

Abstract
We investigated activation of β-adrenergic receptor–adenylyl cyclase–cAMP cascade on the whole-cell voltage-dependent Ca2+ currents (ICa) in acutely isolated rat basolateral amygdala neurons. Application of β-receptor agonist isoproterenol (Iso) caused a long-term enhancement ofICa. The effect of Iso was blocked by concurrent application of β-receptor antagonist propranolol. However, delayed application of propranolol after theICa enhancement did not affect Iso-induced potentiation, suggesting that the sustained effect was not caused by a slow washout of Iso. Nimodipine and ω-conotoxin-GVIA reduced theICa by ∼35 and ∼29%, respectively, without reducing enhancement of ICa by Iso significantly. The modulation appeared to involve P-type current, because the enhancement was abolished after pretreatment with ω-agatoxin-IVA. Forskolin, an adenylyl cyclase activator, mimicked the action of Iso in enhancing ICa, and this effect was blocked by an inhibitor of cAMP cascade, indicating a cAMP-dependent mechanism. Iso also induced a long-term potentiation (LTP) of synaptic transmission, which could be prevented by P-type Ca2+ channel blockers. These results suggest that P-type Ca2+ channels were selectively upregulated in the basolateral amygdala neurons, and enhancement of P-type currents could contribute to presynaptic form of LTP.
Keywords: isoproterenol, calcium channel, synaptic transmission, long-term potentiation, amygdala, cAMP
Protein phosphorylation is a widespread mechanism for signal transduction in the CNS (Nestler and Greengard, 1984; Levitan, 1985). The actions of presynaptic protein kinases in the modulation of transmitter release are particularly important and are probably involved in short- and long-term synaptic plasticity (Byrne and Kandel, 1996). In mammalian hippocampus, a role for cAMP and cAMP-dependent protein kinase (PKA) in the induction of long-term potentiation (LTP), which is a likely mechanism for learning and memory (Bliss and Lomo, 1973; Bliss and Collingridge, 1993) at the mossy fiber→CA3 synapses has been demonstrated (Huang et al., 1994;Weisskopf et al., 1994). Mossy fiber LTP required a rise in presynaptic Ca2+ that activated calmodulin-sensitive adenylyl cyclase and the consequent rise in cAMP, resulting in a long-lasting enhancement of glutamate release. In contrast, in the Schaffer collateral→CA1 synapses, activation of β-adrenergic receptor and adenylyl cyclase increased transmitter release in a reversible manner (Chavez-Noriega and Stevens, 1992; Gereau and Conn, 1994). In addition, cAMP does not act presynaptically to evoke LTP but instead functions as a gate by regulating the activity of phosphoprotein phosphatases postsynaptically (Blitzer et al., 1995; Thomas et al., 1996).
In the amygdala, we have previously shown that activation of β-adrenergic receptor by isoproterenol (Iso) caused a long-term enhancement of EPSP that was accompanied by a decrease in paired pulse facilitation (Huang et al., 1996). The sensitivity of postsynaptic neurons to the glutamate receptor agonist was unchanged, consistent with a presynaptic site of action. Furthermore, the effect of Iso was prevented by low concentrations of ω-agatoxin-IVA (ω-AgTX), suggesting that enhancement of presynaptic P-type Ca2+ currents may underlie the action of Iso. Because the presynaptic terminals are small and difficult to record from, in the present study we used whole-cell patch-clamp recording techniques in acutely dissociated amygdalar neurons to directly examine the effects of Iso and forskolin on the Ca2+currents.
MATERIALS AND METHODS
Slice preparation and intracellular recordings.Male Sprague Dawley rats (125–150 gm) were decapitated, and transverse slices (500 μm) were cut from tissue block of the brain using a Vibroslice (Campden Instruments, Silbey, UK). The slices were placed in a beaker of artificial CSF (ACSF) oxygenated with 95% O2-5% CO2 and kept at room temperature for at least 1 hr before recording. ACSF solution had the following composition (in mm): NaCl 117, KCl 4.7, CaCl22.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2, and glucose 11.
A single slice then was transferred to the recording chamber, in which it was held submerged between two nylon nets and maintained at 32 ± 1°C. The chamber consisted of a circular well of low volume (1–2 ml) and was perfused constantly at a rate of 2–3 ml/min. A bipolar stimulating electrode (SNE-200X; Kopf Instruments, Bern, Germany) was placed in the ventral endopyriform nucleus, near the recording electrode (1–2.5 mm apart). Orthodromic stimuli were delivered with monophasic constant-voltage pulses at 0.033 Hz from a Grass stimulator with an isolation unit.
Intracellular recording microelectrodes were pulled from 1.0 mm microfiber capillary tubing on a Brown–Flaming electrode puller (Sutter Instruments, San Rafael, CA). The electrodes were filled with 4m potassium acetate with resistance ranging from 60 to 120 MΩ. The microelectrode tips were positioned into the basolateral subdivision of the amygdala. Electrical signals were amplified by using an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) and recorded on a Gould 3200 chart recorder. For data acquisition and analysis, pClamp 6.0 running on a 486 personal computer was used. All data are expressed as mean ± SE. Statistical analysis was performed using Student’s t test, and p < 0.05 was considered statistically significant.
Preparation of isolated amygdalar neurons and whole-cell recordings.Amygdalar neurons were isolated using a technique adapted from Kay and Wong (1986). Male Sprague Dawley rats aged 2–3 weeks were decapitated, and serial transverse slices were cut. Amygdalar regions were dissected under a dissection microscope and then transferred to 10 ml of 1,4-piperazinethanesulfonic acid (PIPES) saline solution in a spinner flask. The temperature was maintained at 32 ± 1°C, and 2–3 mg of protease XIV (Sigma, St. Louis, MO) was added. The amygdalar minislices were stirred gently at a rate sufficient to prevent them from settling, and the solution was bubbled continuously with 95% O2-5% CO2. The PIPES saline solution contained (in mm): NaCl 120, KCl 5, CaCl2 1, MgCl2 1, glucose 25, and PIPES 20, pH 7.0.
One hour later, slices were washed with 10 ml of PIPES saline solution (three times) and transferred for storage to a holding chamber filled with continuously bubbled PIPES saline solution at room temperature. When needed, a slice was transferred to 1 ml of external solution, and the neurons were dissociated by trituration using a fire-polished Pasteur pipette with an ∼1 mm tip diameter. The suspension of dissociated amygdalar neurons then was transferred to a 0.5–1 ml recording chamber mounted on an Olympus IMT-2 inverted microscope. Neurons were allowed to settle on poly-l-lysine-coated coverslips.
Whole-cell Ca2+ currents were recorded using a patch-clamp amplifier (EPC-7, List Electronic) and filtered at 10 KHz. Patch pipettes were pulled from borosilicate glass and fire-polished with a resistance of 2–5 MΩ. The external solution contained (in mm): CaCl2 3, glucose 10, tetraethylammonium hydrochloride (TEA) 120, 4-aminopyridine (4-AP) 5, and PIPES 10; 2 μm tetrodotoxin was always added. Pipette solution contained (in mm): MgCl2 2, PIPES 10, glucose 10, TEA 20, cesium methanesulfonate 100, EGTA 10, Mg-ATP 4, Na-GTP 0.3, phosphocreatine 20, and leupeptin 0.2.
RESULTS
To confirm the involvement of P-type Ca2+channel in the action of Iso, we investigated the effect of ω-AgTX on Iso-induced potentiation. Slices were incubated in 10 nmω-AgTX for at least 30 min before being transferred to the recording chamber where the drug was present at the same concentration. Figure1 shows that Iso no longer induced LTP in slices pretreated with ω-AgTX (103 ± 10%, n = 8). In contrast, the EPSP amplitude was increased to 183 ± 8% (n = 6) by Iso in slices pretreated with 1 μm nimodipine (Fig. 1). These results suggest the involvement of P-type currents in the action of Iso.
Fig. 1.

Inhibition of Iso-induced synaptic potentiation by P-type Ca2+ channel blockers. The EPSP amplitude was plotted against time. Bars denote periods of application of Iso (15 μm), ω-AgTX (10 nm), or nimodipine (1 μm). The slices were incubated in 10 nm ω-AgTX or 1 μm nimodipine for at least 30 min before being transferred to the recording chamber where the drug was present at the same concentration.
To determine whether the increase in EPSP was in fact caused by a decrease in IPSP, we examined the effect of Iso on the monosynaptic IPSP. Monosynaptic IPSPs were evoked by focal stimulation in the presence of glutamate receptor antagonists: 10 μm6-cyano-7-nitroquinoxaline-2,3-dione and 50 μmd-2-amino-5-phosphonovalerate. Depolarization of the membrane potential increased the amplitude of IPSP, whereas hyperpolarization of the membrane potential decreased the amplitude of IPSP. In six neurons, IPSP reversed polarity at −69 ± 2 mV (data not shown) (Rainnie et al., 1991). Figure2 shows that Iso had no effect on the IPSP, in agreement with previous results described by Washburn and Moises (1992).
Fig. 2.

Iso does not affect the IPSP. Monosynaptic IPSP was evoked in the presence of 10 μm6-cyano-7-nitroquinoxaline-2,3-dione and 50 μmd-2-amino-5-phosphonovalerate. At the end of experiments, 20 μm bicuculline (Bic) was added to confirm that the IPSP was indeed mediated by GABAAreceptors.
We tested the effect of Iso on the whole-cell Ca2+currents (ICa) directly in dissociated amygdalar neurons. A sustained inward ICa was normally activated at approximately −40 mV, peaked at −10 mV, and reversed between +30 and +40 mV (Foehring and Scroggs, 1994; Viana and Hille, 1996; Wang et al., 1996) when elicited by 200 msec step commands from a holding potential of −70 mV. We used pharmacological agents to dissect the ICa. L-type current was defined as the current blocked by a saturating concentration of 1 μmnimodipine. On average, nimodipine blocked 35.0 ± 4.2% (n = 9) of the whole-cell current. N-type current was defined as the current blocked by 1 μm ω-conotoxin-GVIA (ω-CgTX). On average, 29.5 ± 1.1% (n = 5) of the ICa was blocked by ω-CgTX. P-type current was defined as the current blocked by a low concentration of ω-AgTX (10 nm) (Mintz et al., 1992; Randall and Tsien, 1995). On average, 10 nm ω-AgTX blocked 20.0 ± 1.3% of theICa, which was consistent with previous results showing a similar degree of ICa blockage by this concentration of neurotoxin (Foehring and Scroggs, 1994).
Figure 3 illustrates the effects of nimodipine, nimodipine plus ω-CgTX, and nimodipine plus ω-CgTX plus ω-AgTX in an amgydalar neuron. Nimodipine (1 μm) alone blocked 27.6% of the current, and nimodipine plus ω-CgTX blocked 58%. Simultaneous application of nimodipine plus ω-CgTX plus ω-AgTX blocked 71.3% of the current, leaving 28.7% unblocked. The current that remained in the presence of all three antagonists may contain Q- and R-type Ca2+ currents that were subsequently blocked by 100 μm Cd++. A similar degree of inhibition was observed in five additional cells.
Fig. 3.

Pharmacological separation ofICa in amygdalar neurons. Plot of peakICa versus time is shown for an experiment in which 1 μm nimodipine, 1 μm nimodipine plus ω-CgTX, 10 nm nimodipine plus ω-CgTX plus ω-AgTX, and 100 μm Cd++ were sequentially applied to an amygdala neuron.
Figure 4A shows that bath application of 15 μm Iso persistently enhancedICa. On average, Iso increased the current by 54 ± 7% (n = 11; p < 0.001). Iso apparently did not change the kinetics ofICa; the current at the beginning of a voltage command (the peak current) was enhanced by the same degree as the current at the end of the command. In addition, as can be seen in Figure 4A, concurrent application of 1 μm propranolol with Iso prevented the effect of Iso (102 ± 4%, n = 6), indicating the involvement of β-adrenergic receptors. Furthermore, to exclude the possibility that the sustained effect was caused by a slow washout of Iso from the slices, we applied propranolol during the washout period. In six neurons, delayed application of 1 μm propranolol did not affect the Iso-induced sustained enhancement ofICa (Fig. 4B), suggesting that the long-term effect was not the result of a continued activation of β-adrenergic receptors by Iso.
Fig. 4.
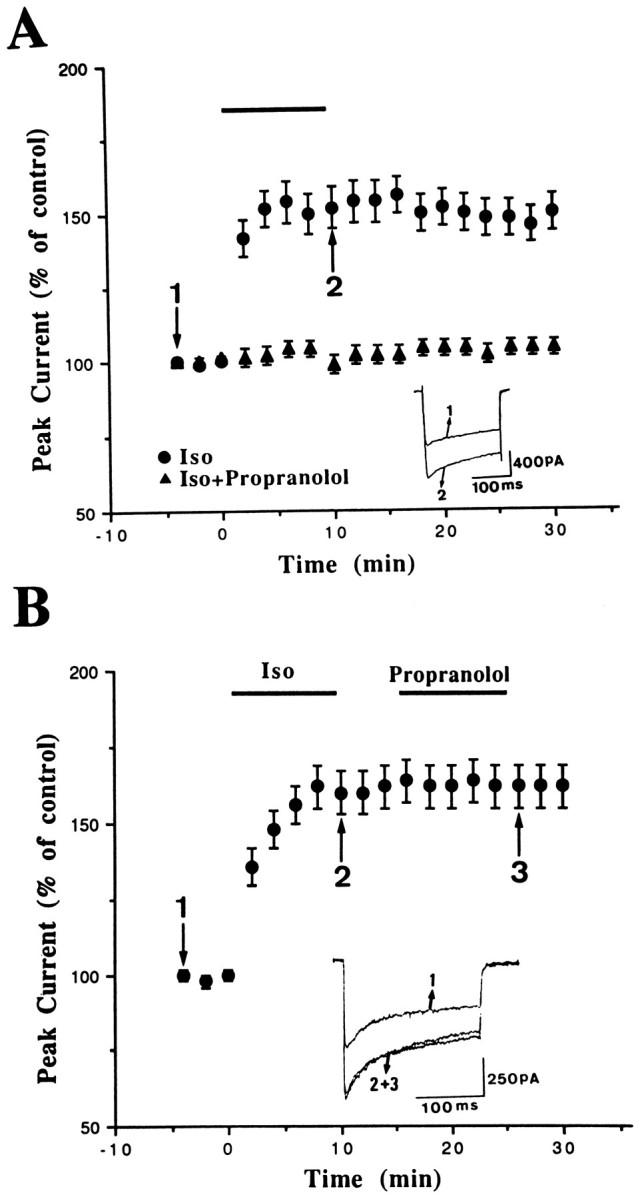
Persistent enhancement ofICa by Iso. A, The percent change of peak ICa was plotted as a function of time.Bar denotes the periods of delivery of Iso (15 μm) or Iso plus propranolol (1 μm). The Iso-induced potentiation was prevented by concurrent application of propranolol. B, Delayed application of propranolol afterICa enhancement did not affect Iso-induced potentiation significantly.
We next examined whether Iso affects a particular type of channel. If the Iso modulation was of N-type channel exclusively, then no modulation should be evident in the presence of 1 μmω-CgTX. However, this is not the case. In six cells pretreated with ω-CgTX, Iso increased the current by 68 ± 4% (Fig.5A). The same experimental design was performed by using 1 μm L-type blocker nimodipine. As illustrated in Figure 5B, Iso increased the current by 58 ± 10% (n = 6) in the presence of nimodipine. These data suggest that N- and L-type channels are not involved in the modulation by Iso. We next tested the hypothesis that P-type channels were modulated by Iso. Figure 5A clearly shows that the effect of Iso was completely eliminated by 10 nm ω-AgTX. The current remained 102 ± 6% (n = 8) of its baseline.
Fig. 5.
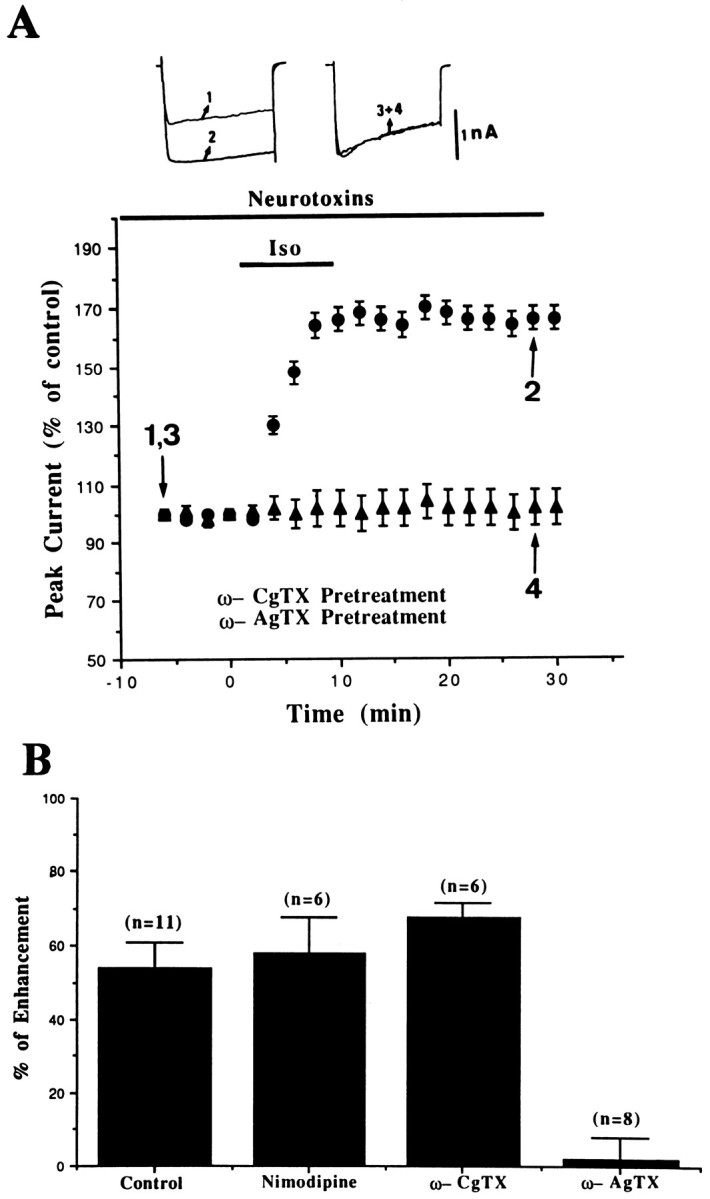
Selective blockade of Iso-induced enhancement ofICa by ω-AgTX. A, The percent change of ICa was plotted as a function of time. The isolated amygdalar neurons were incubated in 1 μm ω-CgTX or 10 nm ω-AgTX for at least 10 min before recordings were made. B, Thebars represent the enhancement ofICa (mean ± SEM) by Iso in control and after application of various types of Ca2+ channel blockers.
If activation of β-adrenergic receptors enhancedICa via PKA activation, then a direct activator of adenylyl cyclase should mimic the effect of Iso. Figure6A shows that bath application of 25 μm forskolin produced an effect similar to that of Iso. On average, ICa was increased by 38 ± 6% (n = 5). Rp-adenosine 3′,5′-cyclic monophosphothioate (Rp-cAMPS), a membrane-permeable PKA inhibitor, effectively blocked forskolin-induced enhancement ofICa. In the presence of 25 μmRp-cAMPS, the ICa remained 101 ± 1% of baseline (n = 6; Fig. 6B). Pretreatment of cells with ω-AgTX similarly abolished the effect of forskolin (100 ± 6% of control; n = 6; Fig.6A).
Fig. 6.
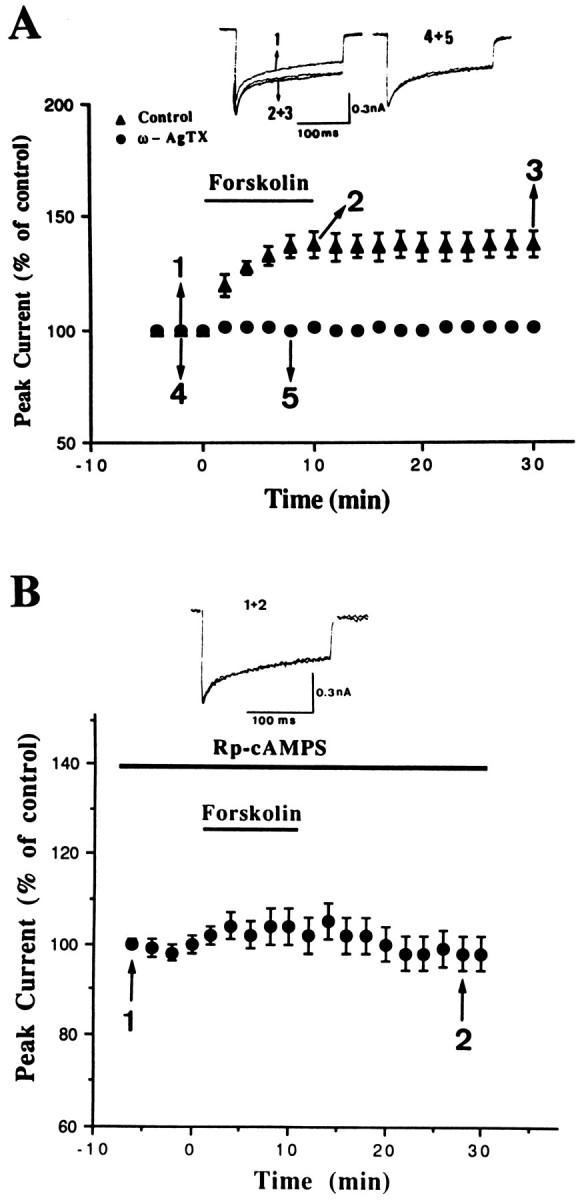
Sustained enhancement ofICa by forskolin and its inhibition by ω-AgTX and Rp-cAMPS. A, The percent change of peakICa was plotted as a function of time.Bar denotes the period of application of 25 μm forskolin. In 10 nm ω-AgTX-treated neurons, the neurotoxin was present for at least 10 min before and during the period of recording. B, The effect of forskolin was abolished by Rp-cAMPS. The isolated neurons were incubated with 25 μm Rp-cAMPS for at least 20 min before and during the period of recording.
TEA at a concentration that blocks the delayed rectifier K+ channels has been shown to produce a long-lasting enhancement of synaptic transmission (Aniksztejn and Ben-Ari, 1991;Huang and Malenka, 1993; Hanse and Gustafsson, 1994). Therefore, the delayed rectifier K+ channels could be a possible target for modulation. The delayed rectifier K+currents were elicited by depolarization from a holding potential of −70 to +60 mV in the presence of 100 μmCd++ and 5 mm 4-AP. This K+ current was activated on depolarization positive to −50 mV, and its magnitude increased on further depolarization. In all six cells tested, Iso had no affect on the delayed rectifier K+ currents (Fig.7).
Fig. 7.

Iso does not affect the delayed rectifier K+ currents. Delayed rectifier K+currents were elicited by depolarizing pulses over the potential ranges of −60 to +60 mV from a holding potential of −70 mV in the presence of 100 μm Cd++ and 5 mm4-aminopyridine.
DISCUSSION
In many excitatory synapses, activation of the PKA pathway has been shown to increase transmitter release, persistently or reversibly, depending on the preparations studied. In hippocampal mossy fiber→CA3 synapses (Huang et al., 1994; Weisskopf et al., 1994), cerebellar parallel fiber synapses (Salin et al., 1996), and basolateral amygdala neurons (Huang et al., 1996), an increase in cAMP and activation of PKA resulted in a persistent facilitation of transmitter release, a phenomenon that may have contributed to a presynaptic form of LTP. In contrast, in Schaffer collateral→CA1 synapses, transmitter release was enhanced in a reversible manner (Chavez-Noriega and Stevens, 1992;Gereau and Conn, 1994). Thus, it is likely that the excitation–secretion coupling processes are differentially regulated by PKA in different brain areas.
Voltage-dependent Ca2+ channels are unique in neuronal communication in terms of transduction of an electrical signal (membrane depolarization) via Ca2+ entry into a chemical signal (neurotransmitter release) (Llinas et al., 1981). Theoretically, modification of Ca2+ channels could be an effective means of regulating neurotransmission. However, it is suggested that reversible enhancement of glutamate release by forskolin in cultured hippocampal neurons was not attributable to an alteration in depolarization-evoked increase in intracellular Ca2+ but was accompanied by a direct modulation of secretory machinery downstream from Ca2+ influx (Trudeau et al., 1996). It is not known whether the same mechanism underlies PKA-induced long-term enhancement of glutamate release in other synapses.
In the present study, we have demonstrated that activation of β-adrenergic receptor and the enzyme adenylyl cyclase persistently increased the whole-cell Ca2+ currents that paralleled a long-term enhancement of transmitter release. Both effects were blocked by the P-type Ca2+ channel blocker ω-AgTX but not by L- or N-type blockers. These results indicate that both presynaptic and postsynaptic P-type Ca2+channels are subject to neuromodulation by PKA, and that long-term enhancement of Ca2+ currents may underlie the Iso-induced presynaptic form of LTP in the amygdala. However, unless presynaptic Ca2+ currents and PSPs could be recorded simultaneously in the basolateral amygdala, it is difficult to prove directly any causal relationship between P-type channel modulation and synaptic potentiation. Therefore, we cannot exclude the possibility that the enhancement of transmission may involve additional mechanisms such as a direct modulation of secretory machinery downstream from Ca2+ influx (Trudeau et al., 1996).
The mechanisms involved in Iso potentiation of P-type Ca2+ channels in amygdalar neurons remain to be determined. Phosphorylation by PKA modulates voltage-dependent Ca2+ channels in several excitable cells (Dolphin, 1996). The modulation of L-type Ca2+ channels has been repeatedly demonstrated in the central neurons (Gray and Johnston, 1987; Kavalali et al., 1997). Only in two studies, one in hippocampal CA3 neurons and the other in Xenopus oocytes expressed with human β subunits, was selective enhancement of P-type currents via stimulation of adenylyl cyclase reported (Mogul et al., 1993; Fukuda et al., 1996), although the reversibility of the drug effect was not addressed. In the present study, we found that the increase inICa outlasted the duration of application of Iso and administration of propranolol before but not afterICa enhancement prevented the effect of Iso. These results suggest that the long-term effect was not attributable to a slow washout of Iso. In the future, it would be of interest to determine whether long-term effect was caused by a continued kinase activity by delayed application of Rp-cAMPS or inhibitors of the PKA catalytic site.
In light of the significant contribution of P-type channels to presynaptic Ca2+ entry and transmitter release (Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994), persistent enhancement of these channels will have a profound influence on neuronal activity in the amygdala. Indeed, behavioral studies have shown that post-training intra-amygdala administration of β-adrenergic agonists enhances the retention of an inhibitory avoidance task (Liang et al., 1986). More importantly, in normal humans, propranolol, a β-adrenergic blocker, selectively impaired long-term memory for an emotionally arousing short story (Cahill et al., 1994). In addition, the patient with bilateral brain damage confined to the amygdala failed to show normal memory associated with emotional arousal (Cahill et al., 1995).
Footnotes
This study was supported by the National Science Council of Taiwan Grant NSC86-2314-B006-002 M10. We thank Drs. M. D. Lai and T. H. Leu for their insightful comments on this manuscript.
Correspondence should be addressed to Dr. Po-Wu Gean at the above address.
REFERENCES
- 1.Aniksztejn L, Ben-Ari Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature. 1991;349:67–69. doi: 10.1038/349067a0. [DOI] [PubMed] [Google Scholar]
- 2.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blitzer RD, Wong T, Nouranifar R, Lyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- 5.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cahill L, Prins B, Weber M, McGaugh JL. β-adrenergic activation and memory for emotional events. Nature. 1994;371:702–704. doi: 10.1038/371702a0. [DOI] [PubMed] [Google Scholar]
- 7.Cahill L, Babinsky R, Markowitsch HJ, McGaugh JL. The amygdala and emotional memory. Nature. 1995;377:295–296. doi: 10.1038/377295a0. [DOI] [PubMed] [Google Scholar]
- 8.Chavez-Noriega LE, Stevens CF. Modulation of synaptic efficacy in field CA1 of the rat hippocampus by forskolin. Brain Res. 1992;574:85–92. doi: 10.1016/0006-8993(92)90803-h. [DOI] [PubMed] [Google Scholar]
- 9.Dolphin AC. Facilitation of Ca++ current in excitatory cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- 10.Foehring RC, Scroggs RS. Multiple high-threshold calcium currents in acutely isolated rat amygdaloid pyramidal cells. J Neurophysiol. 1994;71:433–436. doi: 10.1152/jn.1994.71.1.433. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda K, Kaneko S, Yada N, Kikuwaka M, Akaike A, Satoh M. Cyclic AMP-dependent modulation of N- and Q-type Ca++ channels expressed in Xenopus oocytes. Neurosci Lett. 1996;217:13–16. doi: 10.1016/0304-3940(96)13055-x. [DOI] [PubMed] [Google Scholar]
- 12.Gereau RW, Conn PJ. Presynaptic enhancement of excitatory synaptic transmission by β-adrenergic receptor activation. J Neurophysiol. 1994;72:1438–1442. doi: 10.1152/jn.1994.72.3.1438. [DOI] [PubMed] [Google Scholar]
- 13.Gray R, Johnston D. Noradrenaline and β-adrenoreceptor agonists increase activity of voltage-dependent calcium channels in hippocampal neurons. Nature. 1987;327:620–622. doi: 10.1038/327620a0. [DOI] [PubMed] [Google Scholar]
- 14.Hanse E, Gustafsson B. TEA elicits two distinct potentiations of synaptic transmission in the CA1 region of the hippocampal slices. J Neurosci. 1994;14:5028–5034. doi: 10.1523/JNEUROSCI.14-08-05028.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang Y-Y, Malenka RC. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca++ channels in the induction of LTP. J Neurosci. 1993;13:568–576. doi: 10.1523/JNEUROSCI.13-02-00568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y-Y, Li X-C, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalent-mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- 17.Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kavalali ET, Hwang KS, Plummer MR. cAMP-dependent enhancement of dihydropyridine-sensitive calcium channel availability in hippocampal neurons. J Neurosci. 1997;15:5334–5348. doi: 10.1523/JNEUROSCI.17-14-05334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kay AR, Wong RKS. Isolation of neurons suitable for patch-clamping from adult mammalian central nervous system. J Neurosci Methods. 1986;16:227–238. doi: 10.1016/0165-0270(86)90040-3. [DOI] [PubMed] [Google Scholar]
- 20.Levitan IB. Phosphorylation of ion channels. J Membr Biol. 1985;87:177–190. doi: 10.1007/BF01871217. [DOI] [PubMed] [Google Scholar]
- 21.Liang KC, Juler RG, McGaugh JL. Modulating effects of post-training epinephrine on memory: involvement of the amygdala noradrenergic system. Brain Res. 1986;368:125–133. doi: 10.1016/0006-8993(86)91049-8. [DOI] [PubMed] [Google Scholar]
- 22.Llinas R, Steinberg IZ, Walton K. Presynaptic calcium currents in squid giant synapse. Biophys J. 1981;33:289–322. doi: 10.1016/S0006-3495(81)84898-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luebke JI, Dunlap K, Turner TJ. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 1993;11:895–902. doi: 10.1016/0896-6273(93)90119-c. [DOI] [PubMed] [Google Scholar]
- 24.Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- 25.Mogul DJ, Adams ME, Fox AP. Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca++ current in hippocampal CA3 neurons. Neuron. 1993;10:327–334. doi: 10.1016/0896-6273(93)90322-i. [DOI] [PubMed] [Google Scholar]
- 26.Nestler EJ, Greengard P. Protein phosphorylation in the nervous system. Wiley; New York: 1984. [Google Scholar]
- 27.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Inhibitory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:999–1009. doi: 10.1152/jn.1991.66.3.999. [DOI] [PubMed] [Google Scholar]
- 28.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca++ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- 31.Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 32.Trudeau LE, Emery DG, Haydon PG. Direct modulation of the secretory machinery underlies PKA-dependent synaptic facilitation in hippocampal neurons. Neuron. 1996;17:789–797. doi: 10.1016/s0896-6273(00)80210-x. [DOI] [PubMed] [Google Scholar]
- 33.Viana F, Hille B. Modulation of high voltage-activated calcium channels by somatostatin in acutely isolated rat amygdaloid neurons. J Neurosci. 1996;16:6000–6011. doi: 10.1523/JNEUROSCI.16-19-06000.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang SJ, Huang CC, Hsu KS, Tsai JJ, Gean PW. Inhibition of N-type calcium currents by lamotrigine in rat amygdalar neurones. NeuroReport. 1996;7:3037–3040. doi: 10.1097/00001756-199611250-00048. [DOI] [PubMed] [Google Scholar]
- 35.Washburn MS, Moises HC. Inhibitory responses of rat basolateral amygdaloid neurons recorded in vitro. Neuroscience. 1992;50:811–830. doi: 10.1016/0306-4522(92)90206-h. [DOI] [PubMed] [Google Scholar]
- 36.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 37.Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca++ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]