral ischemia. NeuroReport. 1996;8:143–148. doi: 10.1097/00001756-199612200-00029. [DOI] [PubMed] [Google Scholar]
- 46.Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, Fishman MC, Moskowitz MA. Attenuated hippocampal damage after global cerebral ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- 47.Schell MJ, Brady RO, Molliver ME, Snyder SH. d-Serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci. 1997;17:1604–15. doi: 10.1523/JNEUROSCI.17-05-01604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schuman EM, Madison DV. Locally distributed synaptic potentiation in the hippocampus. Science. 1994;263:532–536. doi: 10.1126/science.8290963. [DOI] [PubMed] [Google Scholar]
- 49.Schurr A, Tseng MT, West CA, Rogor BM. Taurine improves recovery of neuronal function following cerebral hypoxia. Life Sci. 1987;40:2059–2066. doi: 10.1016/0024-3205(87)90098-1. [DOI] [PubMed] [Google Scholar]
- 50.Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABA-A receptors. Science. 1995;269:977–981. doi: 10.1126/science.7638623. [DOI] [PubMed] [Google Scholar]
- 51.Sternau LL, Lust WD, Ricci AJ, Ratcheson R. Role for GABA in selective vulnerability in gerbils. Stroke. 1989;20:281–287. doi: 10.1161/01.str.20.2.281. [DOI] [PubMed] [Google Scholar]
- 52.Tossman U, Jonsson G, Ungerstadt U. Regional distribution and extracellular levels of amino acids in rat central nervous system. Acta Physiol Scand. 1986;127:533–545. doi: 10.1111/j.1748-1716.1986.tb07938.x. [DOI] [PubMed] [Google Scholar]
- 53.van den Pol AN, Obrietan K, Chen G. Excitatory actions of GABA after neuronal trauma. J Neurosci. 1996;16:4283–4292. doi: 10.1523/JNEUROSCI.16-13-04283.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood J, Garthwaite J. Models of the diffusional spread of nitric oxide: implications for neural nitric oxide signaling and its pharmacological properties. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 55.Wroblenski TJ, Fadda E, Mazzetta J, Lagarewicz WJ, Crosta EI. Glycine and d-serine as positive modulators of signal transduction at NMDA receptors. Neuropharmacology. 1989;28:447–452. doi: 10.1016/0028-3908(89)90077-4. [DOI] [PubMed] [Google Scholar]
- 56.Yamamoto S, Golanov EV, Berger SB, Reis DJ. Inhibition of NO synthesis increases focal ischemic infarction in rat. J Cereb Blood Flow Metab. 1992;12:717–726. doi: 10.1038/jcbfm.1992.102. [DOI] [PubMed] [Google Scholar]
- 57.Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor 7-nitroindazole decreases focal infarct volume but not the response to topical actylcholine in pial vessels. J Cereb Blood Flow Metab. 1994;14:924–930. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]