

Abstract
We studied how metabotropic glutamate receptor (mGluR) activation modifies the synaptic and intrinsic membrane properties of neonatal rat trigeminal motoneurons using the broad-spectrum mGluR agonist (1S,3R)-1-amino-1,3-cyclopentane-dicarboxylic acid [(1S,3R)-ACPD], group I/II antagonist (±)-α-methyl-4-carboxy-phenylglycine (MCPG), and group III agonist l-2-amino-4-phosphonobutanoate (l-AP4). (1S,3R)-ACPD depressed excitatory transmission to trigeminal motoneurons presynaptically and postsynaptically via presynaptic inhibition and by reducing the currents carried by ionotropic glutamate receptors selective for AMPA. (1S,3R)-ACPD also depolarized trigeminal motoneurons and increased input resistance by suppressing a Ba2+-sensitive leakage K+ current. These effects were not mimicked byl-AP4 (100–200 μm). High-threshold Ca2+ currents were also suppressed by (1S,3R)-ACPD. Repetitive stimulation of excitatory premotoneurons mimicked the postsynaptic effects of (1S,3R)-ACPD. The postsynaptic effects of (1S,3R)-ACPD and repetitive stimulation were both antagonized by MCPG, suggesting that mGluRs were similarly activated in both experiments. We conclude that mGluRs can be recruited endogenously by glutamatergic premotoneurons and that mGluR-mediated depression of excitatory transmission, combined with increased postsynaptic excitability, enhances the signal-to-noise ratio of oral-related synaptic input to trigeminal motoneurons during rhythmical jaw movements.
Keywords: (1S,3R)-ACPD; MCPG; leakage K+currents; Ca2+ currents; mEPSCs; mGluR
To understand how the brain generates jaw movements, we must characterize the electrophysiological properties of oral-related brain stem trigeminal neurons and their synaptic connections and determine whether these properties are modulated by endogenous neuromessengers. The anatomy and electrophysiology of the trigeminal motoneurons, which innervate jaw musculature, have been studied (Moore and Appenteng, 1990, 1991; Curtis and Appenteng, 1993; Chandler et al., 1994; Turman and Chandler, 1994;Appenteng et al., 1995). However, neuromodulation of these properties has only been examined for serotonin (Trueblood et al., 1996; Hsiao et al., 1997, 1998).
The endogenous excitatory transmitter glutamate, acting at metabotropic receptors, can modulate neuronal systems by regulating the intrinsic and synaptic properties of constituent neurons (for review, see Gerber and Gähwiler, 1994; Glaum and Miller, 1994; Pin and Duvoisin, 1995). The neurons controlling jaw movements might also be influenced by metabotropic glutamate receptor (mGluR) activation. First, trigeminal motoneurons express mGluRs (Turman et al., 1997) and receive glutamatergic input (Turman and Chandler, 1994; Appenteng et al., 1995) from rhythmically active premotoneurons during masticatory activity (Katakura and Chandler, 1990). Also, continuous high-frequency stimulation of the masticatory cortex is the most effective method to evoke rhythmical jaw movements in vivo (Lund, 1976;Nakamura, 1980; Goldberg et al., 1982). Such repetitive, high-frequency recruitment of excitatory synapses activates mGluRs endogenously (Scanziani et al., 1997).
In specific brain and spinal systems, the diverse effects of mGluR activation modify neuronal function. For instance, in the hippocampus, mGluR activation presynaptically inhibits transmission to the dentate gyrus and CA1 region and enhances pyramidal cell excitability. These effects may amplify the signal-to-noise ratio of transmission through the hippocampus (Conn et al., 1994). In brain stem–spinal networks that generate respiratory rhythms (Smith and Feldman, 1987), (1S,3R)-1-amino-1,3-cyclopentane-dicarboxylic acid [(1S,3R)-ACPD] reduces inspiratory motor output at low agonist concentrations yet enhances inspiratory motor output at high agonist concentrations. These paradoxical effects of mGluR activation were attributed, in part, to differential effects of (1S,3R)-ACPD on the synaptic and intrinsic properties of diaphragmatic motoneurons (Dong et al., 1996).
Unfortunately, the same level of understanding has not been established for the neurons controlling jaw movements. Therefore, we tested the hypothesis that mGluR activation influences the synaptic and intrinsic properties of trigeminal motoneurons in vitro. We examined the regulation of excitatory synaptic transmission to trigeminal motoneurons at presynaptic and postsynaptic sites of action and the modulation of intrinsic membrane properties by mGluR activation. Finally, we provide evidence for the endogenous activation of mGluRs by excitatory trigeminal premotoneurons.
MATERIALS AND METHODS
Whole-cell patch-clamp experiments were performed using a neonatal rat brain stem slice preparation. Rats (0–7 d) were anesthetized by halothane inhalation (Halocarbon Laboratories, River Edge, NJ) and dissected in oxygenated ice-cold cutting solution (see below). Coronal slices (300 μm) were cut (DSK Microslicer, Ted Pella, Redding, CA) and placed into incubation solution (see below) at 37°C for 40 min.
Cutting solution contained (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 1 CaCl2, 5 MgCl2, and 4 lactic acid (Schurr et al., 1988). Recording solution contained (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, and 2 MgCl2. Incubation solution was identical to recording solution with additional 4 mm lactic acid. Solutions were bubbled with 95% O2–5% CO2 and maintained at pH ≅7.3 (22–24°C). When recording Ca2+ currents we used a phosphate-free solution containing (in mm): 105.25 NaCl, 3 KCl, 26 NaHCO3, 10 glucose, 2 CaCl2, 20 tetraethylammonium chloride (TEA-Cl), and 3 CsCl.
Normally, patch electrode solution contained (in mm): 9 NaCl, 140 KCl, 1 MgCl2, 10 HEPES buffer, 0.2 EGTA, 10 phosphocreatine, 0.1 leupeptin, 5 K2-ATP, 1 Na3-GTP, pH ≅7.25 (osmolarity 280–290 mm). When recording synaptic or Ca2+ currents we used a modified patch solution to block K+ currents containing (in mm): 125 cesium methanesulfonate (CsMeSO4), 4 NaCl, 3 KCl, 1 MgCl2, 8 HEPES, 9 EGTA, 10 phosphocreatine, 0.1 leupeptin, 5 K2-ATP, 1 Na3-GTP, pH ≅7.25. Lucifer yellow (0.1%) was added to patch solutions for fluorescent viewing.
Drugs were bath-applied at the following concentrations: (1S,3R)-ACPD (10–100 μm; RBI, Natick, MA), MCPG (1 mm; RBI), l-AP4 (0.1–0.2 mm), bicuculline methiodide [20 μm; Sigma (St. Louis, MO)], strychnine (5 μm; Sigma), 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) (10 μm; RBI),d,l-2-amino-5-phosphonovaleric acid (APV) (10 μm; Sigma), 6-chloro-3,4-dihydro-3-[2-norbornen-5-y]-2H-1,2–4-benzothiadiazine-7-sulfonamide 1,1-dioxide (cyclothiazide) (10 μm; Sigma), glycine (2 μm; Sigma), and tetrodotoxin (TTX) (0.5 μm; Sigma). AMPA (0.5 mm; RBI) and NMDA (1 mm; RBI) were applied by micropressure ejection. Ba2+ (2 mm) and Cd2+ (50–200 μm) were added directly to the bath in some experiments to reduce leakage K+ and Ca2+ currents, respectively.
Slices were perfused by oxygenated recording solution at room temperature and visualized by differential interference contrast microscopy (Edwards et al., 1989; Stuart et al., 1993). The trigeminal motor nucleus was identified bilaterally in the coronal slice under low magnification (5×) as an ellipsoid region, equidistant from the midline and lateral border, ventral to the mesencephalic trigeminal sensory nucleus. The trigeminal motor nucleus could also be distinguished at 40× for visual selection of motoneurons. In some experiments, the trigeminal motor and sensory nuclei were retrogradely and anterogradely labeled, respectively, using Texas red (10%, Molecular Probes, Eugene, OR) for fluorescent viewing. Microinjections of Texas red into the masseter and temporalis jaw closer muscles were performed at least 24 hr before the experiment.
We fabricated patch electrodes from capillary glass (3–7 MΩ; Sutter Instruments P-97, Novato, CA), and recorded using an Axopatch-1D amplifier and pCLAMP software (Axon Instruments, Burlingame, CA). Signals were grounded (Ag/AgCl wire) using a 3 m KCl agar bridge; a 1 mV junction potential was not compensated.
Cell capacitance (CM) for each trigeminal motoneuron recorded in voltage clamp was determined from the integral of capacity current in response to 15 msec hyperpolarizing commands. Uncompensated series resistance (RS) was calculated from the decay time constant (τ) of the transient (τ ≅RSCM). It averaged 17.1 ± 0.9 MΩ and was routinely compensated to 5.3 ± 0.3 MΩ (n = 66) through the amplifier.
We recorded excitatory synaptic currents in the presence of bicuculline and strychnine to block inhibitory ionotropic receptors and used patch solution containing intracellular Cs+ to block K+ currents and improve space clamp. Motoneuronal membrane properties were examined in the presence of bicuculline, strychnine, CNQX, APV, and usually TTX to block inhibitory and excitatory ionotropic receptors and Na+ channels, respectively. During these experiments we used normal patch solution because motoneuronal K+ channels were of interest.
To measure miniature EPSCs (mEPSCs), cells were held at −60 mV in the presence of TTX, and spontaneous synaptic events were recorded at 5 kHz and analog-filtered at 2 kHz for a duration of 2 min. A noise histogram was generated from the baseline current during periods containing no synaptic events (>16,000 points), and a Gaussian distribution was fitted to the histogram to determine the SD of baseline noise. Synaptic events were selected automatically by a threshold-crossing algorithm, with detection level set at, or greater than, two times the SD of baseline noise (Datapac III, Run Technologies, Irvine, CA). To test whether (1S,3R)-ACPD modified the amplitude or frequency of miniature synaptic currents, we used cumulative probability histograms, for amplitude (bin size 0.1–0.5 pA) and interevent interval (minimum bin size 10 msec), and the Kolmogorov–Smirnov statistic (SYSTAT 7.0, SPSS, Chicago, IL). Significance was assessed at p < 0.05.
To evoke excitatory synaptic responses, individual interneurons (0.1–15 μA) or clusters of interneurons (10–150 μA) surrounding the trigeminal motor nucleus were stimulated extracellularly (Grass Instruments, Quincy, MA) using saline-filled pipettes. Ionotropic glutamate receptor currents were measured in trigeminal motoneurons using intracellular Cs+ patch solution in the presence of bicuculline, strychnine, and TTX. Glutamate agonists were pressure-applied locally by a Picospritzer II ejection system (General Valve, Fairfield, NJ) using patch-like pipettes (diameter = 10 μm) and ejection times of 300–750 msec. Differences in the means of evoked synaptic currents and their number of failures and ionotropic glutamate receptor agonist-evoked currents were tested using Student’s t statistic (SYSTAT); significance was assessed at p < 0.05. Means are expressed ± SE of the mean (except for Fig. 1A; see legend).
Fig. 1.
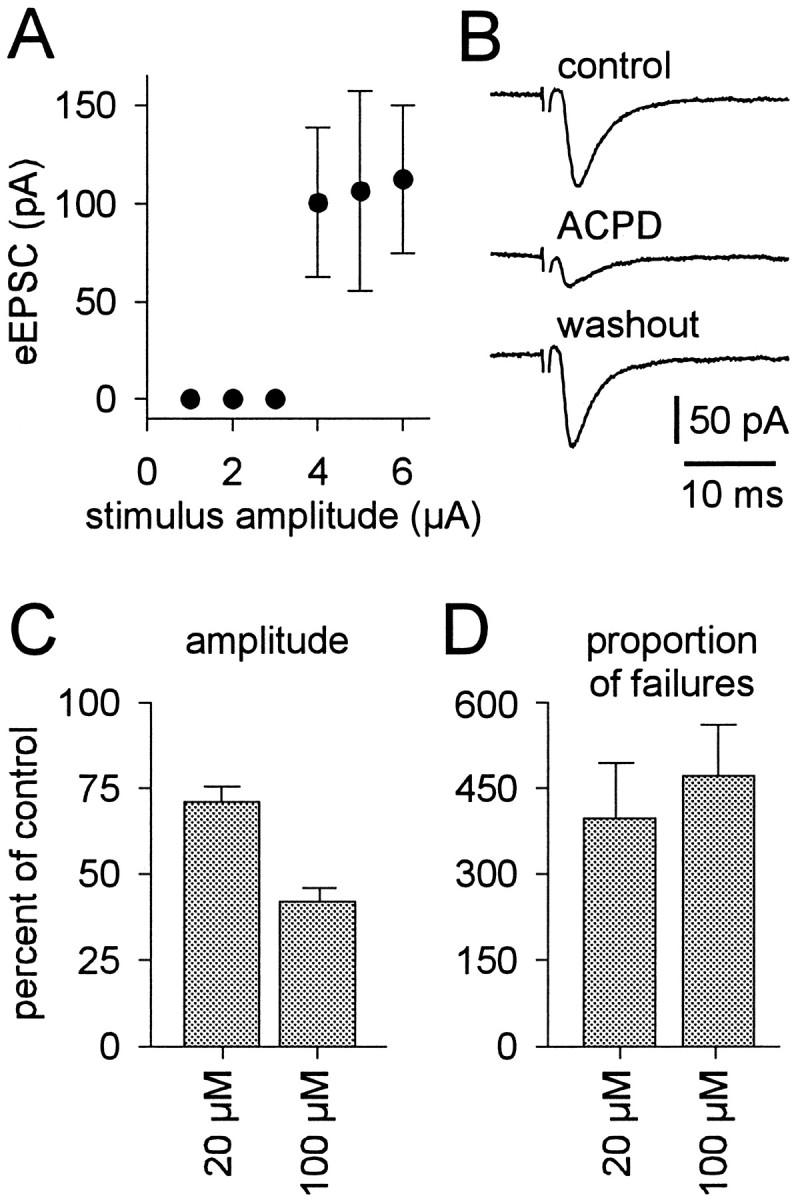
The effects of (1S,3R)-ACPD on unitary evoked synaptic transmission. A, A plot of the relationship between evoked EPSC amplitude (mean ± SD) and stimulus intensity.B, Evoked EPSCs from the cell in A during control (top), 100 μm (1S,3R)-ACPD (middle), and washout (bottom) conditions. Trigeminal motoneurons were recorded with patch solution containing intracellular Cs+ in the presence of bicuculline and strychnine. Holding potential was −60 mV; time and current calibrations are shown. The stimulus artifact is partially deleted. C, D, A summary of the effects of 20 and 100 μm (1S,3R)-ACPD on evoked EPSC amplitude (C) and proportion of synaptic failures relative to control (D). Both ordinate axes reflect percentage of control; bars reflect the mean of all cells tested (± SE).
RESULTS
Depression of excitatory synaptic transmission
We identified unitary connections between excitatory trigeminal premotoneurons and trigeminal motoneurons by stimulating visually identified, putative premotoneurons (0.1–15 μA). If a synaptic connection existed with the target motoneuron, an evoked EPSC was observed only when the stimulus intensity was increased beyond threshold (Fig. 1A, 3 μA). Retracting the stimulating pipette 2–5 μm from the cell soma elevated the threshold. Suprathreshold stimulus intensities did not induce graded responses in evoked EPSC amplitude (Fig.1A). These data indicate that trigeminal premotoneuron–motoneuron synaptic connections were unitary, that is, the result of stimulation of a single presynaptic neuron (Stern et al., 1992).
To test the effects of mGluR activation on evoked synaptic transmission, 20–50 suprathreshold stimuli were applied at 0.3 Hz during control, (1S,3R)-ACPD, and washout conditions. The mean amplitude of the evoked EPSC was calculated from the average of all traces containing a synaptic event, and the number of synaptic failures was noted. (1S,3R)-ACPD reversibly reduced the mean amplitude of the evoked EPSC and increased the number of synaptic failures (Fig.1B, same cell as A). In general, 100 μm (1S,3R)-ACPD significantly decreased the mean amplitude of the evoked EPSC from 59.1 ± 16.0 pA to 24.1 ± 5.0 pA (n = 6). Evoked EPSC amplitude was also significantly reduced by 20 μm (1S,3R)-ACPD from 43.0 ± 9.8 pA to 28.9 ± 4.6 pA (n = 9) (Fig. 1C). The extent to which 20 and 100 μm(1S,3R)-ACPD suppressed the evoked EPSC also differed significantly.
The effect of (1S,3R)-ACPD on synaptic failures was evaluated by the ratio of the number of failures in (1S,3R)-ACPD to the number of failures in control. Failures increased significantly by 472 ± 90% in the presence of 100 μm (1S,3R)-ACPD (n = 6) and by 397 ± 95% in the presence of 20 μm (1S,3R)-ACPD (n = 9) (Fig.1D). The elevation in synaptic failures suggests that mGluR activation lowered the probability of presynaptic release. However, these data could not establish whether the (1S,3R)-ACPD-induced reduction in evoked EPSC amplitude resulted from inhibition of presynaptic release or from postsynaptic modification of excitatory amino acid receptors, or both.
To distinguish presynaptic and postsynaptic effects of mGluR activation, we recorded mEPSCs in trigeminal motoneurons using TTX (Figs. 2,3). (1S,3R)-ACPD (100 μm) reversibly and significantly decreased mEPSC frequency by 2.6 ± 0.9 Hz (57 ± 16% of control) in four of five cells tested; in one cell mEPSC frequency increased by 2.3 Hz. (1S,3R)-ACPD (20 μm) significantly decreased mEPSC frequency by 2.3 ± 1.2 Hz (60 ± 8% of control) in all cells tested (n = 3). The (1S,3R)-ACPD-mediated reduction in mEPSC frequency is shown in sample traces (Fig. 2A) and mEPSC amplitude histograms from the same cell (Fig.2B). These data also suggest that (1S,3R)-ACPD caused presynaptic inhibition.
Fig. 2.

The effects of (1S,3R)-ACPD on miniature synaptic currents. A, Sample traces from a representative TMN in control (top), 100 μm (1S,3R)-ACPD (middle), and washout (bottom) conditions. Trigeminal motoneurons were recorded using patch solution containing intracellular Cs+ in the presence of bicuculline, strychnine, and TTX. Holding potential was −60 mV; time and current calibrations are shown. B, mEPSC amplitude histograms from the cell in A. Histograms for control (open bars) and (1S,3R)-ACPD (closed bars) conditions are superimposed (left). The histogram for washout is shown at right. The ordinate axis (left) applies to both histograms.
Fig. 3.

The effects of (1S,3R)-ACPD on cumulative probability mEPSC amplitude histograms. A, In the same cell as Figure 2, 100 μm (1S,3R)-ACPD caused a leftward shift of the cumulative probability histogram. B, An example of a TMN whose cumulative probability histograms superimposed for control and 20 μm (1S,3R)-ACPD conditions. For bothA and B, control and (1S,3R)-ACPD conditions are distinguished by ○ and •, respectively. Bin size was adjusted to 1 pA for display purposes.
We examined postsynaptic contributions to (1S,3R)-ACPD-mediated synaptic depression using cumulative probability mEPSC amplitude histograms (Fig. 3). These distributions are obtained by integrating the mEPSC amplitude histogram (e.g., Fig. 2B) and normalizing the ordinate range to the interval zero to one. In the majority of trigeminal motoneurons tested, (1S,3R)-ACPD caused a significant leftward shift of the cumulative probability amplitude histogram, indicating a postsynaptic reduction in mEPSC amplitude (Fig.3A; same cell as Fig. 2). This result was observed in four of five cells tested at 100 μm (1S,3R)-ACPD and in one of three cells tested at 20 μm (1S,3R)-ACPD. In all other cells tested, at 20 or 100 μm (1S,3R)-ACPD, the cumulative probability histograms superimposed for control and (1S,3R)-ACPD conditions, and the Kolmogorov–Smirnov statistic was not significant, suggesting no postsynaptic modification of mEPSC amplitude (Fig. 3B). All motoneurons whose cumulative probability histograms superimposed in control and (1S,3R)-ACPD conditions also exhibited an (1S,3R)-ACPD-induced reduction in mEPSC frequency. Therefore, it is unlikely that the reduction in mEPSC frequency could have been an artifact of a reduction in mEPSC amplitude caused by mEPSCs falling below detection levels.
The leftward shift of the cumulative probability histogram could be caused by postsynaptic modulation of excitatory amino acid receptors. To test this possibility, specific excitatory amino acid receptor agonists were locally applied to trigeminal motoneurons in control, (1S,3R)-ACPD (20 or 100 μm), and washout conditions. We used AMPA to activate non-NMDA receptors, while trigeminal motoneurons were held at −60 mV in the presence of the NMDA receptor antagonist APV and cyclothiazide to prevent AMPA receptor desensitization (Hall et al., 1993; Partin et al., 1993; Sharp et al., 1994). (1S,3R)-ACPD (100 μm) significantly and reversibly reduced the peak AMPA-induced current from 2.7 ± 0.5 nA to 1.8 ± 0.3 nA (n = 5) (Fig.4A). (1S,3R)-ACPD (20 μm) reduced the AMPA current from 1.5 ± 0.5 nA to 1.2 ± 0.2 nA (n = 4 of 5), although this was not statistically significant.
Fig. 4.
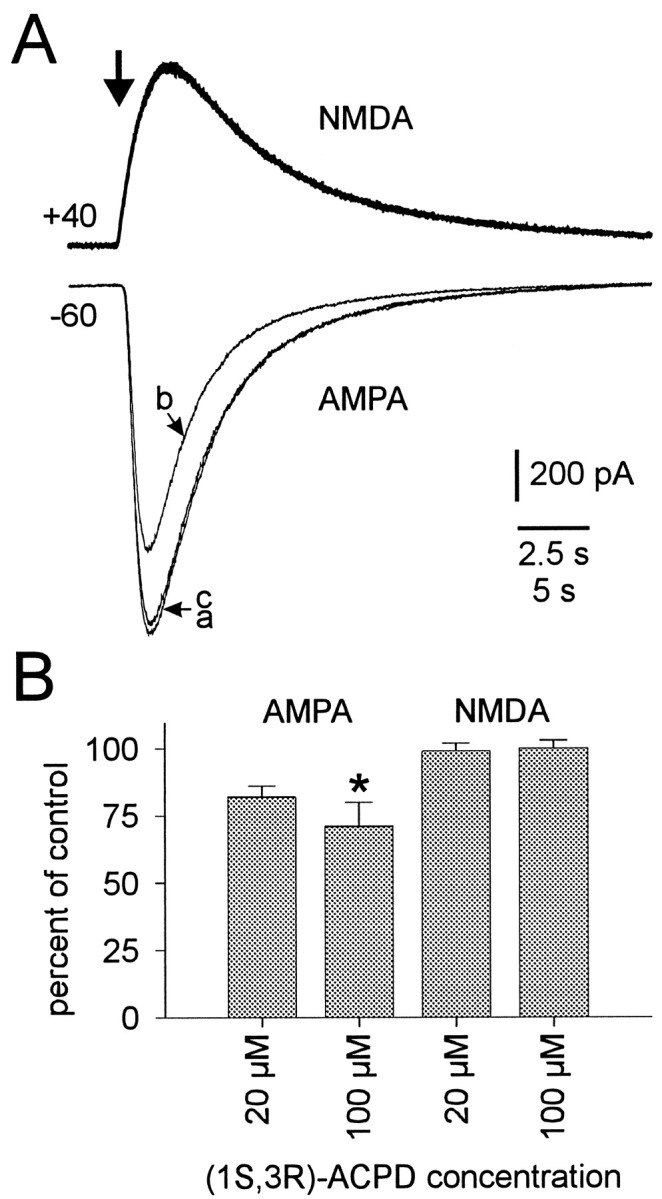
(1S,3R)-ACPD modulation of ionotropic glutamate receptors. A, Current responses of trigeminal motoneurons to specific excitatory amino acid receptor agonists: NMDA (top) and AMPA (bottom). Agonists were applied at the time indicated by the large arrow(duration 300–750 msec). All trigeminal motoneurons were recorded using a patch solution containing intracellular Cs+in the presence of bicuculline and strychnine and other drugs needed to isolate ionotropic glutamate receptor subtypes (see Materials and Methods). Holding potentials and current calibrations are shown. The 2.5 sec time calibration applies to the NMDA traces; the 5 sec time calibration applies to the AMPA traces. Sample NMDA responses during control, 100 μm (1S,3R)-ACPD, and washout conditions are superimposed (top). AMPA responses during control, 100 μm (1S,3R)-ACPD, and washout conditions are differentiated by traces labeled a, b, and c, respectively (bottom).B, A summary of the effects of 20 and 100 μm (1S,3R)-ACPD on ionotropic glutamate receptor currents. A statistically significant change relative to control is indicated by an asterisk.
The potential for modulation of NMDA receptors was tested with NMDA in the presence of glycine and the AMPA receptor antagonist CNQX. Trigeminal motoneurons were held at +40 mV to relieve voltage-dependent block of NMDA channels by Mg2+. Postsynaptic NMDA currents were unaffected by either 20 or 100 μm(1S,3R)-ACPD in all cells tested (n = 3 and 7, respectively) (Fig. 4A, top). The effects of (1S,3R)-ACPD on peak excitatory amino acid agonist responses are summarized in Figure 4B.
To test whether the output of excitatory premotoneurons was affected by mGluR activation, we recorded spontaneous EPSCs in trigeminal motoneurons in the absence of TTX. The recording protocol and statistical tests were identical for spontaneous EPSCs and mEPSCs. In five motoneurons, 100 μm (1S,3R)-ACPD reversibly and significantly increased spontaneous EPSC frequency by 10.4 ± 0.9 Hz (277% of control) (Fig.5A). Because (1S,3R)-ACPD caused presynaptic inhibition (Figs. 1-3), the enhancement of spontaneous EPSC frequency, in the absence of TTX, suggests that the soma–dendritic membranes of premotoneurons were excited by (1S,3R)-ACPD to produce more spikes and augment synaptic output. In contrast, in two of seven cells tested, 100 μm(1S,3R)-ACPD reversibly and significantly decreased spontaneous EPSC frequency by 3.4 Hz (52% of control) (Fig. 5B), suggesting either presynaptic inhibition or disfacilitation of excitatory input to trigeminal motoneurons (TMNs).
Fig. 5.

The effects of (1S,3R)-ACPD on spontaneous synaptic currents. Trigeminal motoneurons were recorded with intracellular Cs+ patch solution and held at −60 mV, in the presence of bicuculline and strychnine. Sample traces during control (top), 100 μm (1S,3R)-ACPD (middle), and washout (bottom) are shown.A, (1S,3R)-ACPD (100 μm) increased spontaneous EPSC frequency. B, Spontaneous EPSC frequency was reversibly reduced by (1S,3R)-ACPD in this cell. Time and current calibrations apply to A andB.
Modulation of TMN excitability
In current clamp, using TTX to prevent excitation of premotoneurons by (1S,3R)-ACPD (e.g., Fig. 5A), 100 μm (1S,3R)-ACPD reversibly depolarized trigeminal motoneurons and increased input resistance (Fig.6A) (n= 3). At a lower dose (20 μm), (1S,3R)-ACPD depolarized the majority of trigeminal motoneurons tested (n = 5 of 7); at 10 μm, (1S,3R)-ACPD was ineffective (n = 3; data not shown). Therefore, 20 μmapproximated the minimum dose required to depolarize trigeminal motoneurons. To examine the effects of (1S,3R)-ACPD in more detail, we measured the input resistance of motoneurons from the slope of the steady-state voltage–current (V–I) relationship constructed from membrane potential responses to 600 msec current step commands. (1S,3R)-ACPD (100 μm) reversibly increased input resistance, most likely by decreasing a tonic K+ conductance, because the steady-stateV–I curves intersected at −90 mV (close to the Nernst potential for K+, −97 mV) (Fig.6B, same cell as A).
Fig. 6.

The effects of (1S,3R)-ACPD on membrane properties of trigeminal motoneurons. All cells were recorded with a normal patch solution. A, (1S,3R)-ACPD (100 μm) caused reversible depolarization and increase in input resistance. Current steps command of 300 msec, −350 pA were delivered at 0.1 Hz;inset shows membrane potential deflections before, during, and after (1S,3R)-ACPD application. B, Steady-state V–I curves for the cell inA before, during, and after a subsequent application of 100 μm (1S,3R)-ACPD. Steady-state voltage deflections were measured at the end of 600 msec current step commands. Control (○), (1S,3R)-ACPD (•), and washout (▪) are shown with regression lines. C, Membrane potential responses to 20 μm (1S,3R)-ACPD in the presence of TTX. The duration of (1S,3R)-ACPD application is shown by bars overlying the voltage trace. Bath application of MCPG is also illustrated by abar overlying the voltage trace. Time breaks in the record are indicated; time and voltage calibrations apply only toC. D, E, Steady-state I–Vcurves obtained from slow voltage ramp protocols (8 mV/sec) were used to calculate the current induced by (1S,3R)-ACPD (IACPD). Cells were bathed in bicuculline, strychnine, CNQX, APV, and TTX. The abscissa and ordinate apply to both D and E.IACPD (○) is shown during control (D) and 1 mm MCPG conditions (E). The reversibility of (1S,3R)-ACPD is indicated by the subtracted current (washout − control;D, •). The inset in Eshows IACPD on an expanded ordinate axis; the abscissa is identical to the main plot (below).
The postsynaptic depolarizing effects of (1S,3R)-ACPD were significantly and reversibly antagonized by preapplication (>6 min) of the group I/II mGluR antagonist MCPG (n = 5) (Eaton et al., 1993; Jane et al., 1993). (1S,3R)-ACPD (20 μm) normally depolarized trigeminal motoneurons by 7.8 ± 1.2 mV. The (1S,3R)-ACPD-induced depolarization decreased significantly and reversibly to 1.0 ± 0.6 mV in the presence of MCPG (Fig.6C), suggesting that (1S,3R)-ACPD depolarized trigeminal motoneurons by recruiting a group I or II mGluR to directly modulate postsynaptic membrane properties.
To further examine the effects of (1S,3R)-ACPD, we measured the steady-state V–I relationship of trigeminal motoneurons in voltage clamp in the presence of bicuculline, strychnine, CNQX, APV, and TTX to synaptically isolate cells and block spikes. We used slow voltage ramps (8 mV/sec) to obtain quasi-steady-state I–V data. To justify the use of ramps, which approximate steady state, theI–V curve was obtained using a series of 2 sec voltage-step commands; this curve superimposed with theI–V relationship obtained from the slow voltage ramp (verified in five trigeminal motoneurons; data not shown). The ramp protocol was preferable because it could collectI–V data more rapidly, thereby minimizing the periods of drug application.
The current induced by 20 μm (1S,3R)-ACPD (IACPD) was obtained by subtracting the current obtained in control from that obtained during (1S,3R)-ACPD application (Fig. 6D, ○).IACPD, when plotted versus membrane potential, was U-shaped in all cells tested at 20 or 100 μm (1S,3R)-ACPD (n = 13 andn = 8, respectively). These data suggested that (1S,3R)-ACPD induced a voltage-dependent inward current in the range −100 to −30 mV, which appeared to reverse at a membrane potential less than −65 mV, and another voltage-dependent current from −30 mV to more depolarized levels, which reversed at a potential greater than −20 mV. The effects of (1S,3R)-ACPD were largely reversible, as shown by the difference current obtained from subtracting control from the washout conditions (Fig. 6D, •).
In separate cells, we measured IACPD in the presence of the group I/II mGluR antagonist MCPG (Fig.6E). Normally, the peak inwardIACPD, measured near −30 mV was −210 ± 31 pA (n = 13). Pre-incubation (>6 min) in MCPG significantly reduced the peakIACPD to −27 ± 14 pA (n = 8) (Fig. 6E). The inset of Figure6E shows that IACPD, plotted versus membrane potential, was still U-shaped, but the magnitude of IACPD was greatly reduced because of MCPG antagonism. Because IACPD was negligible in the presence of MCPG (Fig. 6E), the data are consistent with the current-clamp experiments in which MCPG antagonized the (1S,3R)-ACPD-induced depolarization (Fig. 6C).
To test whether a group III mGluR could also be involved in modulation of motoneuronal membrane properties, we applied the group III agonistl-AP4 at 100 and 200 μm (n = 5 and 3, respectively) in the presence of bicuculline, strychnine, CNQX, APV, and TTX. In current clamp, neither dose of l-AP4 depolarized trigeminal motoneurons or had any affect on the steady-state V–I relationship, suggesting no involvement of group III mGluRs (Fig.7).
Fig. 7.

The effect of 200 μml-AP on membrane properties. Trigeminal motoneurons were recorded with normal patch solution in the presence of bicuculline, strychnine, CNQX, APV, and TTX. Steady-state V–Icurves, obtained from 600 msec current pulses, are superimposed for control (○), l-AP4 (•), and washout (▵) conditions.
Next we identified the intrinsic conductances modulated by (1S,3R)-ACPD. IACPD in the range of −100 to −30 mV reversed near −90 mV (Fig. 6D). Because the reversal potential for K+ is −97 mV (in our conditions) and (1S,3R)-ACPD caused depolarization and increased input resistance, we hypothesized that (1S,3R)-ACPD reduced a leakage K+ current. To test this, Ba2+was added to the bath before (1S,3R)-ACPD in an attempt to occlude modulation of leakage currents. For these and all subsequent experiments, 3 mm Cs+ was used to block the hyperpolarization-activated, mixed cationic, inwardly rectifying current Ih, which is active at potentials more negative than rest in trigeminal (Chandler et al., 1994) and other motoneurons (Takahashi, 1990; Bayliss et al., 1994). The current induced by Ba2+ (IBa) was obtained by subtraction: the difference between Ba2+ conditions and control.IBa was a linear inward current at potentials greater than −100 mV and appeared to reverse at −100 mV (n = 5) (Fig. 8, ○). This is consistent with the blockade of leakage K+channels (Hsiao et al., 1997). We then applied 100 μm(1S,3R)-ACPD. In the voltage range −100 to −30 mV, Ba2+ occluded the effects of (1S,3R)-ACPD (IACPD is negligible at potentials more negative than −30 mV) (Fig. 8, •).
Fig. 8.

The effect of extracellular Ba2+ on IACPD. Trigeminal motoneurons recorded with normal patch solution in the presence of extracellular Cs+, bicuculline, strychnine, CNQX, APV, and TTX. The current induced by Ba2+(IBa) was obtained by subtraction (○). IACPD (100 μm) in the presence of Ba2+ is also shown (•).
In the presence of Ba2+, 100 μm(1S,3R)-ACPD still induced outward current at potentials greater than −30 mV (Fig. 8, •). The outwardly rectifying current induced by (1S,3R)-ACPD could be explained by enhancement of a voltage-dependent K+ current, reduction of a voltage-dependent, noninactivating inward current, or both. To test this, 20 mm TEA-Cl was substituted for NaCl (in normal recording solution containing ionotropic receptor antagonists, TTX, Ba2+, and Cs+) in an attempt to occlude the (1S,3R)-ACPD-induced outward current. The block of TEA-sensitive outward currents exposed a prominent region of inward current (presumably attributable to Ca2+ channels) in the steady-state I–V relationship.IACPD (Fig.9A, bottom) was not occluded by TEA, because under these conditions (1S,3R)-ACPD reversibly reduced the region of inward current at potentials more positive than −30 mV (Fig. 9A, top) (n = 5). However, the outward current induced by (1S,3R)-ACPD was blocked by previous application of 100 μm Cd2+(n = 3) (Fig. 9B). These data suggested that (1S,3R)-ACPD reduced a Cd2+-sensitive, noninactivating, high-threshold Ca2+ current (ICa).
Fig. 9.

The effects of (1S,3R)-ACPD on high-threshold Ca2+ currents. A, Steady-stateI–V curves (not subtracted currents) from a representative cell during control (○), 100 μm(1S,3R)-ACPD (•), and washout conditions (line) are displayed in the top plot. The bottom plot shows the subtracted current,IACPD (ACPD − control).B, The top plot shows raw membrane current. Preapplication of 100 μm Cd2+(•) blocked the effects of 100 μm (1S,3R)-ACPD (gray circles). The subtracted current,IACPD, is shown in the bottom plot. Motoneurons in A and B were recorded with normal patch solution in the presence of TEA, Ba2+, extracellular Cs+, bicuculline, strychnine, CNQX, APV, and TTX. C, The sequence of a typical experiment using intracellular Cs+ patch solution and phosphate-free recording solution with TEA to test mGluR modulation of Ca2+currents. The results are qualitatively identical to Aand B but were obtained in a continuous experiment in one cell.
Modulation of ICa by (1S,3R)-ACPD was further examined under conditions that isolate Ca2+ currents (Fig. 9C). Normal patch electrode solution was replaced by patch solution containing intracellular Cs+, and recording solution was replaced by phosphate-free solution. Under these conditions, intracellular Cs+ minimizes the leakage K+ current (and Ih), and Ca2+ was the only charge carrier for Ca2+ channels. The recording solution also contained TEA to block active K+ currents, including Ca2+-dependent K+ currents (Chandler et al., 1994; Kobayashi et al., 1997). Figure 9Cillustrates a typical experiment in one cell. (1S,3R)-ACPD (100 μm) reversibly reduced ICa. After recovery, 50 μm Cd2+ was applied to block ICa. In the presence of Cd2+, (1S,3R)-ACPD reapplication was ineffective;IACPD was nearly zero over the range of potentials tested (n = 3). These effects were reversible (Fig. 9C).
Endogenous activation of mGluRs
On the basis of the sensitivity of trigeminal motoneurons to (1S,3R)-ACPD, we hypothesized that mGluRs would be activated endogenously during repetitive stimulation of excitatory premotoneurons. To test this, we identified a compound excitatory synaptic connection between several trigeminal premotoneurons and a motoneuron. The compound synaptic connection was more advantageous than a unitary connection because it provided a larger amplitude, more consistent synaptic response. Therefore, it was more likely to activate a larger proportion of postsynaptic glutamate receptors. Trigeminal motoneurons were recorded using normal intracellular patch solution in the presence of bicuculline and strychnine. Groups of cells, visually identified outside the borders of the trigeminal motor nucleus, were stimulated extracellularly. Evoked compound EPSCs were observed at all stimulus intensities. The amplitude of the compound EPSC was graded (until the maximal response), and no synaptic failures were observed (Fig. 10A), demonstrating that the synaptic connection was compound, as opposed to unitary (compare Fig. 1A).
Fig. 10.

Endogenous activation of mGluRs by high-frequency activity in excitatory synapses onto trigeminal motoneurons.A, Compound excitatory synaptic currents in a motoneuron held at −60 mV. Stimulus intensity is indicated by each sample trace (left). Time and current calibrations apply to all traces in A. Stimulus artifacts were partially deleted.B, Schematic diagram of a typical experiment. Pharmacological conditions are indicated by the barsoverlying the protocol. Time breaks are shown. Ramp icons represent the acquisition of a steady-state I–V curve via slow voltage ramp commands (8 mV/sec). The inverted comb icon represents 30 Hz stimulation (40 μA intensity) of premotoneurons for 1 min (as labeled). C, Subtracted currents from the protocols illustrated in B. Abscissa and ordinate axes are identical for all three plots.
Next, the compound EPSC was eliminated by CNQX and APV application (Fig. 10A, bottom). In the presence of ionotropic glutamate receptor antagonists, the effects of mGluR activation are unlikely to be obscured by temporal summation of fast synaptic currents. Figure 10B shows the protocol. The steady-state I–V relationship was obtained as a control (Fig. 10Ba). Premotoneurons were then stimulated at 30 Hz for 1 min to induce glutamate accumulation and mGluR activation (Scanziani et al., 1997). The I–V relationship was acquired immediately after termination of the 30 Hz protocol (Fig.10Bb) to assess whether the protocol modified postsynaptic membrane properties. The current induced by 30 Hz stimulation (I30Hz) was obtained by subtraction (post-30 Hz − control) and is illustrated in Figure10C (b-a). I30Hz was U-shaped, similar to IACPD (e.g., Fig.6D). The I–V relationship was then obtained 1 min after the stimulation protocol to assess the reversibility of these effects (Fig. 10Bc). These effects were partially reversible, with some rundown [60 sec post-30 Hz − control; Fig. 10C (c-a)].
This experiment was then repeated in the presence of MCPG (Fig.10B). Here, the effects of 30 Hz stimulation were antagonized over the entire voltage range, althoughI30Hz was still slightly U-shaped (Fig.10C, e-d). These data resemble the MCPG antagonism of IACPD (Fig. 6E). The stimulation protocol mimicked (1S,3R)-ACPD application, andI30Hz was antagonized by MCPG (n= 6 of 8). This suggests that high-frequency activation of excitatory synapses recruited mGluRs endogenously.
DISCUSSION
In trigeminal motoneurons, mGluR activation depresses excitatory transmission via presynaptic inhibition and postsynaptic depression of ionotropic glutamate receptors selective for AMPA. Also, mGluRs enhance postsynaptic excitability by reducing leakage K+currents and high-threshold Ca2+ currents. How can these seemingly disparate effects influence the neurophysiology of jaw movements?
We propose that mGluR activation enhances the signal-to-noise ratio of oral-related motor commands. The presynaptic and postsynaptic effects of mGluRs inhibit asynchronous sources of synaptic “noise” and accentuate rhythmical oral-related signals composed of slow and synchronous synaptic potentials. Finally, motoneuronal output is augmented via mGluR enhancement of postsynaptic excitability. We elaborate on this hypothesis in our discussion of the data.
According to our hypothesis, mGluRs are activated endogenously by high-frequency recruitment of excitatory synapses. mGluR activation will decrease the amplitude and frequency of fast excitatory synaptic inputs to trigeminal motoneurons. However, excitatory transmission using non-NMDA receptors will be more strongly depressed than transmission mediated by NMDA receptors, because NMDA receptors are not modulated by mGluRs postsynaptically (Fig. 4). Therefore, this favors slow and synchronous sources of synaptic input that, through temporal summation, depolarize trigeminal motoneurons away from the resting potential (−65 mV) and recruit NMDA receptors, to a greater extent, by relieving the voltage-dependent Mg2+ blockade (Mayer et al., 1984; Nowak et al., 1984; Mayer and Westbrook, 1985). The relative effectiveness of fast and asynchronous sources of excitatory input, which are mediated primarily by AMPA receptors near resting potential, will decrease in comparison. This may represent a mechanism to depress sporadic and asynchronous synaptic “noise” and favor rhythmical signals composed of the sum of slow and synchronous synaptic potentials.
The postsynaptic effectiveness of “favored,” synchronous synaptic signals will be amplified by the effects of mGluRs on the intrinsic properties of trigeminal motoneurons. The reduction in leakage K+ current by mGluR activation causes depolarization and increases input resistance. Depolarization brings the cell closer to spike threshold and the threshold for activation of NMDA receptors. Increased input resistance amplifies the gain between synaptic current and membrane voltage response. Therefore, the synaptic currents received by trigeminal motoneurons will cause greater depolarization, leading to NMDA receptor activation and spike discharge. The reduction of high-threshold Ca2+ currents may further augment the frequency–current relationship in trigeminal motoneurons by suppressing Ca2+-dependent K+currents.
Depression of excitatory transmission
The reversible reduction of mEPSC frequency by (1S,3R)-ACPD indicated presynaptic inhibition. Presynaptic inhibition was also indicated by the reversible increase in the number of evoked EPSC failures during (1S,3R)-ACPD application. We believe that this latter effect represents a reduction in presynaptic release probability, as opposed to a failure to excite premotoneurons. Our conclusion is based on indirect evidence, shown in Figure 5A, that excitability was enhanced by (1S,3R)-ACPD in the majority of excitatory premotoneurons.
The mean amplitude of unitary evoked EPSCs in trigeminal motoneurons was consistently depressed by (1S,3R)-ACPD. We attribute this depression to presynaptic and postsynaptic effects, because presynaptic inhibition reduces the quantal content of evoked synaptic events and AMPA receptor depression reduces the postsynaptic effectiveness of the released transmitter.
In contrast to other reports of solely presynaptic mechanisms for (1S,3R)-ACPD-induced depression of excitatory transmission (Gereau and Conn, 1995; Dong et al., 1996; Zorumski et al., 1996), mGluR activation in trigeminal motoneurons also regulates transmission postsynaptically by modulating AMPA receptors. This conclusion is based on the (1S,3R)-ACPD-induced leftward shift of the cumulative probability amplitude histogram and the reversible reduction of AMPA-evoked currents. mGluR-mediated presynaptic inhibition and postsynaptic regulation of AMPA receptors has been reported, but in contrast to trigeminal motoneurons, AMPA receptor currents were enhanced in these cells (Glaum and Miller, 1992, 1993; Glaum et al., 1992, 1993). In these neurons, the differential effects of mGluR activation may sustain the level of excitatory drive by enhancing AMPA receptor currents during presynaptic inhibition.
In contrast, in neostriatal cells and trigeminal motoneurons, presynaptic inhibition is coupled with depression of ionotropic glutamate receptors (Lovinger, 1991; Lovinger et al., 1993; Colwell and Levine, 1994; Lovinger and Tyler, 1996). However, in neostriatal neurons, the NMDA receptor is the postsynaptic target of mGluR regulation, as opposed to the AMPA receptor in trigeminal motoneurons. Therefore, mGluR activation in neostriatal neurons (Colwell and Levine, 1994) and trigeminal motoneurons suppresses excitatory transmission presynaptically and postsynaptically, but by acting at different ionotropic receptors.
We believe that differential regulation of ionotropic glutamate receptors influences the production of rhythmical jaw movements. In trigeminal motoneurons, non-NMDA receptors primarily mediate fast synaptic potentials evoked in vivo by brief cortical stimulation (Katakura and Chandler, 1990) or reflex activation (Chandler, 1989). However, non-NMDA and NMDA receptors are used during cortically induced, sustained rhythmical masticatory activity (Katakura and Chandler, 1990). This cyclical masticatory drive, recorded in vivo in trigeminal motoneurons (Chandler and Goldberg, 1982;Goldberg et al., 1982), closely resembles our proposed favored synaptic signal. This, we argue, is a rhythmical signal, composed of many slow and synchronous synaptic potentials, that recruits both non-NMDA as well as NMDA receptors. Therefore, mGluR activation may help discriminate synaptic noise from fundamental masticatory, or oral-related, motor patterns based on the temporal characteristics of synaptic inputs and the ionotropic receptors they preferentially activate. This signal/noise discrimination is achieved postsynaptically by differentially modulating the ionotropic glutamate receptors.
When synaptic transmission from trigeminal premotoneurons to motoneurons occurred spontaneously in the absence of TTX, (1S,3R)-ACPD increased synaptic activity (spontaneous EPSCs) in the majority of cells. These results imply that the soma–dendritic membranes of most premotoneurons are excited by (1S,3R)-ACPD and overcome presynaptic inhibition through enhanced spike output. In another subset of premotoneurons, spontaneous synaptic activity was reduced by (1S,3R)-ACPD, which we attribute, in part, to presynaptic inhibition. If premotoneuronal mGluRs are endogenously activated during the production of jaw movements, then a subset of cells will be excited. Their elevated spike output could be an additional mechanism for sculpting excitatory input to trigeminal motoneurons, favoring inputs with strong modulation (excitation) by mGluRs.
Modulation of intrinsic properties
The final integration of synaptic input is controlled by the intrinsic membrane properties of trigeminal motoneurons. (1S,3R)-ACPD reduced a Ba2+-sensitive leakage K+ current. Reduction in the leakage current causes depolarization and increased input resistance in trigeminal motoneurons and other neurons (McCormick and von Krosigk, 1992; Crépel et al., 1994; Guérineau et al., 1994; Dong et al., 1996; Mercuri et al., 1996). (1S,3R)-ACPD also reversibly induced a sustained outward current, which activated near −30 mV and was blocked by Cd2+. We conclude that mGluR activation caused a transient reduction in a noninactivating high-threshold Ca2+ current (Sayer et al., 1992; Swartz and Bean, 1992; Trombley and Westbrook, 1992; Sahara and Westbrook, 1993; Stefani et al., 1994). The depression of high-threshold Ca2+currents could indirectly suppress Ca2+-dependent K+ currents in trigeminal motoneurons (Kobayashi et al., 1997). This would increase the excitability of trigeminal motoneurons by reducing spike afterhyperpolarizations and elevating the frequency–current relationship.
These postsynaptic effects of mGluR activation were mediated by a group I or II mGluR because the effects of (1S,3R)-ACPD were antagonized by the group I/II antagonist MCPG, and the postsynaptic effects of (1S,3R)-ACPD were not mimicked by the selective group III agonistl-AP4, even at 200 μm (Fig. 7).
Endogenous activation of mGluRs during jaw movements
Our hypothesis regarding the role of mGluRs in jaw movements requires endogenous activation of the metabotropic receptors during oral-motor activity. The evidence suggests that this probably occurs. First, trigeminal motoneurons express mGluRs (Turman et al., 1997) and receive glutamatergic input (Turman and Chandler, 1994; Appenteng et al., 1995) from rhythmically active premotoneurons during masticatory activity (Katakura and Chandler, 1990). Second, high-frequency activity in excitatory synapses causes glutamate accumulation and endogenous mGluR activation (Scanziani et al., 1997), and the most effective method to evoke rhythmical jaw movements in vivo is continuous high-frequency stimulation of the masticatory cortex (Dellow and Lund, 1971; Lund, 1976; Nakamura, 1980; Goldberg et al., 1982). Last, in vitro, high-frequency activity in excitatory premotoneurons mimicked exogenous mGluR activation and was antagonized by MCPG.
During periods of endogenous mGluR activation, we propose that the presynaptic and postsynaptic regulatory effects of mGluRs work together to discriminate the favored, synchronous synaptic signal from asynchronous synaptic noise and amplify its postsynaptic effectiveness via increased motoneuronal excitability. Thus, mGluRs help sculpt the final oral-related motor output of trigeminal motoneurons.
Footnotes
This work was supported by National Institute of Dental Research Grant RO1 DE-06193. We thank Dr. M. Levine for critical reading of this manuscript and Marvin Z. Castillo for technical contributions.
Correspondence should be addressed to Dr. Scott H. Chandler, Department of Physiological Science, 2851 Slichter Hall, University of California at Los Angeles, Los Angeles, CA 90095-1568.
REFERENCES
- 1.Appenteng K, Curtis JC, Grimwood PD, Min M-Y, Yang H-W. Excitatory synaptic transmission in the rat trigeminal motor nucleus. In: Morimoto T, Matsuya T, Takada K, editors. Brain and oral function: oral motor function and dysfunction. Elsevier; Amsterdam: 1995. pp. 107–114. [Google Scholar]
- 2.Bayliss DA, Viana F, Bellingham MC, Berger AJ. Characteristics and postnatal development of a hyperpolarization-activated inward current in rat hypoglossal motoneurons in vitro. J Neurophysiol. 1994;71:119–128. doi: 10.1152/jn.1994.71.1.119. [DOI] [PubMed] [Google Scholar]
- 3.Chandler SH. Evidence for excitatory amino acid transmission between mesencephalic nucleus of V afferents and jaw-closer motoneurons in the guinea pig. Brain Res. 1989;477:252–264. doi: 10.1016/0006-8993(89)91413-3. [DOI] [PubMed] [Google Scholar]
- 4.Chandler SH, Goldberg LJ. Intracellular analysis of synaptic mechanisms controlling spontaneous and cortically induced rhythmical jaw movements in the guinea pig. J Neurophysiol. 1982;48:126–138. doi: 10.1152/jn.1982.48.1.126. [DOI] [PubMed] [Google Scholar]
- 5.Chandler SH, Hsiao CF, Inoue T, Goldberg LJ. Electrophysiological properties of guinea pig trigeminal motoneurons recorded in vitro. J Neurophysiol. 1994;71:129–145. doi: 10.1152/jn.1994.71.1.129. [DOI] [PubMed] [Google Scholar]
- 6.Colwell CS, Levine MS. Metabotropic glutamate receptors modulate N-methyl-d-aspartate receptor function in neostriatal neurons. Neuroscience. 1994;61:497–507. doi: 10.1016/0306-4522(94)90429-4. [DOI] [PubMed] [Google Scholar]
- 7.Conn PJ, Winder DG, Gereau RW. Regulation of neuronal circuits and animal behavior by metabotropic glutamate receptors. In: Conn PJ, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 195–229. [Google Scholar]
- 8.Crépel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca(2+)-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 9.Curtis JC, Appenteng K. The electrical geometry, electrical properties and synaptic connections onto rat V motoneurones in vitro. J Physiol (Lond) 1993;465:85–119. doi: 10.1113/jphysiol.1993.sp019668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dellow PG, Lund JP. Evidence for central timing of rhythmical mastication. J Physiol (Lond) 1971;215:1–13. doi: 10.1113/jphysiol.1971.sp009454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong XW, Morin D, Feldman JL. Multiple actions of 1S,3R-ACPD in modulating endogenous synaptic transmission to spinal respiratory motoneurons. J Neurosci. 1996;16:4971–4982. doi: 10.1523/JNEUROSCI.16-16-04971.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eaton SA, Jane DE, Jones PL, Porter RH, Pook PC, Sunter DC, Udvarhelyi PM, Roberts PJ, Salt TE, Watkins JC. Competitive antagonism at metabotropic glutamate receptors by (S)-4-carboxyphenylglycine and (RS)-alpha-methyl-4-carboxyphenylglycine. Eur J Pharmacol. 1993;244:195–197. doi: 10.1016/0922-4106(93)90028-8. [DOI] [PubMed] [Google Scholar]
- 13.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 14.Gerber U, Gähwiler BH. Modulation of ionic currents by metabotropic glutamate receptors in the CNS. In: Conn JP, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 125–146. [Google Scholar]
- 15.Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaum SR, Miller RJ. Metabotropic glutamate receptors mediate excitatory transmission in the nucleus of the solitary tract. J Neurosci. 1992;12:2251–2258. doi: 10.1523/JNEUROSCI.12-06-02251.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glaum SR, Miller RJ. Activation of metabotropic glutamate receptors produces reciprocal regulation of ionotropic glutamate and GABA responses in the nucleus tractus solatarius of the rat. J Neurosci. 1993;13:1636–1641. doi: 10.1523/JNEUROSCI.13-04-01636.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glaum SR, Miller RJ. Acute regulation of synaptic transmission by metabotropic glutamate receptors. In: Conn JP, Patel J, editors. The metabotropic glutamate receptors. Humana; Totowa, NJ: 1994. pp. 147–172. [Google Scholar]
- 19.Glaum SR, Slater NT, Rossi DJ, Miller RJ. Role of metabotropic glutamate (ACPD) receptors at the parallel fiber-Purkinje cell synapse. J Neurophysiol. 1992;68:1453–1462. doi: 10.1152/jn.1992.68.4.1453. [DOI] [PubMed] [Google Scholar]
- 20.Glaum SR, Sunter DC, Udvarhelyi PM, Watkins JC, Miller RJ. The actions of phenylglycine derived metabotropic glutamate receptor antagonists on multiple (1S,3R)-ACPD responses in the rat nucleus of the tractus solitarius. Neuropharmacology. 1993;32:1419–1425. doi: 10.1016/0028-3908(93)90039-6. [DOI] [PubMed] [Google Scholar]
- 21.Goldberg LJ, Chandler SH, Tal M. Relationship between jaw movements and trigeminal motoneuron membrane-potential fluctuations during cortically induced rhythmical jaw movements in the guinea pig. J Neurophysiol. 1982;48:110–138. doi: 10.1152/jn.1982.48.1.110. [DOI] [PubMed] [Google Scholar]
- 22.Guérineau NC, Gähwiler BH, Gerber U. Reduction of resting K+ current by metabotropic glutamate and muscarinic receptors in rat CA3 cells: mediation by G-proteins. J Physiol (Lond) 1994;474:27–33. doi: 10.1113/jphysiol.1994.sp019999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hall RA, Kessler M, Quan A, Ambros-Ingerson J, Lynch G. Cyclothiazide decreases [3H]AMPA binding to rat brain membranes: evidence that AMPA receptor desensitization increases agonist affinity. Brain Res. 1993;628:345–348. doi: 10.1016/0006-8993(93)90978-v. [DOI] [PubMed] [Google Scholar]
- 24.Hsiao CF, Trueblood PR, Levine MS, Chandler SH. Multiple effects of serotonin on membrane properties of trigeminal motoneurons in vitro. J Neurophysiol. 1997;77:2910–2924. doi: 10.1152/jn.1997.77.6.2910. [DOI] [PubMed] [Google Scholar]
- 25.Hsiao C-F, Del Negro CA, Trueblood PR, Chandler SH. The ionic basis for serotonin-induced bistable membrane properties in guinea pig trigeminal motoneurons. J Neurophysiol. 1998;79:2847–2856. doi: 10.1152/jn.1998.79.6.2847. [DOI] [PubMed] [Google Scholar]
- 26.Jane DE, Jones PL, Pook PC, Salt TE, Sunter DC, Watkins JC. Stereospecific antagonism by (+)-alpha-methyl-4-carboxyphenylglycine (MCPG) of (1S,3R)-ACPD-induced effects in neonatal rat motoneurones and rat thalamic neurones. Neuropharmacology. 1993;32:725–727. doi: 10.1016/0028-3908(93)90088-k. [DOI] [PubMed] [Google Scholar]
- 27.Katakura N, Chandler SH. An iontophoretic analysis of the pharmacologic mechanisms responsible for trigeminal motoneuronal discharge during masticatory-like activity in the guinea pig. J Neurophysiol. 1990;63:356–369. doi: 10.1152/jn.1990.63.2.356. [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi M, Inoue T, Matsuo R, Masuda Y, Hidaka O, Kang Y, Morimoto T. Role of calcium conductances on spike afterpotentials in rat trigeminal motoneurons. J Neurophysiol. 1997;77:3273–3283. doi: 10.1152/jn.1997.77.6.3273. [DOI] [PubMed] [Google Scholar]
- 29.Lovinger DM. Trans-1-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD) decreases synaptic excitation in rat striatal slices through a presynaptic action. Neurosci Lett. 1991;129:17–21. doi: 10.1016/0304-3940(91)90710-b. [DOI] [PubMed] [Google Scholar]
- 30.Lovinger DM, Tyler E. Synaptic transmission and modulation in the neostriatum. Int Rev Neurobiol. 1996;39:77–111. doi: 10.1016/s0074-7742(08)60664-9. [DOI] [PubMed] [Google Scholar]
- 31.Lovinger DM, Tyler E, Fidler S, Merritt A. Properties of a presynaptic metabotropic glutamate receptor in rat neostriatal slices. J Neurophysiol. 1993;69:1236–1244. doi: 10.1152/jn.1993.69.4.1236. [DOI] [PubMed] [Google Scholar]
- 32.Lund JP. Evidence for a central neural pattern generator regulating the chewing cycle. In: Anderson DJ, Matthews B, editors. Mastication. John Wright and Sons; Bristol, UK: 1976. pp. 204–212. [Google Scholar]
- 33.Mayer ML, Westbrook GL. The action of N-methyl-d-aspartic acid on mouse spinal neurones in culture. J Physiol (Lond) 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 35.McCormick DA, von Krosigk M. Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci USA. 1992;89:2774–2778. doi: 10.1073/pnas.89.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mercuri NB, Bonci A, Calabresi P, Bernardi G. Characterization of a barium-sensitive outward current following glutamate application on rat midbrain dopaminergic cells. Eur J Neurosci. 1996;8:1780–1786. doi: 10.1111/j.1460-9568.1996.tb01321.x. [DOI] [PubMed] [Google Scholar]
- 37.Moore J, Appenteng K. The membrane properties and firing characteristics of rat jaw-elevator motoneurones. J Physiol (Lond) 1990;423:137–153. doi: 10.1113/jphysiol.1990.sp018015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore JA, Appenteng K. The morphology and electrical geometry of rat jaw-elevator motoneurones. J Physiol (Lond) 1991;440:325–343. doi: 10.1113/jphysiol.1991.sp018711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura Y. Brainstem neuronal mechanisms controlling the trigeminal motoneuron activity. In: Desmedt JE, editor. Spinal and supraspinal mechanisms of voluntary motor control and locomotion, Vol. 8. Karger; Basel: 1980. pp. 181–202. [Google Scholar]
- 40.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 41.Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- 42.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 43.Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayer RJ, Schwindt PC, Crill WE. Metabotropic glutamate receptor-mediated suppression of L-type calcium current in acutely isolated neocortical neurons. J Neurophysiol. 1992;68:833–842. doi: 10.1152/jn.1992.68.3.833. [DOI] [PubMed] [Google Scholar]
- 45.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 46.Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240:1326–1328. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- 47.Sharp RL, May PC, Mayne NG, Snyder YM, Burnett JP. Cyclothiazide potentiates agonist responses at human AMPA/kainate receptors expressed in oocytes. Eur J Pharmacol. 1994;266:R1–2. doi: 10.1016/0922-4106(94)90216-x. [DOI] [PubMed] [Google Scholar]
- 48.Smith JC, Feldman JL. In vitro brainstem-spinal cord preparations for study of motor systems for mammalian respiration and locomotion. J Neurosci Methods. 1987;21:321–333. doi: 10.1016/0165-0270(87)90126-9. [DOI] [PubMed] [Google Scholar]
- 49.Stefani A, Pisani A, Mercuri NB, Bernardi G, Calabresi P. Activation of metabotropic glutamate receptors inhibits calcium currents and GABA-mediated synaptic potentials in striatal neurons. J Neurosci. 1994;14:6734–6743. doi: 10.1523/JNEUROSCI.14-11-06734.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stern P, Edwards FA, Sakmann B. Fast and slow components of unitary EPSCs on stellate cells elicited by focal stimulation in slices of rat visual cortex. J Physiol (Lond) 1992;449:247–278. doi: 10.1113/jphysiol.1992.sp019085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 52.Swartz KJ, Bean BP. Inhibition of calcium channels in rat CA3 pyramidal neurons by a metabotropic glutamate receptor. J Neurosci. 1992;12:4358–4371. doi: 10.1523/JNEUROSCI.12-11-04358.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takahashi T. Inward rectification in neonatal rat spinal motoneurones. J Physiol (Lond) 1990;423:47–62. doi: 10.1113/jphysiol.1990.sp018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trombley PQ, Westbrook GL. l-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J Neurosci. 1992;12:2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trueblood PR, Levine MS, Chandler SH. Dual-component excitatory amino acid-mediated responses in trigeminal motoneurons and their modulation by serotonin in vitro. J Neurophysiol. 1996;76:2461–2473. doi: 10.1152/jn.1996.76.4.2461. [DOI] [PubMed] [Google Scholar]
- 56.Turman JE, Jr, Chandler SH. Immunohistochemical localization of glutamate and glutaminase in guinea pig trigeminal premotoneurons. Brain Res. 1994;634:49–61. doi: 10.1016/0006-8993(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 57.Turman JE, Castillo MZ, Hiyama LS, Chandler SH (1997) Expression of metabotropic glutamate receptors in neonatal trigeminal neurons. Soc Neurosci Abstr 255.11. [DOI] [PubMed]
- 58.Zorumski CF, Mennerick S, Que J. Modulation of excitatory synaptic transmission by low concentrations of glutamate in cultured rat hippocampal neurons. J Physiol (Lond) 1996;494:465–477. doi: 10.1113/jphysiol.1996.sp021506. [DOI] [PMC free article] [PubMed] [Google Scholar]