

Abstract
Amphetamine (AMPH) inhibits uptake and causes release of dopamine (DA) from presynaptic terminals. AMPH can act on both vesicular storage of DA and directly on the dopamine transporter (DAT). To assess the relative importance of these two processes, we have examined the releasing actions of AMPH in mice with a genetic deletion of the DAT. The sequence of actions of AMPH has been determined by following the real time changes of DA in the extracellular fluid of intact tissue with fast scan cyclic voltammetry. In striatal slices from wild-type mice, AMPH causes a gradual (∼30 min) increase in extracellular DA, with a concomitant disappearance of the pool of DA available for depolarization-evoked release. Conversely, in slices from mice lacking the DAT, although a similar disappearance of electrically stimulated DA release occurs, extracellular DA does not increase. Similarly, microdialysis measurements of DA after AMPH in freely moving animals show no change in mice lacking the DAT, whereas it increases 10-fold in wild-type mice. In contrast, redistribution of DA from vesicles to the cytoplasm by the use of a reserpine-like compound, Ro4–1284, does not increase extracellular DA in slices from wild-type animals; however, subsequent addition of AMPH induces rapid (<5 min) release of DA. Thus, the DAT is required for the releasing action, but not the vesicle-depleting action, of AMPH on DA neurons, and the latter represents the rate-limiting step in the effects of AMPH. Furthermore, these findings suggest that in the absence of pharmacological manipulation, such as the use of amphetamine, endogenous cytoplasmic DA normally does not reach sufficient concentrations to reverse the DAT.
Keywords: amphetamine, dopamine transporter, knockout, mice, voltammetry, Ro4–1284, tetrabenazine, synaptic vesicles, microdialysis, dopamine
The effects of psychostimulants are thought to result from increased extracellular dopamine (DA) concentrations in limbic regions of the brain, including the striatum (Koob and Bloom, 1988; Self and Nestler, 1995; Wise, 1996). Some psychostimulants, such as cocaine, increase extracellular DA by inhibiting the reuptake of released DA by the plasma membrane DA transporter (DAT) (Heikkila et al., 1975; Horn, 1990; Amara and Kuhar, 1993; Giros and Caron, 1993). Others, including the amphetamines, cause release of DA from presynaptic nerve terminals in addition to inhibiting reuptake (Heikkila et al., 1975; Seiden et al., 1993). DA can be released by two mechanisms (Raiteri et al., 1979): vesicular release, which is calcium and impulse-dependent, and transporter-mediated release, which is impulse-independent and has little calcium dependence (Hurd and Ungerstedt, 1989; Pierce and Kalivas, 1997). DA release in response to AMPH occurs by the second mechanism. This process has been called reverse transport (Sulzer et al., 1995).
Several specific sites of action of AMPH on DA neurons have been identified. AMPH can cross plasma membranes via lipophilic diffusion (Mack and Bonisch, 1979; Liang and Rutledge, 1982a; Zaczek et al., 1991a,b), and it is also a substrate for the DAT (Liang and Rutledge, 1982a,b; Zaczek et al., 1991a,b; Seiden et al., 1993). Once inside the cells, AMPH can displace DA from secretory vesicles into the neuronal cytoplasm (Sulzer and Rayport, 1990; Floor et al., 1995; Floor and Meng, 1996), from which DA can then be released into the extracellular space by outward transport by the DAT. The relative importance of these two processes, vesicular depletion and reverse transport, has remained controversial. On the one hand, the exchange diffusion or reverse transport model (Fischer and Cho, 1979; Liang and Rutledge, 1982a,b;Burnette et al., 1996) proposes that AMPH acts primarily at the plasma membrane transporter, which acts as a mobile carrier with a binding site that allows DA transport from one side of the membrane to the other. By acting as a substrate, AMPH increases the number of inward-facing transporter binding sites and thus increases the rate of reverse transport. Consistent with this mechanism, early work showed that the behavioral effects of AMPH were independent of vesicular stores (Scheel-Kruger, 1971). In contrast, the weak base or vesicle depletion model (Sulzer and Rayport, 1990; Sulzer et al., 1992) proposes that the action of AMPH arises primarily from its effects on secretory vesicles. AMPH enters DA vesicles and causes displacement of DA from vesicles into the cytoplasm by disruption of the interior-acidic pH gradient. In this view, the elevated cytoplasmic DA and altered concentration gradient of DA across the plasma membrane causes reverse transport of DA, a process independent of AMPH (Sulzer et al., 1995). The complexity of the actions of AMPH, which has led to the mutual exclusion of these two proposed models, has hampered a satisfactory resolution of the issue.
The recent availability of a genetically modified mouse in which the DAT gene has been deleted, in combination with the electrochemical technique of fast-scan cyclic voltammetry which allows real-time measurements of neurotransmitter release and uptake, has provided information about the requirement of the DAT in the action of AMPH (Giros et al., 1996). Here, we further characterize the model to establish the relative contribution of vesicular depletion and reverse transport to the DA-releasing actions of AMPH. We now demonstrate that although vesicular depletion is rate-limiting, both of these mechanisms are critical in producing DA release by AMPH.
MATERIALS AND METHODS
Animals. Male C57BL/129SvJ wild-type mice (DAT +/+) and their littermates homozygous for DAT deletion (homozygote DAT knockout mice or DAT −/−), 2- to 4-months-old, were used in these experiments. They were housed in an animal care facility at a temperature of 23°C with a 12 hr light cycle and given food and waterad libitum. They were caged with approximately three other littermates of the same sex. Homozygote DAT knockout mice differ markedly from their wild-type littermates, demonstrating a specific phenotype characterized by behavioral hyperactivity and dwarfism (Giros et al., 1996; Bosse et al., 1997). Animal care was in accordance with institutional guidelines.
Cyclic voltammetry. Carbon-fiber electrodes were prepared as described previously (Kawagoe et al., 1993). A potentiostat (EI-400, Ensman Instrumentation, Bloomington, IN) was used for fast-scan cyclic voltammetry. The electrode potential was linearly scanned from −400 to 1000 mV and back to −400 mV at 300 V/sec, repeated every 100 msec. The peak oxidation current for DA was between 500 and 700 mV. Each electrode was calibrated with 10 μm DA at the end of the experiment. Mice were decapitated, and the brain was rapidly removed. Coronal slices (400 μm thick) containing the striatum were prepared, placed in a recording chamber, and superfused with an artificial cerebrospinal fluid (Jones et al., 1995a,b). DA release was evoked by single-pulse stimulations (350 μA, 4 msec) from a stimulating electrode placed 100–200 μm away from the carbon-fiber electrode (Jones et al., 1995a,b). Electrodes were placed in the dorsolateral portion of the caudate-putamen. Each slice served as its own precondition control. Background-subtracted cyclic voltammograms were constructed by subtracting the background current obtained before release (15–50 nA) from the current measured after release. In every reported case, DA was the substance detected and was identified by its characteristic cyclic voltammogram. Data were analyzed with one-way ANOVA using Systat software (Evanston, IL).
Microdialysis. Intracerebral microdialysis (Ungerstedt, 1984; Gainetdinov et al., 1997) was performed using concentric microdialysis probes (2 mm membrane length; cutoff 6000 Da; CMA-11, CMA/Microdialysis, Solna, Sweden). Stereotaxic coordinates were slightly different to correct for the marked size difference between wild-type and DAT −/− mice at 8–10 weeks (DAT −/− 55% of control weight) (Bosse et al., 1997): anterioposterior (AP) 0.0, dorsoventral (DV) −4.4, lateral (L) 2.5 for DAT +/+ mice, and AP 0.0, DV −3.2, L 1.8 for DAT −/−, relative to bregma (Franklin and Paxinos, 1996). The dialysis probes were perfused during implantation into the brain and for 1 hr afterward with artificial cerebrospinal fluid (in mm): Na+ 150, K+ 3.0, Ca2+ 1.4, Mg2+ 0.8, PO4− 31.0, Cl− 155 (ESA Inc., Bedford, MA), pH 7.3. One hour after the operation, animals were returned to their home cages; 24 hr after surgery the dialysis probe was perfused at 1.0 μl/min for 80 min before the experiment. Perfusate samples were collected every 20 min. At least four predrug samples were collected before AMPH was administered.
HPLC. Measurements of DA and DOPAC in microdialysis samples were by HPLC with electrochemical detection (HPLC/EC) with a microbore column (5 μm particles, Unijet C18, 1 × 150 mm; BAS, West Lafayette, IN) and electrochemically detected with a Unijet (3 mm) electrode. The mobile phase contained 50 mm sodium citrate, 10 mm NaH2PO4, 0.5 mm octyl sodium sulfate, 0.1 mm EDTA, and 17% methanol, at pH 3.5.
Drugs.d-AMPH, cocaine, sulpiride, pargyline, and nialamide were from Sigma (St. Louis, MO), Ro4–1284 was a gift from Hoffmann-LaRoche (Nutley, NJ), and tetrabenazine was from Fluka (Buchs, Switzerland).
RESULTS
Effect of AMPH on stimulated and baseline DA
Baseline and stimulated release of DA in striatal brain slices were monitored by cyclic voltammetry. Electrically stimulated release was elicited by a single pulse of electrical current locally applied at 5 min intervals. Figure 1 shows representative profiles of stimulated and baseline DA release over a 50 min period as AMPH (10 μm) was applied to slices from wild-type (DAT +/+) and homozygote DAT knockout (DAT −/−) mice. In slices from wild-type mice, baseline DA release began to increase ∼10 min after AMPH application, as seen by the rise in the recordings. At the same time, the amount of electrically stimulated DA release started to decrease, as seen by the decreasing peak heights of the “spikes” of DA efflux observed at each stimulation. The current changes were confirmed to be caused by increases in DA by examination of the cyclic voltammograms collected during the time the baseline was rising, at the plateau for the baseline release, and also at the peak of the response for the stimulated release (data not shown). After ∼25 min of AMPH, a plateau of baseline release was established between 3 and 10 μm DA, with an average of 4.2 ± 0.8 μm. By 25–35 min of AMPH exposure, electrically stimulated DA release had disappeared completely.
Fig. 1.

Effect of AMPH (10 μm) on DA efflux in striatal slices from wild-type (DAT +/+) and homozygote DAT knockout (DAT −/−) mice. Current was measured by cyclic voltammetry at microelectrodes implanted in the slices. Stimulated DA release was elicited by single electrical pulses applied to the slice at the times indicated by small arrowheads. On the time scale shown, electrically stimulated DA release events appear as sharp spikes. Baseline release was monitored as any increase in the current measured between stimulations that was identified as DA. In the absence of pharmacological or electrical intervention, baseline and electrically stimulated DA recordings were stable for >3 hr. Inset, Single-pulse stimulations in slices from a wild-type and homozygote DAT knockout mouse with the time scale expanded to show the time course of the DA release and clearance. Filled circles are individual measurements of the concentration of DA, collected every 100 msec.
Effect of AMPH on DA in slices from DAT-deficient mice
Application of AMPH to slices from DAT −/− mice did not change baseline DA overflow. A decrease in electrically stimulated DA release commenced ∼15 min after AMPH treatment, and it was eliminated after 45–60 min (Fig. 1). Thus AMPH entered the DA terminals and depleted vesicular stores, as evidenced by the elimination of stimulated release, but did not cause release of DA into the extracellular space in the absence of the DAT. The longer time course for vesicular depletion in homozygote DAT knockout mice compared with the wild type is consistent with AMPH entering the DAT −/− nerve terminals at a reduced rate when only lipophilic diffusion occurs. In contrast, AMPH can accumulate in terminals from wild-type mice by diffusion plus efficient uptake through the DAT (Fischer and Cho, 1979; Liang and Rutledge, 1982a,b; Zaczek et al., 1991a,b).
Modulation of effects of AMPH by sulpiride
Figure 2 shows the amount of electrically stimulated DA release and baseline DA overflow measured in 5 min intervals in slices from wild-type and homozygote DAT knockout mice, with and without sulpiride. In the presence of 2 μmsulpiride, a D2 receptor antagonist, electrically stimulated DA release was abolished by AMPH, with an approximate 10 min delay in slices from wild-type mice. This lengthening of the time course of AMPH by the addition of sulpiride indicates that presynaptic autoreceptor activation by baseline DA overflow played a role in decreasing stimulated release in the slices from wild-type mice. Electrically stimulated release in slices from homozygote DAT knockout mice was not altered by sulpiride, as expected, because there was no increase in extracellular DA by AMPH and thus autoreceptors were not activated. However, baseline DA overflow in slices from wild-type mice, elevated by AMPH as a result of reverse transport, was unaltered by sulpiride, suggesting that under the present conditions DAT-mediated transport is not altered by autoreceptor inhibition. This finding is consistent with results from other laboratories (Kuczenski et al., 1990; Iravani and Kruk, 1995).
Fig. 2.

Effect of AMPH (10 μm) on stimulated release and baseline overflow of DA in the absence and presence of sulpiride (2 μm), a D2 autoreceptor antagonist. Stimulated DA release is measured by cyclic voltammetry as the peak height of DA elicited by a single electrical pulse, and baseline overflow is measured as the average DA concentration change over a 5 min period between stimulations. Filled barsare wild-type data, and open bars are homozygote DAT knockout data. AMPH (top) or AMPH + sulpiride (AMPH + SULP) (bottom) were added to the slice 5 min before the first measurements. Results are the mean ± SEM of at least four independent experiments. *Significantly different from AMPH alone data (p < 0.05).
Microdialysis measurements of AMPH action
The detection limit for cyclic voltammetry is ∼25 nm, and small changes in baseline extracellular levels of DA might not be measurable by this method, whereas microdialysis is more than one order of magnitude more sensitive (Westerink and Justice, 1991). In vivo microdialysis was used to monitor extracellular levels of DA in the striatum of freely moving wild-type and homozygote DAT knockout mice. The results of the microdialysis experiment are shown in Figure 3. After intraperitoneal administration of 10 mg/kg AMPH, wild-type mice showed a significant, 8- to 10-fold increase in extracellular DA, whereas homozygote DAT knockout mice did not show any significant change (Fig.3A). Note that the basal extracellular levels of DA in homozygote DAT knockout mice are about five times higher than in DAT +/+ mice [predrug concentrations of DA in dialysates were 55 ± 15 fmol/20 μl (n = 6) for DAT +/+ and 234 ± 84 fmol/20 μl (n = 6) for DAT −/− mice]. Thesein vivo data confirm the cyclic voltammetry findings, which demonstrate that the DA-releasing actions of AMPH are dependent on the DAT.
Fig. 3.
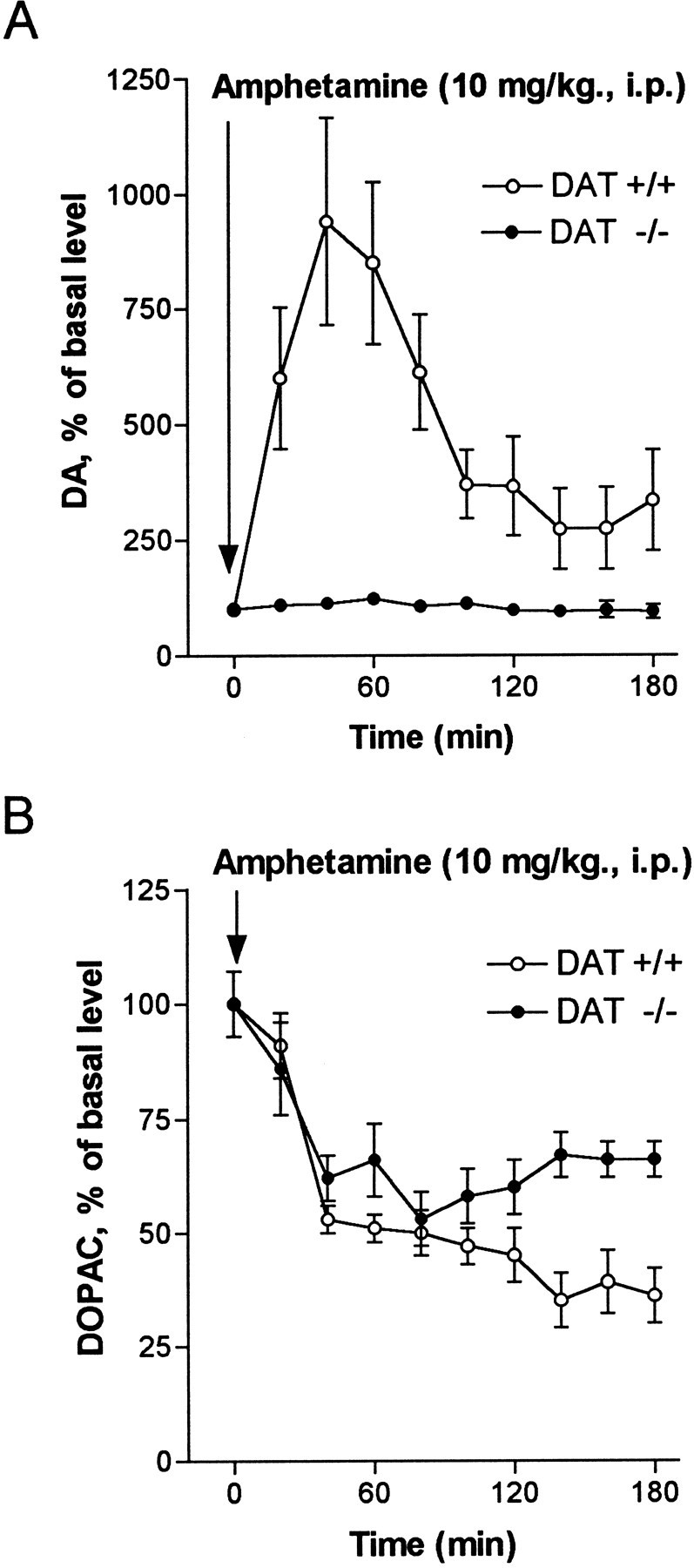
Effect of AMPH (10 mg/kg., i.p.) on extracellular DA and DOPAC measured by microdialysis in the striatum of freely moving mice. Open circles are data from wild-type mice (DAT +/+), and filled circles are data from homozygote DAT knockout mice (DAT −/−). A, Extracellular DA is increased 8- to 10-fold in wild-type mice after AMPH, whereas there is no significant difference in DA levels after AMPH in homozygotes.B, Extracellular DOPAC is decreased to approximately the same degree in both wild-type and homozygote mice. Concentrations of DOPAC in dialysates were 12.3 ± 3.5 pmol/20 μl for DAT +/+ and 5.3 ± 1.8 pmol/20 μl for DAT −/− mice. Results are the mean ± SEM of six independent experiments.
After AMPH administration, dialysate levels of dihydroxyphenylacetic acid (DOPAC) were decreased by ∼50% in both the wild-type and mutant mice (Fig. 3B). This is consistent with AMPH entering nerve terminals and inhibiting monoamine oxidase (MAO), one of the known actions of AMPH (Green and El Haut, 1978; Miller et al., 1980). These data provide additional evidence that AMPH enters the presynaptic terminal in significant amounts by lipophilic diffusion, in the absence of the DAT. Moreover, they indicate that differences in the actions of AMPH in wild-type versus DAT −/− mice are not attributable to their differential effects on MAO.
Effect of inhibition of vesicular transport
To independently examine vesicle depletion and its effect on transport, a vesicular monoamine transporter type 2 (VMAT-2) inhibitor, Ro4–1284, which rapidly depletes secretory vesicles (Colzi et al., 1993; Filinger, 1994), was used to mimic this aspect of AMPH action. Application of Ro4–1284 (10 μm) to slices from wild-type mice resulted in a gradual reduction in electrically stimulated DA release over ∼30 min, with no accompanying increase in baseline DA overflow (Fig. 4). Stimulated DA release remained constant over a similar time interval with no added drug (Jones et al., 1995b). Similar results were also obtained with another short-acting VMAT-2 inhibitor, tetrabenazine (data not shown). Reserpine was not investigated because its actions are very slow (Callaway et al., 1989; Cadoni et al., 1995). After Ro4–1284 caused the disappearance of electrically stimulated DA in a slice from a wild-type mouse, 10 μm AMPH was applied to the slice, and baseline DA overflow increased rapidly (Fig. 4). The magnitude of overflow under these conditions was similar to that induced over 30 min with AMPH alone, but the rate of overflow was much faster and a plateau was reached in 5–10 min (Fig. 5). From these findings, the relative time courses of AMPH actions in a striatal slice were estimated to be 25 min for vesicle depletion and 5 min for reverse transport.
Fig. 4.

Effect of a fast-acting reserpine-like drug, Ro4–1284 (10 μm), and AMPH (10 μm) on stimulated and baseline release of DA. Individual recordings of DA efflux measured by cyclic voltammetry in striatal slices from wild-type (DAT +/+) and homozygote DAT knockout mice (DAT −/−). Drugs were applied to slices at the times indicated by the large arrows. Top, Single-pulse stimulations were applied to a slice from a wild-type mouse at the times indicated by thesmall arrowheads. Inset, Cyclic voltammograms, current (nA) versus potential (mV) plots, recorded during postcalibration with authentic DA (solid line) and during the plateau phase of the release induced by AMPH (filled circles), are plotted for comparison.Bottom, Single-pulse stimulations were applied to a slice from a homozygote DAT knockout mouse at the times indicated by the small arrowheads.
Fig. 5.

Effect of pargyline (PARG, 10 μm, top), an MAO inhibitor, and sulpiride (SULP, 2 μm, bottom), a D2 autoreceptor antagonist, on the effects of Ro4–1284 followed by AMPH in slices from wild-type mice. Stimulated DA release is measured by cyclic voltammetry as the peak height of DA elicited by single electrical pulses, and baseline overflow is measured as the average DA concentration change over a 5 min period between stimulations. PARG or SULP was added to the slice 10 min before the first measurements. Results are the mean ± SEM of at least four independent experiments.
Application of 10 μm Ro4–1284 or tetrabenazine caused a gradual reduction and final disappearance of electrically stimulated DA release in slices from homozygote DAT knockout mice similar to that found in the wild type. However, the addition of AMPH did not induce measurable baseline overflow of DA (Fig. 4). This further confirms the requirement for DAT in the releasing action of AMPH.
Inhibition of MAO was monitored along with Ro4–1284-mediated vesicle depletion to test whether degradation of cytoplasmic DA was the reason for the lack of baseline DA overflow after Ro4–1284. Figure 5 shows that 10 μm pargyline, an MAO inhibitor, did not have any effect on baseline DA overflow or stimulated DA release during Ro4–1284 application. Nialamide (10 μm), another MAO inhibitor, also had no effect (data not shown). Furthermore, the addition of 2 μm sulpiride to slices before Ro4–1284 did not significantly alter the disappearance of stimulated DA release (Fig. 5), indicating that autoreceptor stimulation did not play a major role in the decrease in electrically stimulated DA release in this case.
To duplicate uptake inhibition, another important property of AMPH, cocaine was used in combination with Ro4–1284. Cocaine (10 μm) was applied before Ro4–1284 and did not change the effects of Ro4–1284 (Fig. 6,top). This indicates that rapid reuptake was not masking any Ro4–1284-induced baseline DA overflow. Preadministration of cocaine to the slice, however, did prevent the rapid AMPH-induced baseline overflow normally found after Ro4–1284 application. This was expected because cocaine is known to inhibit the releasing action of AMPH. Finally, cocaine applied after Ro4–1284 and AMPH had the effect of decreasing the plateau of baseline DA overflow (Fig. 6,bottom). Thus, the plateau level of DA reached after AMPH was maintained by continuous release of DA via reverse transport, which can be antagonized by a transporter blocker.
Fig. 6.

Effect of cocaine (COC, 10 μm) on the effects of Ro4–1284 followed by AMPH in slices from wild-type mice. Top, Cocaine was added 10 min before the first measurements were made. Bottom, Cocaine was added 15 min after AMPH. Results are the mean ± SEM of at least four independent experiments. *Significantly different from wild-type values without cocaine (p < 0.05).
DISCUSSION
It has long been recognized that AMPH exerts numerous actions on DA nerve terminals (Seiden et al., 1993). The present results illustrate the central importance of both depletion of DA from secretory vesicles and reversal of DAT-mediated transport in the releasing effects of AMPH. Although both effects are necessary to induce DA release (termed overflow here to distinguish it from electrically stimulated release), the vesicle-depleting action of AMPH is the rate-limiting step. In addition, several other points of controversy are resolved. For example, the results clearly demonstrate that the plasma membrane DAT is absolutely required for the overflow of DA into the extracellular space, but it is not required for the depletion of vesicular DA by AMPH. Moreover, when endogenous, releasable DA was mobilized from vesicles into the cytoplasm, the resulting increased DA concentration gradient across the plasma membrane was not sufficient to reverse the DAT to a measurable degree. Release under these conditions was observed only in the presence of AMPH.
The initial step in the action of AMPH on DA terminals is its entry. As shown previously, this usually occurs via the DAT (Liang and Rutledge, 1982a). Entry by its lipophilic diffusion across the plasma membrane has been demonstrated only at concentrations far above itsKi for DA transport inhibition (Liang and Rutledge, 1982a). However, our data from homozygote DAT knockout mice demonstrate that diffusional entry can occur at lower concentrations, although it is less efficient. In striatal slices from homozygote DAT knockout mice, AMPH (10 μm) induces depletion of electrically stimulated DA release, but it requires a longer time than in tissue that contains transporters. Moreover, in homozygote DAT knockout mice, 10 mg/kg AMPH inhibits MAO, as evidenced by a decrease in extracellular DOPAC measured by microdialysis, in an manner equivalent to that found in wild-type animals. This provides further evidence that AMPH can access intracellular compartments in the absence of the DAT.
Once inside the cell, AMPH can deplete vesicles of releasable DA. This action, manifested as a decrease in electrically stimulated release, is also found with the VMAT-2 inhibitors tested. Although the pool of vesicular DA released by electrical stimulation may comprise only a small fraction of the tissue content of DA, this vesicular DA is of high significance, because it represents the most physiologically active DA. The actions of AMPH and the reserpine-like compounds differ, however, in that only AMPH induces an increase in baseline overflow that accompanies the decrease in electrically stimulated release. We infer that the DA displaced from the vesicles by Ro4–1284 is in the cytoplasm because it is not detected in the extracellular fluid. Thus, the increased concentration gradient across the plasma membrane caused by accumulation of free cytosolic DA from vesicles is insufficient to reverse transport at the levels achieved in this work, in contrast to previous proposals (Sulzer et al., 1993, 1995). The DAT operates on an electrochemical gradient in which the concentrations of substrate, sodium, and chloride, and membrane potential are all important. DAT-mediated transport can be reversed if the cytoplasmic concentrations of substrate are manipulated artificially, as shown by numerous in vitro studies (Eshleman et al., 1993; Pifl et al., 1995; Sulzer et al., 1995) or if the normal ionic gradients are altered (Eshleman et al., 1993). However, our data show that the movement of the endogenous, releasable vesicular pool of DA to the cytoplasm of DA nerve terminals does not provide a sufficient concentration gradient to reverse DAT-mediated transport.
AMPH addition to a slice depleted of releasable vesicular DA leads to a rapid reversal of transport, further supporting our inference that it is cytoplasmic. Apparently, an ongoing supply of DA from the cytoplasm is supplied to the extracellular space via the DAT, because when cocaine is added to the slice after AMPH, the extracellular levels of DA drop. This reverse transport of DA caused by AMPH is consistent with a recently proposed mechanism for the norepinephrine transporter (Burnette et al., 1996), in which any substrate added to the outside of the cell will cause the allosteric translocation of the transporter to the inside of the cell. Such an inward-facing transporter is available to transport free cytosolic substrate to the outside of the cell. The probability of outward-directed transport is a function of the concentration of an external transportable species, AMPH in this case. The probability of reverse transport is lowered by blocking access to the transporter, as demonstrated by the effects of cocaine. This model is essentially the same as the “exchange diffusion model” proposed by Fischer and Cho (1979). New evidence, however, has demonstrated that the transporter can operate in a channel-like mode, in which a pore is opened through the plasma membrane (Sonders and Amara, 1996; Sonders et al., 1997). The exact relationship between these properties, substrate binding, inward transport, and reverse transport, has yet to be elucidated.
The times required for vesicular depletion and reverse transport were measured to be ∼25 min and 5 min, respectively. The total time to induce a plateau level of DA overflow in an untreated slice was ∼30 min, which is consistent with the time course of AMPH-induced DA overflow in vivo as measured by microdialysis (Fig.3A). AMPH-induced depletion of DA from synaptic vesicles can arise from inhibition of monoamine uptake into vesicles through the VMAT-2 followed by leakage from the vesicles (Knepper et al., 1988;Schuldiner et al., 1993; Peter et al., 1994; Floor et al., 1995;Erickson et al., 1996), or by alkalinization of the interior of vesicles (Sulzer and Rayport, 1990; Sulzer et al., 1992; Schuldiner et al., 1993). The 30 min time course may be required either to accumulate sufficient AMPH for vesicle depletion or for the vesicles to “leak” out the stored DA. The rate of reverse transport that follows AMPH after vesicle depletion with reserpine-like drugs is consistent with the rates reported for efflux of preloaded DA from COS7 cells (which do not have secretory vesicles) transfected with the norepinephrine transporter (Burnette et al., 1996).
There is an extensive literature regarding whether AMPH releases cytoplasmic or vesicular DA (for review, see Seiden et al., 1993). Pretreatment of animals with reserpine, a vesicular DA depleter, does not abolish all of the DA release elicited by AMPH. Subsequent treatment with α-methyl-p-tyrosine (αMPT), a DA synthesis inhibitor, does abolish DA release elicited by AMPH (Butcher et al., 1988, Callaway et al., 1989). In addition, DA-mediated, AMPH-induced behaviors are not blocked by reserpine (Creese and Iverson, 1975). These experiments have been interpreted as meaning that AMPH primarily releases DA from the newly synthesized, cytoplasmic pool of DA. The present results and others (Chiueh and Moore, 1975; Liang and Rutledge, 1982b; Parker and Cubeddu, 1986a,b; Cadoni et al., 1995) suggest a different interpretation, one that includes two separate, equally important aspects of DA release by AMPH. The first is reverse transport by the DAT, which will cause the release of any DA from the cytoplasm of the nerve terminal; the second is the slower displacement of vesicular DA to the cytoplasm, followed by release via reverse transport. This conclusion is supported by the finding that in the presence of AMPH alone, electrically stimulated release disappears within the same time frame that baseline overflow appears (Fig. 1). This effect is altered only slightly by the addition of autoreceptor antagonists, since autoreceptors might be expected to decrease electrically stimulated release as extracellular DA increases (Fig. 2). In addition, the finding that AMPH causes the same amount of DA overflow before or after the movement of the readily releasable vesicular stores of DA to the cytoplasm, albeit on different time scales (Fig. 4), suggests that the vesicular pool is the major supplier of DA for the releasing action of AMPH, and that the time frame of DA appearance in the extracellular space is dependent on vesicle depletion.
The observation that DOPAC levels are decreased to a similar extent after AMPH in both wild-type and DAT knockout animals provides evidence for another action of AMPH, MAO inhibition. Other hypotheses have been proposed to explain the DOPAC decrease such as the removal of a major MAO substrate, newly synthesized DA, by AMPH (Zetterstrom et al., 1988). However, the results with homozygote DAT knockout animals show that DOPAC was decreased even when cytoplasmic DA was not released into the extracellular space, and when levels of cytoplasmic DA had probably increased significantly. Under these conditions, MAO inhibition by pargyline had no effect on the cytoplasmic DA generated by VMAT-2 inhibitors, suggesting that cytoplasmic dopamine can persist at least for the time of the experiments before outward transport induced by AMPH.
In conclusion, we have demonstrated that both the vesicle depletion and the reverse transport caused by AMPH are important in its DA-releasing effects. We have shown that the time course of reverse transport is much faster than that of vesicle depletion (5 vs 25 min). In addition, we have demonstrated that when the endogenous, releasable pool of DA is displaced into the cytoplasm, it does not produce a concentration gradient across the plasma membrane large enough for outward transport by the DAT, and the presence of AMPH is required to catalyze this effect.
Footnotes
This work was supported in part by Grant NS 19576 from National Institutes of Health, and unrestricted grants from Bristol Myers Squibb and Zeneca Pharmaceuticals (M.G.C.) and National Institute on Drug Abuse (NIDA) Grant DA 10900 (R.M.W.). S.R.J. is supported by NIDA fellowship 1 F32 DA05749–01. R.R.G. is a visiting fellow from the Institute of Pharmacology Russian Academy of Medical Sciences, Baltiyskaya, 8, 125315, Moscow, Russia, and is supported by a Tourette Syndrome Association fellowship. R.M.W. is the recipient of a Guggenheim Fellowship.
Correspondence should be addressed to Dr. Marc G. Caron, Howard Hughes Medical Institute Laboratories, Box 3287, Duke University Medical Center, Durham, NC 27710.
REFERENCES
- 1.Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 2.Bosse R, Fumagalli F, Jaber M, Giros B, Gainetdinov RR, Wetsel WC, Missale C, Caron MG. Anterior pituitary hypoplasia and dwarfism in mice lacking the dopamine transporter. Neuron. 1997;19:127–138. doi: 10.1016/s0896-6273(00)80353-0. [DOI] [PubMed] [Google Scholar]
- 3.Burnette WB, Bailey MD, Kukoyi S, Blakely RD, Trowbridge CG, Justice JB. Human norepinephrine transporter kinetics using rotating disk electrode voltammetry. Anal Chem. 1996;68:2932–2938. doi: 10.1021/ac960022x. [DOI] [PubMed] [Google Scholar]
- 4.Butcher SP, Fairbrother IS, Kelly JS, Arbuthnott GW. Amphetamine-induced dopamine release in the rat striatum: an in vivo microdialysis study. J Neurochem. 1988;50:346–355. doi: 10.1111/j.1471-4159.1988.tb02919.x. [DOI] [PubMed] [Google Scholar]
- 5.Cadoni C, Pinna A, Russi G, Consolo S, Di Chiara G. Role of vesicular dopamine in the in vivo stimulation of striatal dopamine transmission by amphetamine: evidence from microdialysis and Fos immunohistochemistry. Neuroscience. 1995;65:1027–1039. doi: 10.1016/0306-4522(94)00507-2. [DOI] [PubMed] [Google Scholar]
- 6.Callaway CW, Kuczenski R, Segal DS. Reserpine enhances amphetamine stereotypies without increasing amphetamine-induced changes in striatal dialysate dopamine. Brain Res. 1989;505:83–90. doi: 10.1016/0006-8993(89)90118-2. [DOI] [PubMed] [Google Scholar]
- 7.Chiueh CC, Moore KE. d-amphetamine-induced release of “newly synthesized” and “stored” dopamine from the caudate nucleus in vivo. J Pharmacol Exp Ther. 1975;192:642–653. [PubMed] [Google Scholar]
- 8.Colzi A, D’Agostini R, Cesura AM, Borroni E, Da Prada M. Monoamine oxidase-A inhibitors and dopamine metabolism in rat caudatus: evidence that an increased cytosolic level of dopamine displaces reversible monoamine oxidase-A inhibitors in vivo. J Pharmacol Exp Ther. 1993;265:103–111. [PubMed] [Google Scholar]
- 9.Creese I, Iverson SD. The pharmacological and anatomical substrates of the amphetamine response in the rat. Brain Res. 1975;83:419–436. doi: 10.1016/0006-8993(75)90834-3. [DOI] [PubMed] [Google Scholar]
- 10.Erickson JD, Schafer MK-H, Bonner TI, Eiden LE, Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci USA. 1996;93:5166–5171. doi: 10.1073/pnas.93.10.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eshleman AJ, Henningsen RA, Neve KA, Janowsky A. Release of dopamine via the human transporter. Mol Pharmacol. 1993;45:312–316. [PubMed] [Google Scholar]
- 12.Filinger EJ. Effect of a reserpine-like agent on the release and metabolism of [3H]NA in cell bodies and terminals. Gen Pharmacol. 1994;25:1039–1043. doi: 10.1016/0306-3623(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 13.Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- 14.Floor E, Meng L. Amphetamine releases dopamine from synaptic vesicles by dual mechanisms. Neurosci Lett. 1996;215:53–56. doi: 10.1016/s0304-3940(96)12963-3. [DOI] [PubMed] [Google Scholar]
- 15.Floor E, Leventhal PS, Wang Y, Meng L, Chen W. Dynamic storage of dopamine in rat brain synaptic vesicles in vitro. J Neurochem. 1995;64:689–699. doi: 10.1046/j.1471-4159.1995.64020689.x. [DOI] [PubMed] [Google Scholar]
- 16.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1996. [Google Scholar]
- 17.Gainetdinov RR, Fumagalli F, Jones SR, Caron MG. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem. 1997;69:1322–1325. doi: 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- 18.Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- 19.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 20.Green AL, El Haut MJ. Inhibition of mouse brain monoamine oxidase by (+)-amphetamine in vivo. J Pharm Pharmacol. 1978;30:262–263. doi: 10.1111/j.2042-7158.1978.tb13223.x. [DOI] [PubMed] [Google Scholar]
- 21.Heikkila RE, Orlansky H, Cohen G. Studies on the distinction between uptake inhibition and release of 3H-dopamine in rat brain tissue slices. Biochem Pharmacol. 1975;24:847–852. doi: 10.1016/0006-2952(75)90152-5. [DOI] [PubMed] [Google Scholar]
- 22.Horn AS. Dopamine uptake: a review of progress in the last decade. Prog Neurobiol. 1990;34:387–400. doi: 10.1016/0301-0082(90)90033-d. [DOI] [PubMed] [Google Scholar]
- 23.Hurd YL, Ungerstedt U. Ca2+ dependence of the amphetamine, nomifensine, and Lu 19–005 effect on in vivo dopamine transmission. Eur J Pharmacol. 1989;166:261–269. doi: 10.1016/0014-2999(89)90067-8. [DOI] [PubMed] [Google Scholar]
- 24.Iravani M, Kruk ZL. Effects of AMPH on carrier-mediated and electrically stimulated DA release in slices of rat caudate putamen and nucleus accumbens. J Neurochem. 1995;64:1161–1168. doi: 10.1046/j.1471-4159.1995.64031161.x. [DOI] [PubMed] [Google Scholar]
- 25.Jones SR, Garris PA, Kilts CD, Wightman RM. Comparison of dopamine uptake in the basolateral amygdaloid nucleus, caudate-putamen and nucleus accumbens of the rat. J Neurochem. 1995a;64:2581–2589. doi: 10.1046/j.1471-4159.1995.64062581.x. [DOI] [PubMed] [Google Scholar]
- 26.Jones SR, Garris PA, Wightman RM. Different effects of cocaine and nomifensine on DA uptake in the caudate-putamen and nucleus accumbens. J Pharmacol Exp Ther. 1995b;274:396–403. [PubMed] [Google Scholar]
- 27.Kawagoe KT, Zimmerman JB, Wightman RM. Principles of voltammetry and microelectrode surface states. J Neurosci Methods. 1993;48:225–240. doi: 10.1016/0165-0270(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 28.Knepper SM, Grunewald GL, Rutledge CO. Inhibition of norepinephrine transport into synaptic vesicles by amphetamine analogs. J Pharmacol Exp Ther. 1988;247:487–494. [PubMed] [Google Scholar]
- 29.Koob GR, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- 30.Kuczenski R, Segal DS, Manley LD. Apomorphine does not alter amphetamine-induced dopamine release measured in striatal dialysates. J Neurochem. 1990;54:1492–1499. doi: 10.1111/j.1471-4159.1990.tb01196.x. [DOI] [PubMed] [Google Scholar]
- 31.Liang NY, Rutledge CO. Comparison of the release of [3H]dopamine from isolated corpus striatum by amphetamine, fenfluramine and unlabelled dopamine. Biochem Pharmacol. 1982a;31:983–992. doi: 10.1016/0006-2952(82)90332-x. [DOI] [PubMed] [Google Scholar]
- 32.Liang NY, Rutledge CO. Evidence for carrier-mediated efflux of dopamine from corpus striatum. Biochem Pharmacol. 1982b;31:2479–2484. doi: 10.1016/0006-2952(82)90057-0. [DOI] [PubMed] [Google Scholar]
- 33.Mack F, Bonisch H. Dissociation constants and lipophilicity of catecholamines and related compounds. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:1–9. doi: 10.1007/BF00499868. [DOI] [PubMed] [Google Scholar]
- 34.Miller HH, Shore PA, Clarke DE. In vivo monoamine oxidase inhibition by d-amphetamine. Biochem Pharmacol. 1980;29:1347–1354. doi: 10.1016/0006-2952(80)90429-3. [DOI] [PubMed] [Google Scholar]
- 35.Parker EM, Cubeddu LX. Effects of d-amphetamine and dopamine synthesis inhibitors on dopamine and acetylcholine neurotransmission in the striatum. I. Release in the absence of vesicular transmitter stores. J Pharmacol Exp Ther. 1986a;237:179–192. [PubMed] [Google Scholar]
- 36.Parker EM, Cubeddu LX. Effects of d-amphetamine and dopamine synthesis inhibitors on dopamine and acetylcholine neurotransmission in the striatum. II. Release in the presence of vesicular transmitter stores. J Pharmacol Exp Ther. 1986b;237:193–203. [PubMed] [Google Scholar]
- 37.Peter D, Jimenez J, Liu Y, Kim J, Edwards RH. The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. J Biol Chem. 1994;269:7231–7237. [PubMed] [Google Scholar]
- 38.Pierce RC, Kalivas PW. Repeated cocaine modifies the mechanism by which amphetamine releases dopamine. J Neurosci. 1997;17:3254–3262. doi: 10.1523/JNEUROSCI.17-09-03254.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pifl C, Drobny H, Reither H, Hornykiewicz O, Singer EA. Mechanism of the dopamine-releasing actions of amphetamine and cocaine: plasmalemmal dopamine transporter versus vesicular monoamine transporter. Mol Pharmacol. 1995;47:368–373. [PubMed] [Google Scholar]
- 40.Raiteri M, Cerrito F, Cervoni AM, Levi G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J Pharmacol Exp Ther. 1979;208:195–202. [PubMed] [Google Scholar]
- 41.Scheel-Kruger J. Comparative studies of various amphetamine analogues demonstrating different interactions with the metabolism of the catecholamines in the rat brain. Eur J Pharmacol. 1971;14:47–59. doi: 10.1016/0014-2999(71)90121-x. [DOI] [PubMed] [Google Scholar]
- 42.Schuldiner S, Steiner-Mordoch S, Rodrigo Y, Wall SC, Rudnick G. Amphetamine derivatives interact with both plasma membrane and secretory vesicle biogenic amine transporters. Mol Pharmacol. 1993;44:1227–1231. [PubMed] [Google Scholar]
- 43.Seiden LS, Sabol KE, Ricaurte GA. Amphetamine: effects on catecholamine systems and behavior. Annu Rev Pharmacol Toxicol. 1993;32:639–677. doi: 10.1146/annurev.pa.33.040193.003231. [DOI] [PubMed] [Google Scholar]
- 44.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 45.Sonders MS, Amara SG. Channels in transporters. Curr Opin Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- 46.Sonders MS, Zhu SJ, Zahniser NR, Kavanaugh MP, Amara SG. Multiple ionic conductances of the human dopamine transporter: the actions of dopamine and psychostimulants. J Neurosci. 1997;17:960–974. doi: 10.1523/JNEUROSCI.17-03-00960.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sulzer D, Rayport S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: a mechanism of action. Neuron. 1990;5:797–808. doi: 10.1016/0896-6273(90)90339-h. [DOI] [PubMed] [Google Scholar]
- 48.Sulzer D, Pothos E, Sung HM, Maidment NT, Hoebel BG, Rayport S. Weak base model of amphetamine action. Ann NY Acad Sci. 1992;654:525–528. doi: 10.1111/j.1749-6632.1992.tb26020.x. [DOI] [PubMed] [Google Scholar]
- 49.Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- 50.Sulzer D, Chen T-K, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ungerstedt U. Measurement of neurotransmitter release by intracranial dialysis. In: Marsden CA, editor. Measurement of neurotransmitter release in vivo. Wiley; Chichester, UK: 1984. pp. 81–105. [Google Scholar]
- 52.Westerink BHC, Justice JB., Jr . Microdialysis compared with other in vivo release models. In: Robinson TE, Justice JB Jr, editors. Microdialysis in the neurosciences. Elsevier; Amsterdam: 1991. pp. 23–43. [Google Scholar]
- 53.Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- 54.Zaczek R, Culp S, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. II. Active transport. J Pharmacol Exp Ther. 1991a;257:830–835. [PubMed] [Google Scholar]
- 55.Zaczek R, Culp S, Goldberg H, McCann DJ, De Souza EB. Interactions of [3H]amphetamine with rat brain synaptosomes. I. Saturable sequestration. J Pharmacol Exp Ther. 1991b;257:820–829. [PubMed] [Google Scholar]
- 56.Zetterstrom T, Sharp T, Collin AK, Ungerstedt U. In vivo measurement of extracellular dopamine and DOPAC in rat striatum after various dopamine-releasing drugs: implications for the origin of extracellular DOPAC. Eur J Pharmacol. 1988;148:327–334. doi: 10.1016/0014-2999(88)90110-0. [DOI] [PubMed] [Google Scholar]