

Abstract
Early cellular development was studied in the neocortex of the tish rat. This neurological mutant is seizure-prone and displays cortical heterotopia similar to those observed in certain epileptic patients. The present study demonstrates that a single cortical preplate is formed in a typical superficial position of the developing tish neocortex. In contrast, two cortical plates are formed: one in a normotopic position and a second in a heterotopic position in the intermediate zone. As the normotopic cortical plate is formed, it characteristically separates the subplate cells from the superficial Cajal-Retzius cells. In contrast, the heterotopic cortical plate is not intercalated between the preplate cells because of its deeper position in the developing cortex. Cellular proliferation occurs in two zones of the developing tish cortex. One proliferative zone is located in a typical position in the ventricular/subventricular zone. A second proliferative zone is located in a heterotopic position in the superficial intermediate zone, i.e., between the two cortical plates. This misplaced proliferative zone may contribute cells to both the normotopic and heterotopic cortical plates. Taken together, these findings indicate that misplaced cortical plate cells, but not preplate cells, comprise the heterotopia of the tish cortex. Heterotopic neurogenesis is an early developmental event that is initiated before the migration of most cortical plate cells. It is concluded that misplaced cellular proliferation, in addition to disturbed neuronal migration, can play a key role in the formation of large cortical heterotopia.
Keywords: heterotopia, neurogenesis, cortical development, epilepsy, preplate, tish rat
Malformations of the human neocortex are present in >1% of the general population and in ∼20–40% of intractable epileptics (Meencke and Janz, 1984; Hardiman et al., 1988;Farrell et al., 1992; Meencke and Veith, 1992; Mischel et al., 1995). These malformations range in severity from minor displacements of a few neurons to massive rearrangements of cortical structure. Traditionally, clinical classification systems have lumped most of these malformations into the category of “neuronal migration disorders” (e.g., Palmini et al., 1993). However, it has become increasingly evident that disturbances in other developmental events, such as cellular proliferation and programmed cell death, also contribute to cortical malformations (Rakic, 1988; Evrard et al., 1989; Barkovich et al., 1992, 1996; Rorke, 1994; Eksioglu et al., 1996; Kuida et al., 1996;Brunstrom et al., 1997).
One of the most profound types of human cortical malformation is subcortical band heterotopia (SBH), or double cortex. Subcortical band heterotopia are characterized by a large collection of heterotopic neurons located below the normotopic neocortex (Matell, 1893; Jacob, 1936; Barkovich et al., 1989). Individuals affected with this disorder are subject to intractable epilepsy and mental retardation (Barkovich et al., 1989; Livingston and Aicardi, 1990; Vahldiek et al., 1990;Palmini et al., 1991; Ricci et al., 1992; Soucek et al., 1992;Hashimoto et al., 1993; Iannetti et al., 1993; Battaglia et al., 1994). The developmental mechanisms responsible for many human cortical malformations, including band heterotopia, remain largely unknown. Moreover, the paucity of appropriate animal models of human cortical malformations makes it difficult to examine such issues experimentally. Recently, a seizure-prone mutant rat (tish) was discovered that displays inherited band heterotopia similar to those of the human malformation of SBH (Lee et al., 1997). The heterotopic cells in tish display various features that are characteristic of neocortical cells, including cellular morphology, afferent connectivity, efferent connectivity, and contemporaneous neurogenesis with the neocortex (Lee et al., 1997; Schottler et al., 1998). The tish rat thus represents a genetic animal model for (1) characterizing the developmental mechanisms involved in the formation of a human-like cortical malformation and (2) evaluating the roles of heterotopic neurons in the genesis and maintenance of epilepsy.
To identify potential mechanisms participating in the formation of cortical heterotopia, a series of experiments was undertaken to examine the distribution of Cajal-Retzius and subplate cells. These cells comprise the preplate during early cortical development (Marin-Padilla, 1978) and are believed to influence cortical organization through effects on cellular lamination (D’Arcangelo et al., 1995; Ogawa et al., 1995) and afferent/efferent connectivity (McConnell et al., 1989; Ghosh et al., 1990; DeCarlos and O’Leary, 1992; Ghosh and Shatz, 1992, 1993; Allendoerfer and Shatz, 1994). Disturbances in these cells could have severe consequences for proper cortical development. A second series of experiments examined cellular proliferation and migration in the developing tish cortex. Although band heterotopia are generally regarded as neuronal migration disorders, alterations in cellular proliferation could also contribute to the misplacement of neurons.
MATERIALS AND METHODS
Breeding. All procedures were approved by the University of Virginia Animal Research Committee. Female animals were placed in the home cage of a male animal at 5 P.M. and removed the following morning at 9 A.M. The tish phenotype is inherited in an autosomal recessive pattern (Lee et al., 1997), but this phenotype cannot be recognized easily by visual inspection of the brain until approximately embryonic day 17 (E17). Consequently, experiments in which animals were killed before E18 involved the pairing of (1) two homozygous tish animals to ensure all homozygous (tish/tish) affected offspring or (2) a tish animal and a wild-type (+/+) animal to ensure all heterozygous (tish/+) unaffected offspring. Homozygous breeders were identified by either magnetic resonance imaging of the brain to verify the tish phenotype (Lee et al., 1997) or by histological verification of the tish phenotype in both parents of both of the breeders. In experiments in which animals were killed at later stages of development, one homozygous affected animal was paired with a heterozygous unaffected animal, producing a mix of ∼1:1 affected and unaffected offspring. Heterozygous animals, which exhibit a normal phenotype, were used as controls in all experiments. Vaginal smears were taken the morning after the pairing, and sperm-positive females were placed in maternity cages and given food and waterad libitum. The day on which a sperm-positive smear was identified was defined as E1. The day of birth was defined as postnatal day 1 (P1).
Bromodeoxyuridine labeling. Pregnant dams were injected intraperitoneally on either E15 or E18 with 5-bromo-2′-deoxyuridine (BrdU) [50 μg/g body weight, in 0.007N NaOH with 0.9% NaCl (Sigma, St. Louis, MO)]. Dams were anesthetized with ketamine/xylazine (66:7 mg/kg) at 2 hr or 24 hr after BrdU injection, and pups were removed by hysterotomy. Pups were decapitated, and their heads were fixed by immersion in 70% ethanol overnight. The brains were removed, dehydrated in graded ethanols, cleared in xylenes, and embedded in paraffin. The brains were sectioned coronally at a thickness of 6 μm with a microtome, and sections were mounted on microscope slides. The sections were deparaffinized, rehydrated, and processed for BrdU immunohistochemistry using the avidin–biotin complex (ABC) technique as described in detail by Takahashi et al. (1992). Briefly, sections were immersed in 2N HCl for 30 min, neutralized in PBS for 3 min, and then incubated with primary antibody [anti-BrdU, mouse monoclonal (Becton Dickinson, Cockeysville, MD)] at a dilution of 1:100 in PBS with 0.5% Tween 20 for 60 min. Sections were rinsed in PBS and incubated in biotinylated secondary antibody [Elite anti-mouse IgG kit, 1:200 (Vector Laboratories, Burlingame, CA)] for 30 min. The sections were then processed with ABC (Vector) for 30 min, rinsed in PBS, and visualized with VIP (Vector) or DAB (Sigma). Sections were dehydrated in graded ethanols, cleared in xylenes, and coverslipped using Permount.
In another set of experiments, pregnant dams were injected with BrdU on late E13 or early E14. On P1, the offspring were anesthetized by hypothermia and decapitated, and the brains were immersed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were then sectioned and prepared for BrdU histochemistry as described above, except that the sections were treated with trypsin (0.1% in 0.1 m Tris buffer, pH 7.5, with 0.1% CaCl2) after rehydration.
Calretinin immunohistochemistry. Calretinin-positive neurons were identified in the developing tish and control cortices at E15, E18, and E20. Dams were anesthetized, and the brains of pups were fixed in ethanol, embedded in paraffin, and sectioned coronally as described in the preceding paragraphs. The sections were deparaffinized in xylenes, rehydrated through a series of graded ethanols, and rinsed in distilled water. Incubation with primary antibody [anti-calretinin; rabbit polyclonal (Chemicon, Temecula, CA)] was performed at a dilution of 1:500 for 60 min in the presence of 1% goat serum and 0.5% Tween 20. Sections were rinsed in PBS and incubated in biotinylated secondary antibody (Elite anti-rabbit IgG kit, 1:200; Vector) for 30 min. The sections were then treated with ABC (Vector) for 30 min, rinsed in PBS, and visualized using VIP (Vector) for 5 min. Sections were counterstained with methyl green. Sections were dehydrated in graded ethanols, cleared in xylenes, and coverslipped using Permount.
CR-50 immunohistochemistry. Dams were anesthetized and E20 pups were removed by hysterotomy. The pups were perfused intracardially with heparinized saline and then a fixative of 4% paraformaldehyde in phosphate buffer. The heads were placed in the fixative solution overnight at 4°C. The brains were removed and placed in 25% sucrose in PBS until they sank. The brains were then frozen rapidly and sectioned coronally at a thickness of 20 μm with a cryostat. The sections were placed on microscope slides and processed for CR-50 immunoreactivity in a manner similar to that described by Ogawa et al. (1995). Briefly, sections were preincubated in a blocking solution of 10% horse serum for 30 min. Incubation with primary antibody (CR-50, mouse monoclonal) was performed at a dilution of 1:500 in blocking solution for 60 min. The CR-50 antibody was provided by Dr. Masaharu Ogawa (Kochi Medical School). Sections were rinsed in PBS and incubated in biotinylated secondary antibody (Elite anti-mouse IgG kit, 1:200; Vector) for 30 min. The sections were then treated with ABC (Vector) for 30 min, rinsed in PBS, and visualized using VIP (Vector) for 30 sec. Sections were dehydrated and cleared in graded ethanols and xylenes and then coverslipped using Permount.
Proliferating cell nuclear antigen immunohistochemistry. Proliferating cell nuclear antigen (PCNA)-positive neurons were identified in the developing neocortices of tish and control embryos at E15 and E18. Pregnant dams were anesthetized with ketamine/xylazine (66:7 mg/kg), and pups were removed by hysterotomy. Pups were decapitated, and their heads were fixed overnight by immersion in 70% ethanol. The brains were removed, dehydrated through graded ethanols, cleared in xylenes, and paraffin-embedded. Coronal sections were cut at a thickness of 6 μm with a microtome and slide-mounted. Tissue sections were then deparaffinized in xylenes and rehydrated through graded ethanols and treated with 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4, for 30 min at room temperature. Slides were rinsed and placed in Coplin jars with 1× saline sodium citrate buffer (SSC), pH 6.0, brought to a boil in a microwave oven, and allowed to cool to room temperature. Tissue was neutralized by two rinses in phosphate buffered saline (PBS). The sections were blocked and permeabilized with PBS, pH 7.4, containing 10% horse serum/0.25% Triton X-100 for 30 min. Incubation with primary antibody (anti-PCNA, mouse monoclonal; Novocastra, Newcastle upon Tyne, UK) was performed at a dilution of 1:200 in the above medium for 60 min. Sections were rinsed in PBS and incubated in biotinylated secondary antibody (Elite anti-mouse IgG kit, 1:200; Vector) for 30 min. After a PBS rinse, the sections were processed with ABC (Vector) for 30 min, rinsed in PBS again, and visualized with VIP (Vector). Tissue sections were then dehydrated through graded ethanols, cleared in xylenes, and coverslipped using Permount.
RESULTS
Development of the cortical preplate
The distribution of the calcium-binding protein calretinin was examined in the neocortex of tish and control animals on E15, E18, and E20. Previous work has shown that anti-calretinin antibodies label both Cajal-Retzius cells and subplate cells during early cortical development (Fonseca et al., 1995). At later stages of cortical development (i.e., when subplate cells become separated from the Cajal-Retzius cells by the developing cortical plate), calretinin labeling is most prominent in the subplate, whereas progressively fewer cells are labeled in the marginal zone (Fonseca et al., 1995). In the present study, large- and medium-sized calretinin-positive cells are located in the superficial aspect of the E15 control cortex (Fig.1). The placement and sizes of these cells correspond to those of cortical preplate cells. In E18 control animals, calretinin-positive cells are most prominent at the base of the developing cortical plate, and some labeled cells are present in the developing cortical plate and marginal zone (Fig. 1). By E20, most of the cell labeling is found in the subplate, with a few cells scattered in the cortical plate and marginal zone (Fig. 1). These findings in control animals are consistent with previous observations of calretinin immunoreactivity in the developing rat cortex (Fonseca et al., 1995).
Fig. 1.

Calretinin-positive neurons in the developing cortex. Anti-calretinin immunohistochemistry is shown for control and tish animals at E15, E18, and E20. In each frame, the pial surface is toward the top of the micrograph, and the ventricle is toward the bottom. Calretinin-positive cells (dark purple) are present in the cortical preplate at E15 in both control and tish animals. At E18 and E20, cell staining is most prominent in the subplate at the base of the normotopic cortical plate in both control and tish animals. A few stained cells are present in the marginal zone and the overlying cortical plate in both animals. The arrows in tish E20 indicate the heterotopic cortical plate, and the asterisks indicate the normotopic cortical plate. Scale bars: E15, 50 μm;E18, 100 μm; E20, 200 μm.
The developing tish cortex exhibits a pattern of calretinin immunoreactivity that is generally similar to that of control animals. At E15, cell labeling is concentrated in a superficial position in the developing tish cortex (Fig. 1). By E18, the architectonic organization of the developing tish cortex can be differentiated from that of control animals by the presence of two cortical plates (Fig.2). One cortical plate is located in a normotopic position between the marginal zone and the subplate (Figs.1, 2); this normotopic cortical plate is thinner than that of control animals. A second, heterotopic cortical plate (Figs. 1, 2) is located below the subplate in the intermediate zone. The subventricular and ventricular zones appear thinner in the tish rat than in controls (Fig.2). Calretinin-positive cells are most prominent in the subplate at E18 and E20, with a few scattered cells in the overlying cortical plate and marginal zone (Fig. 1). There are few, if any, calretinin-positive cells in the developing heterotopic cortical plate (Fig. 1). These findings indicate that subplate neurons are restricted to a normal position at the base of the normotopic cortical plate in tish animals.
Fig. 2.
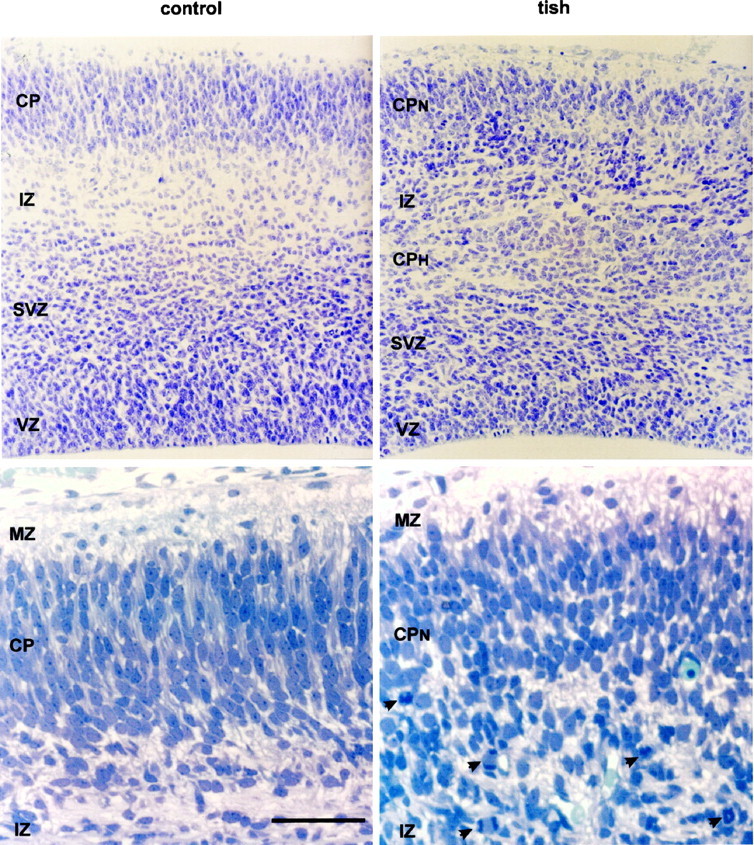
Cytoarchitecture and heterotopic mitotic profiles in the developing tish cortex. Nissl-stained sections are shown fromcontrol and tish animals at E18. Thetop frames are cresyl violet-stained paraffin sections that extend from the pial to the ventricular surfaces. The cortical plate (CP), ventricular zone (VZ), and subventricular zone (SVZ) are thinner in the developing tish cortex than in the developing control cortex. Two unique features of the tish cortex are a heterotopic cortical plate (CPH) and a heterotopic zone of mitotic profiles. The CPH is located in the middle of the area that is normally the intermediate zone (IZ) in control animals. Heterotopic mitotic profiles are located between the CPH and the overlying normotopic cortical plate (CPN) and are positioned in the area that is normally the superficial aspect of the intermediate zone in control animals. The bottom framesshow higher-magnification photomicrographs of thionin-stained semithin sections of the tish and control cortices at E18. The photomicrographs cover the superficial aspect of the developing cortex, including the marginal zone (MZ), cortical plate, and superficial intermediate zone. The marginal zone is relatively cell-sparse, whereas the developing cortical plate contains densely packed neurons in both animals. The superficial intermediate zone of the tish animal contains more cell bodies than the corresponding area of the control animal. In addition, the superficial intermediate zone of tish animals contains mitotic profiles (arrowheads) that exhibit both symmetric and asymmetric cleavage planes. These profiles are indicative of ongoing mitosis in the superficial intermediate zone of the tish cortex. In contrast, mitotic profiles are relatively rare in the superficial intermediate zone of control animals. Scale bar: top frames, 90 μm; bottom frames, 40 μm.
Cajal-Retzius cells produce an extracellular matrix protein (reelin) that is thought to form part of a signaling cascade regulating cortical lamination. A failure in the expression of reelin in Cajal-Retzius cells of the reeler mutant mouse (D’Arcangelo et al., 1995; Ogawa et al., 1995) is associated with improper cortical lamination in which later-generated neurons are incapable of migrating past earlier-generated neurons (Caviness et al., 1988). Ogawa and associates (1995) have developed a monoclonal antibody (CR-50) that recognizes reelin, and immunohistochemical studies demonstrate that this antibody can be used to selectively label Cajal-Retzius cells. In the present experiments, CR-50 immunohistochemistry was used to determine the location of Cajal-Retzius cells in the tish cortex of E20 animals. In control animals, a band of immunoreactivity is present in the marginal zone, with little staining in other layers of the cortex (Fig.3). This pattern of staining is consistent with previous observations in which CR-50 selectively labeled Cajal-Retzius cells in the marginal zone of the mouse cortex (Ogawa et al., 1995). A similar pattern of staining is observed in the marginal zone of tish animals, with little staining in other portions of the normotopic or heterotopic cortices (Fig. 3). These findings suggest that Cajal-Retzius cells are restricted to a normotopic position in the marginal zone of the developing tish cortex.
Fig. 3.
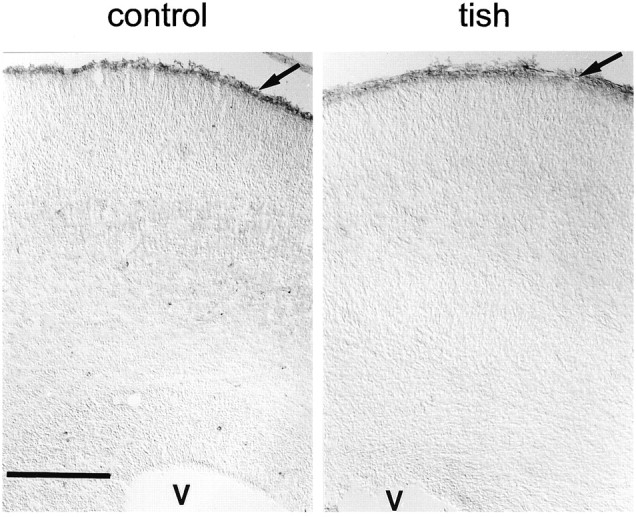
Distribution of reelin in the developing cortex. CR-50 immunohistochemistry is shown for control andtish animals at E20. A band of immunoreactivity is observed in the marginal zone (arrow) of control animals, with little staining seen in other layers of the cortex. A similar band of immunoreactivity is present in the marginal zone (arrow) of tish animals, with little staining in other portions of the normotopic or heterotopic cortices. These sections are not counterstained, and V indicates the position of the ventricle. Scale bar, 200 μm.
It is conceivable that the expression of a particular protein, such as calretinin or reelin, could be disturbed in a neuron, but that the neuron could still be present in appropriate and inappropriate positions. For instance, reelin is not expressed in Cajal-Retzius cells in the cortex of the reeler mouse, although these neurons are present in their appropriate position (Ogawa et al., 1995). The possibility that early-generated preplate neurons are actually present in the heterotopic cortex, but do not express calretinin or reelin, was therefore examined. Pregnant rats were injected with BrdU on late E13 or early E14; this time frame corresponds to the primary period of neurogenesis for Cajal-Retzius and subplate cells in rats (Valverde et al., 1989; Bayer and Altman, 1990, 1991). Offspring were killed on P1 to determine the location of BrdU-positive neurons at a time point when the Cajal-Retzius cells are normally located in the marginal zone and subplate cells are positioned in the subplate. Darkly stained, BrdU-positive cells are concentrated in the marginal zone and subplate of control P1 animals after E13/E14 injections (Fig.4). A similar pattern of BrdU staining is observed in the marginal zone and subplate of tish animals (Fig. 4). In contrast, few if any darkly stained cells are observed in the heterotopic cortex. These findings reinforce the concept that early-generated preplate cells are restricted to their normotopic positions in the tish cortex.
Fig. 4.

Placement of early-generated cells in the developing cortex. The left frames show BrdU-positive cells (stained dark black) in the cortices ofcontrol and tish animals that were labeled in utero with BrdU on late E13 or early E14 and killed on P1. Darkly labeled cells are concentrated in the marginal zone and subplate of both control and tish animals. The heterotopic cortex of the tish animal displays few, if any, BrdU-labeled cells. The right frames show cresyl violet-stained sections from the same animals to provide a reference to the cytoarchitecture. Scale, 200 μm. M, Marginal zone;CP, cortical plate; SP, subplate;WM, white matter; CPN, normotopic cortical plate; CPH, heterotopic cortical plate. Scale bar, 200 μm.
Cellular proliferation and initial migration
Cellular proliferation and migration were investigated in the tish cortex by injecting BrdU into dams on E15 or E18 and killing embryos at early time points after injection. By killing embryos at 2 hr or 24 hr after injection, it is possible to identify the sites of neuronal proliferation and their initial paths of migration, respectively. Two hours after an injection on E15, BrdU-labeled cells in the cortex of control rats are concentrated in a band in the ventricular zone of the developing cortex (Fig.5; E15–2h); this finding is consistent with previous studies of neurogenesis in the rat brain (Bayer and Altman, 1991). In contrast, labeled cells in the E15 tish brain are found in the superficial aspect of the developing cortex, as well as in their typical position in the ventricular zone (Fig. 5;E15–2h). Two hours after an injection on E18, both control and tish rats exhibit characteristic labeling of cells in the ventricular and subventricular zones (Fig. 5; E18–2h). However, in the tish cortex, BrdU-labeled cells are also present in the intermediate zone. A distinct band of labeled cells is present in the superficial aspect of the intermediate zone, which is located below the subplate and above the developing heterotopic cortical plate.
Fig. 5.
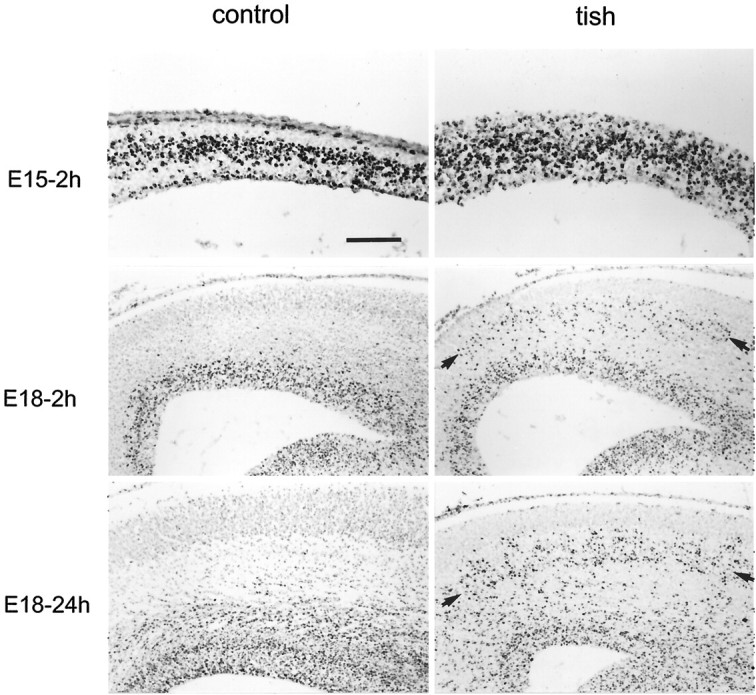
Cellular proliferation and initial migration in the cortex. BrdU-positive cells (stained dark black) are shown in sections from animals that were labeled in utero on E15 or E18 with BrdU. The pial surface of the developing cortex is at the top of each micrograph. Two hours after an injection of BrdU on E15 (E15–2h), labeled cells in control animals are found in a restricted band in the ventricular zone. In the tish rat, BrdU-positive cells are present in both the ventricular zone and in the more superficial aspect of the developing cortex at E15–2h. Two hours after an injection of BrdU on E18 (E18–2h), bothcontrol and tish brains exhibit labeled cells that are characteristically positioned in the vicinity of the ventricular zone. In the tish brain, an additional, heterotopic zone of labeled cells is present in the intermediate zone, with a band of cells in the superficial intermediate zone (arrows). Twenty-four hours after an E18 injection of BrdU (E18–24h), some of the labeled cells have begun to migrate outward from the normotopic (ventricular) proliferative zone in both normal and tish brains. In the tish brain, labeled cells also appear to migrate out of the heterotopic proliferative zone; labeled cells are found in the deep aspect of the normotopic cortical plate and in the superficial aspect of the heterotopic cortical plate (E18–24h). Scale bar: E15–2h, 100 μm;E18–2h, E18–24h, 200 μm.
Twenty-four hours after an E18 injection of BrdU, many labeled cells are moving outward from the normal (ventricular) proliferative zone in both control and tish animals (Fig. 5; E18–24h). These observations are similar to previous findings in the normal rat brain, in which labeled cells are found primarily in the subventricular zone and outer portion of the ventricular zone 24 hr after an injection of [3H]thymidine on E18 (Bayer and Altman, 1991). In tish animals, BrdU labeling in the vicinity of the heterotopic proliferative zone is more dispersed 24 hr after an E18 injection (Fig.5). Some cells from the heterotopic proliferative zone appear to migrate into the overlying normotopic cortical plate, whereas others appear to migrate into the underlying heterotopic cortical plate (Fig.5; E18–24h). However, the possibility cannot be ruled out that rapidly migrating cells originating in the normotopic proliferative zone contribute to these populations of cells. In addition, the heterotopic proliferative zone is relatively broad at E18/E19, making it rather difficult to identify unequivocally the direction of migration of the cells in this area. Longer (i.e., >24 hr) survival times after injection were not examined in detail because the convergence of labeled cells from the two proliferative zones makes it impossible to distinguish the zone of origin of a given labeled cell.
The preceding findings are consistent with the concept that heterotopic cellular proliferation occurs in the developing tish cortex. However, an alternative explanation is that a subset of neurons generated in the normal proliferative zone migrates rapidly into the superficial intermediate zone (Takahashi et al., 1996). Consequently, two additional indices of cellular proliferation were examined to investigate the possibility that heterotopic cellular proliferation occurs in the developing tish cortex. PCNA is present in mitotically competent cells, with the highest levels of expression occurring during S-phase (Morris and Mathews, 1989; Waseem and Lane, 1990; Takahashi and Caviness, 1993). The distribution of PCNA immunoreactivity was examined at E15 and E18 in the developing control and tish brains to identify the sites of cellular proliferation. In E15 control animals, PCNA-positive cells are located in a typical band in the ventricular zone of the developing cortex (Fig. 6), similar to the distribution of BrDU-labeled cells at E15–2h(Fig. 5). In the E15 tish brain, PCNA-positive cells are found in both the ventricular zone and the superficial aspect of the developing cortex. It is unclear whether the heterotopic proliferating cells represent a distinct subpopulation of the progenitor cells; the PCNA-positive cells located in normotopic and heterotopic positions cannot be distinguished from one another on the basis of size or general appearance in the present material. At E18, PCNA-positive cells are restricted to the ventricular and subventricular zones of the control brain (Fig. 6). In contrast, PCNA-positive cells in the tish brain are located in the ventricular and subventricular zones, and in the intermediate zone, with a prominent band of cells in the superficial intermediate zone (Fig. 6). Another index of proliferating cells is the presence of mitotic profiles. The distribution of mitotic profiles was investigated in the E18 neocortex by examining semithin sections stained with thionin. Mitotic profiles in the E18 neocortex are observed in a characteristic position near the cerebral ventricle in both control and tish brains. Additional mitotic profiles are present in the superficial intermediate zone of the tish neocortex but not the control neocortex (Fig. 2). Taken together, these findings confirm the presence of heterotopic cellular proliferation in the developing tish cortex.
Fig. 6.

Distribution of PCNA-positive cells in the developing cortex. In E15 control animals, PCNA-positive cells (purple) are located in a typical band in the ventricular zone. In E18 control animals, labeled cells are also found at typical sites in the ventricular and subventricular zones. In E15 tish animals, PCNA-positive cells are found in both the ventricular zone and in the more superficial aspect of the developing cortex. In E18 tish animals, labeled cells are present in the ventricular and subventricular zones, and in a more superficial (heterotopic) position in the intermediate zone. A prominent band of PCNA-positive cells is present in the superficial aspect of the intermediate zone of the developing tish cortex at this time. Arrows indicate the pial surface of the cortex in the sections from the E15 animals. Scale bars: E15, 50 μm; E18, 200 μm.
DISCUSSION
The widespread use of magnetic resonance imaging in recent years has revealed a high incidence of cortical malformation in patients with epilepsy and cognitive impairment (Barkovich et al., 1989, 1992; Palmini et al., 1991; Kuzniecky et al., 1993; Dobyns et al., 1996; Harding, 1996). Although many human neocortical malformations, including subcortical band heterotopia, are classified as neuronal migration disorders (Barkovich et al., 1989, 1992; Palmini et al., 1993), the etiologies of these anomalies remain poorly understood (Barkovich et al., 1996). Our findings indicate that heterotopic neurogenesis is an early event in the development of a profound malformation in the rat neocortex. Cortical cells proliferate in both heterotopic and normotopic proliferative zones in the developing tish cortex. Cells generated in the heterotopic proliferative zone may migrate into both normotopic and heterotopic cortical plates. These observations indicate that misplaced cellular proliferation, in addition to disturbed neuronal migration, can contribute to the formation of large cortical heterotopia.
Recent evidence indicates that the normotopic and heterotopic cortices of the tish rat exhibit different patterns of neurogenesis (Lee et al., 1997). The normotopic cortex displays a typical inside-out neurogenetic gradient, whereas the heterotopic cortex exhibits a rim-to-core gradient (Fig. 7). The neuronal migration disorder hypothesis for the development of this type of heterotopia (Fig. 7C) predicts that some of the cells originating in the ventricular proliferative zone arrest their migration before reaching appropriate destinations; these cells would then collect to form the heterotopic cortex. The present findings suggest several alternative hypotheses in which heterotopic neurogenesis contributes to the formation of cortical heterotopia. Figure 7D illustrates one variant of the heterotopic neurogenesis hypothesis in which (1) some cells generated in the heterotopic proliferative zone migrate outward to form the normotopic cortex, (2) other cells from the heterotopic proliferative zone migrate inward to form the superficial aspect of the heterotopic cortex, and (3) cells from the normal (ventricular) proliferative zone migrate outward to form the deep aspect of the heterotopic cortex (Fig.7D). This specific hypothesis also predicts that each of the three groups of migrating cells constructs a proximal-to-distal neurogenetic pattern with respect to its initial direction of migration, i.e., later-formed cells migrate past earlier-born cells in each case. In this scenario, inside-out neurogenetic gradients are formed in both the normotopic cortex and deep aspect of the heterotopic cortex, whereas an outside-in pattern is formed in the superficial aspect of the heterotopic cortex. This variant of the heterotopic neurogenesis hypothesis (Fig. 7D) is thus parsimonious with the observations of the present study and with previous observations indicating that earlier-generated cortical plate cells (i.e., corticothalamic and corticospinal neurons) tend to be concentrated in the rim area of the heterotopic cortex (Lee et al., 1997).
Fig. 7.

Two hypotheses of early cellular development in the tish cortex. In A, the general appearance of the adult tish cortex is shown using a Nissl-stained coronal section of the dorsal cortex. B–D are drawings of the tish cortex. Neurogenetic gradients in the tish cortex are illustrated in B. The normotopic cortical plate is constructed in a typical inside-out manner, with earlier-generated cells occupying a deeper position than later-generated cells (Lee et al., 1997). In contrast, the heterotopic cortex develops from its rim toward its core, with earlier-generated cells located in the peripheral aspect (rim) of the structure and later-generated cells located in the central aspect (core) of the structure. In C and D, the proliferative zones have been superimposed onto the drawing of the adult cortex to illustrate their cellular contributions to the developing cortex; this illustration is not intended to represent ongoing proliferation or the position of these zones in the adult brain. The traditionalneuronal migration disorder hypothesis (C) assumes that a subset of neurons derived from a normal (ventricular) proliferative zone fails to reach the normotopic cortical plate, creating a secondary, heterotopic cortical plate. InD, one variant of the heterotopic neurogenesis hypothesis postulates that a heterotopic proliferative zone contributes cells to both the normotopic cortical plate and to the superficial aspect of the heterotopic cortical plate. In this variant of the heterotopic neurogenesis hypothesis, the normotopic (ventricular) proliferative zone provides cells to the deep aspect of the heterotopic cortical plate. It is important to note that all of the possible combinations of contributions from the two proliferative zones are not shown in this figure. For instance, the available data cannot rule out the possibilities that cells from the ventricular proliferative zone migrate into the normotopic cortical plate or that cells from the heterotopic proliferative zone migrate into the deep aspect of the heterotopic cortical plate.
Clearly, a range of plausible variants of the heterotopic neurogenesis hypothesis can be constructed in which the two proliferative zones contribute differentially to the two cortical plates. To discriminate among these possible variants, it will be important for future research to identify directly the relative contributions of the normotopic and heterotopic proliferative zones to the two cortical plates. Moreover, it will be critical to evaluate possible disturbances in glial proliferation and function. The thinning of both the ventricular and subventricular zones in the developing tish cortex suggests that glia, as well as neurons, may be generated in the heterotopic proliferative zone. Heterotopic proliferation of glial cells, in particular radial glial cells, could have a profound effect on cortical development. In the tish rat, it is apparent that many cortical neurons terminate their migration at inappropriate sites and/or migrate in the wrong direction during development. Consequently, any comprehensive hypothesis explaining the development of heterotopia in the tish cortex will ultimately have to account for errors in both migration and proliferation.
Cortical preplate cells, unlike cortical plate cells, are not located heterotopically in the tish cortex. A single preplate is formed in a normotopic position; subplate cells and Cajal-Retzius cells are later separated by the development of the normotopic cortical plate. The absence of a dedicated preplate in the heterotopic cortex could severely impact the development of heterotopic neurons. For instance, subplate cells have been shown to play important roles in establishing appropriate afferent and efferent connectivity of cortical plate cells (Ghosh et al., 1990; DeCarlos and O’Leary, 1992; Ghosh and Shatz, 1992, 1993; Allendoerfer and Shatz, 1994). It is conceivable that heterotopic neurons, deprived of the normal influence of subplate cells, could fail to locate or recognize their synaptic partners. However, despite their heterotopic placement, cortical neurons in the tish rat establish relatively normal afferent and efferent connections (Schottler et al., 1998). These findings reinforce the concept that the relative positioning of subplate cells and cortical plate cells is a rather flexible determinant of regional connectivity in the cortex (Caviness and Yorke, 1976; Steindler and Colwell, 1976; Simmons et al., 1982; Caviness and Frost, 1983; Terashima et al., 1983, 1985; Jensen and Killackey, 1984; Frost et al., 1986; Caviness et al., 1988).
The absence of Cajal-Retzius cells in the heterotopic tish cortex could also affect neuronal organization. During normal cortical development, reelin production in Cajal-Retzius cells is part of a signaling cascade that helps establish proper cortical lamination (D’Arcangelo et al., 1995; Ogawa et al., 1995). Disturbances in this cascade, which may occur after mutations in thereelin, mouse disabled 1 (mdab1), p35, orcdk5 genes, are thought to contribute to cortical lamination errors (D’Arcangelo et al., 1995; Ogawa et al., 1995; Ohshima et al., 1996; Chae et al., 1997; Ware et al., 1997). Animals with mutations of these genes exhibit an inverted lamination of the neocortex (Falconer, 1951; Caviness and Sidman, 1973; Caviness, 1976, 1982; Sweet et al., 1996; Gonzalez et al., 1997). In the case of the reelermutation, this phenotype results from an error in neuronal migration in which later-generated cells fail to migrate past earlier-generated cells (Caviness and Sidman, 1973; Caviness, 1982). The misplacement of cells in the tish cortex could conceivably be caused by an error in this signaling cascade as well. However, the structural malformations in the tish rat differ from those of the reeler, scrambler, and p35-deficient mutant mice in several respects. First, the normotopic cortex of the tish rat exhibits typical lamination, rather than an inversion of lamination. Second, the tish malformation is restricted to only part of the neocortex, whereas the malformations in the other mutants are present in multiple brain regions. Third, the tish rat does not exhibit a generalized defect in the ability of later-generated cells to migrate past earlier-generated cells. Later-generated cells appear to migrate past earlier-generated cells in both the normotopic and heterotopic cortices of the tish rat. Although different influences on the same signaling cascade cannot be ruled out, these findings suggest that the mechanisms responsible for forming cortical heterotopia in the tish rat differ from those involved in the formation of an inverted cortex in the reeler, scrambler, and p35 mutant mice.
The relation between cortical heterotopia in tish rats and SBH in humans warrants consideration. The structural similarities between these two entities are substantial (Barkovich et al., 1989; Lee et al., 1997; Ross et al., 1997). Both are large, bilateral areas of heterotopic gray matter that typically do not extend into the temporal lobe. A thick band of white matter separates the heterotopia from underlying structures, whereas a thinner band of white matter separates the heterotopia from the overlying neocortex. The overlying neocortex retains relatively normal patterns of cellular organization, whereas the heterotopic cells fail to laminate and orient properly. Finally, both tish rats and human SBH patients are prone to exhibit seizures (Barkovich et al., 1989; Lee et al., 1997). Thus, these two disorders bear significant similarities. However, an important feature that differentiates these disorders is their pattern of inheritance. The tish phenotype is inherited as an autosomal recessive trait (Lee et al., 1997), whereas the human SBH syndrome derives from the X-linked gene doublecortin (Pinard et al., 1994; Dobyns et al., 1996;des Portes et al., 1997, 1998; Ross et al., 1997; Gleeson et al., 1998). This difference in chromosomal loci suggests several possibilities, including the following: (1) the same gene is affected in the two disorders, but it is present on different chromosomes in the rat and the human; (2) different genes are affected in the two disorders, but both gene products are essential elements in the same signaling cascade; and (3) different genes are affected and the fundamental mechanisms responsible for forming the heterotopia are different for the two disorders. The initial possibility is unlikely for at least two reasons. First, there exists an extremely high degree of conservation of loci on the X chromosome among mammals. Second, the human syndrome of SBH is observed primarily in females, whereas affected males exhibit a different phenotype with more profound lissencephaly. Because the mutation in the tish rat breeds true as an autosomal recessive, and the profound lissencephalic phenotype is not observed in this animal, it is improbable that the same gene is responsible for the tish rat and human SBH syndromes. The most likely explanation is that two different genes are responsible for the two disorders.
In conclusion, the complex sequence of events required for the development of the neocortex contributes to a surprisingly high incidence of structural malformations in the human brain. Disorders in neuronal proliferation, migration, and programmed cell death undoubtedly play critical roles in many of these anomalies. The present findings in the tish rat suggest a role for misplaced mitotic activity in the development of certain cortical malformations. Heterotopic neurogenesis may be particularly important in the formation of major neocortical malformations (i.e., periventricular heterotopia, band heterotopia, double cortex, and cortical tubers), in which large collections of heterotopic cells underlie the neocortices of epileptic and mentally retarded individuals.
Footnotes
This work was supported by National Institutes of Health Grant NS34124. We thank Dr. Masaharu Ogawa of the Kochi Medical School for kindly providing the CR-50 antibody. We thank Natalie Harrison for excellent technical assistance.
Correspondence should be addressed to Kevin S. Lee, Box 420 HSC, University of Virginia, Charlottesville, VA 22908.
REFERENCES
- 1.Allendoerfer KL, Shatz CJ. The subplate, a transient neocortical structure: its role in the development of connections between thalamus and cortex. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- 2.Barkovich AJ, Jackson DE, Boyer RS. Band heterotopias: a newly recognized neuronal migration anomaly. Radiology. 1989;171:455–458. doi: 10.1148/radiology.171.2.2468173. [DOI] [PubMed] [Google Scholar]
- 3.Barkovich AJ, Gressens P, Evrard P. Formation, maturation, and disorders of brain neocortex. Am J Neuroradiol. 1992;13:423–446. [PMC free article] [PubMed] [Google Scholar]
- 4.Barkovich AJ, Kuzniecky RI, Dobyns WB, Jackson GD, Becker LE, Evrard P. A classification scheme for malformations of cortical development. Neuropediatrics. 1996;27:59–63. doi: 10.1055/s-2007-973750. [DOI] [PubMed] [Google Scholar]
- 5.Battaglia GT, D’Incerti G, Franceschetti S, Zucca C, Savoiardo M, Avanzini G. Ital J Neurol Sci. 1994;15:15–23. doi: 10.1007/BF02343493. [DOI] [PubMed] [Google Scholar]
- 6.Bayer SA, Altman J. Development of layer I and the subplate in rat neocortex. Exp Neurol. 1990;107:48–62. doi: 10.1016/0014-4886(90)90062-w. [DOI] [PubMed] [Google Scholar]
- 7.Bayer SA, Altman J. Neocortical development. Raven; New York: 1991. [Google Scholar]
- 8.Brunstrom JE, Gray-Swain MR, Osborne PA, Pearlman AL. Neuronal heterotopias in the developing cerebral cortex produced by neurotrophin-4. Neuron. 1997;18:505–517. doi: 10.1016/s0896-6273(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 9.Caviness VS., Jr Patterns of cell and fiber distribution in the cortex of the reeler mutant mouse. J Comp Neurol. 1976;170:435–448. doi: 10.1002/cne.901700404. [DOI] [PubMed] [Google Scholar]
- 10.Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon 3H-thymidine autoradiography. Dev Brain Res. 1982;4:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 11.Caviness VS, Jr, Frost DO. Thalamocortical projections in the reeler mutant mouse. J Comp Neurol. 1983;219:182–202. doi: 10.1002/cne.902190205. [DOI] [PubMed] [Google Scholar]
- 12.Caviness VS, Jr, Sidman RL. Time of origin of corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol. 1973;148:141–152. doi: 10.1002/cne.901480202. [DOI] [PubMed] [Google Scholar]
- 13.Caviness VS, Jr, Yorke CH., Jr Interhemispheric neocortical connections of the corpus callosum in the reeler mutant mouse: a study based on anterograde and retrograde methods. J Comp Neurol. 1976;170:449–460. doi: 10.1002/cne.901700405. [DOI] [PubMed] [Google Scholar]
- 14.Caviness VS, Jr, Crandall JE, Edwards MA. The reeler malformation: implications for neocortical histogenesis. In: Peters A, Jones E, editors. Cerebral cortex, Vol 7. Plenum; New York: 1988. pp. 59–89. [Google Scholar]
- 15.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 16.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 17.DeCarlos JA, O’Leary DDM. Growth and targeting of subplate axons and establishment of major cortical pathways. J Neurosci. 1992;12:1194–1211. doi: 10.1523/JNEUROSCI.12-04-01194.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.des Portes V, Pinard JM, Smadja D, Motte J, Boespflug-Tanguy O, Moutard ML, Desguerre I, Billuart P, Carrie A, Bienvenu T, Vinet MC, Bachner L, Beldjord C, Dulac O, Kahn A, Ponsot G, Chelly J. Dominant X-linked subcortical laminar heterotopia and lissencephaly syndrome (X-SCLH/LIS): evidence for the occurrence of mutation in male and mapping of a potential locus in Xq22. J Med Genet. 1997;34:177–183. doi: 10.1136/jmg.34.3.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 20.Dobyns W, Andermann E, Andermann F, Czapansky-Beilman D, Dubeau F, Dulac O, Guerrini R, Hirsch B, Ledbetter D, Lee N, Motte J, Pinard J-M, Radtke RA, Ross ME, Tampieri D, Walsh CA, Truwit CL. X-linked malformations of neuronal migration. Neurology. 1996;47:331–339. doi: 10.1212/wnl.47.2.331. [DOI] [PubMed] [Google Scholar]
- 21.Eksioglu YZ, Scheffer IE, Cardenas P, Knoll J, DiMario F, Ramsby G, Berg M, Kamuro K, Berkovic SF, Duyk GM, Parisi J, Huttenlocher PR, Walsh CA. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77–87. doi: 10.1016/s0896-6273(00)80025-2. [DOI] [PubMed] [Google Scholar]
- 22.Evrard P, de Saint-Georges P, Kadhim H, Gadisseux J-F. Pathology of prenatal encephalopathies. In: French J, editor. Child neurology and developmental disabilities. Paul H Brookes; Baltimore: 1989. pp. 153–176. [Google Scholar]
- 23.Falconer DS. Two new mutants “trembler” and “reeler,” with neurological actions in the house mouse (mus musculus). J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- 24.Farrell MA, De Rosa MJ, Curran JG, Secor DL, Cornford ME, Comair YG, Peacock WJ, Shields WD, Vinters HV. Neuropathologic findings in cortical resections (including hemispherectomies) performed for the treatment of intractable childhood epilepsy. Acta Neuropathol. 1992;83:246–259. doi: 10.1007/BF00296786. [DOI] [PubMed] [Google Scholar]
- 25.Fonseca M, Del Rio J, Martinez A, Gomez S, Soriano E. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol. 1995;361:177–192. doi: 10.1002/cne.903610114. [DOI] [PubMed] [Google Scholar]
- 26.Frost DO, Edwards MA, Sachs GM, Caviness VS., Jr Retinotectal projection in reeler mutant mice: relationships among axon trajectories, arborization patterns and cytoarchitecture. Dev Brain Res. 1986;28:109–120. doi: 10.1016/0165-3806(86)90070-2. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh A, Shatz CJ. Involvement of subplate neurons in the formation of ocular dominance columns. Science. 1992;255:1441–1443. doi: 10.1126/science.1542795. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh A, Shatz CJ. A role for subplate neurons in the patterning of connections from thalamus to neocortex. Development. 1993;117:1031–1047. doi: 10.1242/dev.117.3.1031. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh A, Antonini A, McConnell SK, Shatz CJ. Requirement for subplate neurons in the formation of thalamocortical connections. Nature. 1990;347:179–181. doi: 10.1038/347179a0. [DOI] [PubMed] [Google Scholar]
- 30.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardiman O, Burke T, Phillips J, Murphy S, O’Moore B, Staunton H, Farrell MA. Microdysgenesis in resected temporal neocortex: incidence and clinical significance in focal epilepsy. Neurology. 1988;38:1041–1047. doi: 10.1212/wnl.38.7.1041. [DOI] [PubMed] [Google Scholar]
- 33.Harding B. Gray matter heterotopia. In: Guerrini R, Andermann F, Canapicchi R, Roger J, Zilfkin B, Pfanner P, editors. Dysplasias of cerebral cortex and epilepsy. Lippincott-Raven; Philadelphia: 1996. pp. 81–88. [Google Scholar]
- 34.Hashimoto R, Seki T, Takuma Y, Suzuki N. The “double cortex” syndrome on MRI. Brain Dev. 1993;15:57–59. doi: 10.1016/0387-7604(93)90007-u. [DOI] [PubMed] [Google Scholar]
- 35.Iannetti P, Raucci U, Basile LA, Spalice A, DiBiasi C, Trasimeni G, Gualdi GF. Neuronal migrational disorders: diffuse cortical dysplasia or the “double cortex” syndrome. Acta Paediatr. 1993;82:501–503. doi: 10.1111/j.1651-2227.1993.tb12735.x. [DOI] [PubMed] [Google Scholar]
- 36.Jacob H. Faktoren bei der Entstehung der normalen und entwicklungsgestorten Hirnrinde. Z Neurol u Psych. 1936;155:1–39. [Google Scholar]
- 37.Jensen KF, Killackey HP. Subcortical projections from ectopic neocortical neurons. Proc Natl Acad Sci USA. 1984;81:964–968. doi: 10.1073/pnas.81.3.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasyuama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 39.Kuzniecky R, Murro A, King D, Morawetz R, Smith J, Powers R, Yaghmai F, Faught E, Gallagher B, Snead OC. Magnetic resonance imaging in childhood intractable partial epilepsy: pathologic correlations. Neurology. 1993;43:681–687. doi: 10.1212/wnl.43.4.681. [DOI] [PubMed] [Google Scholar]
- 40.Lee KS, Schottler F, Collins JL, Lanzino G, Couture D, Rao A, Hiramatsu KI, Goto Y, Hong S-C, Caner H, Yamamoto H, Chen Z-F, Bertram E, Berr S, Omary R, Scrable H, Jackson T, Goble J, Eisenman L. A genetic animal model of human neocortical heterotopia associated with seizures. J Neurosci. 1997;17:6236–6242. doi: 10.1523/JNEUROSCI.17-16-06236.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Livingston J, Aicardi J. Unusual MRI appearance of diffuse subcortical heterotopia or “double cortex” in two children. J Neurol Neurosurg Psychiatry. 1990;53:617–620. doi: 10.1136/jnnp.53.7.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marin-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol. 1978;152:109–126. doi: 10.1007/BF00315920. [DOI] [PubMed] [Google Scholar]
- 43.Matell M. Ein Fall von Heterotopie der grauen Substanz in den beiden Hemispheren des Grosshirns. Arch Psychiatr Nervenkr. 1893;25:124–136. [Google Scholar]
- 44.McConnell SK, Ghosh A, Shatz CJ. Subplate neurons pioneer the first axon pathway from the cerebral cortex. Science. 1989;245:978–982. doi: 10.1126/science.2475909. [DOI] [PubMed] [Google Scholar]
- 45.Meencke H-J, Janz D. Neuropathological findings in primary generalized epilepsy: a study of eight cases. Epilepsia. 1984;25:8–21. doi: 10.1111/j.1528-1157.1984.tb04149.x. [DOI] [PubMed] [Google Scholar]
- 46.Meencke H-J, Veith G. Migration disturbances in epilepsy. Epilepsy Res [Suppl] 1992;9:31–40. [PubMed] [Google Scholar]
- 47.Mischel PS, Nguyen LP, Vinters HV. Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol. 1995;54:137–153. doi: 10.1097/00005072-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 48.Morris GF, Mathews MB. Regulation of proliferating cell nuclear antigen during the cell cycle. J Biol Chem. 1989;264:13856–13864. [PubMed] [Google Scholar]
- 49.Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 50.Ohshima T, Ward JM, Huh C-G, Longenecker G, Veeranna Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmini A, Andermann F, Aicardi J, Dulac O, Chaves F, Ponsot G, Pinard JM, Goutieres F, Livingston J, Tampieri D, Andermann E, Robitaille Y. Diffuse cortical dysplasia, or the “double cortex” syndrome: the clinical and epileptic spectrum in 10 patients. Neurology. 1991;41:1656–1662. doi: 10.1212/wnl.41.10.1656. [DOI] [PubMed] [Google Scholar]
- 52.Palmini A, Andermann F, de Grissac H, Tampieri D, Robitaille Y, Langevin P, Desbiens R, Andermann E. Stages and patterns of centrifugal arrest of diffuse neuronal migration disorders. Dev Med Child Neurol. 1993;35:331–339. doi: 10.1111/j.1469-8749.1993.tb11645.x. [DOI] [PubMed] [Google Scholar]
- 53.Pinard J-M, Motte J, Chiron C, Brian R, Andermann E, Dulac O. Subcortical laminary heterotopia and lissencephaly in two families: a single X-linked dominant gene. J Neurol Neurosurg Psychiatry. 1994;57:914–920. doi: 10.1136/jnnp.57.8.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rakic P. Defects of neuronal migration and the pathogenesis of cortical malformations. Prog Brain Res. 1988;73:15–37. doi: 10.1016/s0079-6123(08)60494-x. [DOI] [PubMed] [Google Scholar]
- 55.Ricci S, Cusmai R, Fariello G, Fusco L, Vigevano F. Double cortex. A neuronal migration anomaly as a possible cause of Lennox-Gastaut syndrome. Arch Neurol. 1992;49:61–64. doi: 10.1001/archneur.1992.00530250065017. [DOI] [PubMed] [Google Scholar]
- 56.Rorke LB. A perspective: the role of disordered genetic control of neurogenesis in the pathogenesis of migration disorders. J Neuropathol Exp Neurol. 1994;53:105–117. doi: 10.1097/00005072-199403000-00001. [DOI] [PubMed] [Google Scholar]
- 57.Ross E, Allen K, Srivastava A, Featherstone T, Gleeson J, Hirsch B, Harding B, Andermann E, Abdullah R, Berg M, Czapansky-Bielman D, Flanders D, Guerrini R, Motté J, Mira A, Scheffer I, Berkovic S, Scaravilli F, King R, Ledbetter D, Schlessinger D, Dobyns W, Walsh C. Linkage and physical mapping of X-linked lissencephaly/SBH (XLIS): a gene causing neuronal migration defects in human brain. Hum Mol Genet. 1997;6:555–562. doi: 10.1093/hmg/6.4.555. [DOI] [PubMed] [Google Scholar]
- 58.Schottler F, Couture D, Rao A, Kahn H, Lee KS. Subcortical connections of normotopic and heterotopic neurons in sensory and motor cortices of the tish mutant rat. J Comp Neurol. 1998;395:29–42. [PubMed] [Google Scholar]
- 59.Simmons PA, Lemmon B, Pearlman AL. Afferent and efferent connections of the striate and extrastriate visual cortex of the normal and reeler mouse. J Comp Neurol. 1982;211:295–308. doi: 10.1002/cne.902110308. [DOI] [PubMed] [Google Scholar]
- 60.Soucek D, Birbamer G, Luef G, Felber S, Kristmann E, Bauer G. Laminar heterotopic grey matter (double cortex) in a patient with late onset Lennox-Gastaut syndrome. Wien Klin Wochenschr. 1992;104:607–608. [PubMed] [Google Scholar]
- 61.Steindler DA, Colwell SA. Reeler mutant mouse: maintenance of appropriate and reciprocal connections in the cerebral cortex and thalamus. Brain Res. 1976;105:386–393. doi: 10.1016/0006-8993(76)90949-5. [DOI] [PubMed] [Google Scholar]
- 62.Sweet HO, Bronson R, Johnson K, Cook S, Davisson MT. Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm Genome. 1996;7:798–802. doi: 10.1007/s003359900240. [DOI] [PubMed] [Google Scholar]
- 63.Takahashi T, Caviness VS., Jr PCNA-binding to DNA at the G1/S transition in proliferating cells of the developing cerebral wall. J Neurocytol. 1993;22:1096–1102. doi: 10.1007/BF01235751. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi T, Nowakowski RS, Caviness VS., Jr BUdR as an S-phase marker for quantitative studies of cytokinetic behaviour in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi T, Nowakowski RS, Caviness VS., Jr Interkinetic and migratory behavior of a cohort of neocortical neurons arising in the early embryonic murine cerebral wall. J Neurosci. 1996;16:5762–5776. doi: 10.1523/JNEUROSCI.16-18-05762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Terashima T, Inoue K, Inoue Y, Mikoshiba K, Tsukada Y. Distribution and morphology of corticospinal tract neurons in reeler mouse by the retrograde HRP method. J Comp Neurol. 1983;218:314–326. doi: 10.1002/cne.902180307. [DOI] [PubMed] [Google Scholar]
- 67.Terashima T, Inoue K, Inoue Y, Mikoshiba K, Tsukada Y. Distribution and morphology of callosal commissural neurons within the motor cortex of normal and reeler mice. J Comp Neurol. 1985;232:83–98. doi: 10.1002/cne.902320108. [DOI] [PubMed] [Google Scholar]
- 68.Vahldiek G, Terwey B, Hanefeld F, Sperner J. Magnetic resonance tomography of laminar heterotopia. Fortschr Roentgenstr. 1990;152:378–383. doi: 10.1055/s-2008-1046889. [DOI] [PubMed] [Google Scholar]
- 69.Valverde F, Facal-Valverde MV, Santancana M, Heredia M. Development and differentiation of early generated cells of sublayer VIb in the somatosensory cortex of the rat: a correlated Golgi and autoradiographic study. J Comp Neurol. 1989;290:118–140. doi: 10.1002/cne.902900108. [DOI] [PubMed] [Google Scholar]
- 70.Ware ML, Fox JW, Gonzalez JL, Davis NM, de Rouvroit CL, Russo CJ, Chua SC, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- 71.Waseem NH, Lane DP. Monoclonal antibody analysis of the proliferating cell nuclear antigen (PCNA): structural conservation and the detection of nucleolar form. J Cell Sci. 1990;96:121–129. doi: 10.1242/jcs.96.1.121. [DOI] [PubMed] [Google Scholar]