

Abstract
Focal brain ischemia induces inflammation, extracellular matrix remodeling, gliosis, and neovascularization. Osteopontin (OPN) is a secreted glycoprotein that has been implicated in vascular injury by promoting cell adhesion, migration, and chemotaxis. To investigate the possible involvement of OPN in brain matrix remodeling after focal stroke, we examined the expression of OPN in ischemic cortex after permanent or temporary occlusion of the middle cerebral artery (MCAO) of the rat. OPN mRNA and protein levels in nonischemic cortex were not detected consistently, although significant induction of OPN was observed in the ischemic cortex. OPN mRNA increased 3.5-fold at 12 hr and reached peak levels 5 d (49.5-fold; p < 0.001) after permanent MCAO. The profile of OPN mRNA induction after transient MCAO (160 min) with reperfusion was essentially the same as that of permanent MCAO. In situ hybridization and immunohistochemical studies demonstrated strong induction of OPN in the ischemic cortex, which was localized primarily in a subset of ED-1-positive macrophages that accumulated in the ischemic zone. Moreover, OPN immunoreactivity was detected in the matrix of ischemic brain, suggesting a functional role of the newly deposited matrix protein in cell–matrix interactions and remodeling. Indeed, using a modified Boyden chamber, we demonstrated a dose-dependent chemotactic activity of OPN in C6 astroglia cells and normal human astrocytes. Taken together, these data suggest that the upregulation of OPN after focal brain ischemia may play a role in cellular (glia, macrophage) migration/activation and matrix remodeling that provides for new matrix–cell interaction.
Keywords: osteopontin, focal stroke, matrix protein, astrocyte, macrophage, rat
Osteopontin (OPN) is a highly acidic, secreted phosphoglycoprotein of 41 kDa containing the adhesive motif arginine–glycine–aspartate (RGD), which interacts with the αvβ3 integrin on the cell surface (Liaw et al., 1995). OPN was found originally in bone matrix and subsequently in kidney, placenta, and blood vessels (Butler, 1989; Giachelli et al., 1995). The induced expression of OPN has been identified in a number of cell lines, including osteoclasts, fibroblasts, activated T-cells, macrophages, epithelial cells, and vascular smooth muscle cells in response to inflammatory cytokines and growth factors (Butler, 1989; Patarca et al., 1989; Singh et al., 1990; Denhardt and Guo, 1993; Giachelli et al., 1995; Wang et al., 1996). Elevated expression of OPN also has been observed in a number of disease processes, including autoimmune disorders (Patarca et al., 1990; Lampe et al., 1991), neointima formation in carotid arteries subjected to balloon angioplasty (Giachelli et al., 1993; Wang et al., 1996), human atherosclerotic plaques (Giachelli et al., 1993; O’Brien et al., 1994), myocardial injury (Murry et al., 1994), and renal tubulointerstitial fibrosis (Giachelli et al., 1994; Pichler et al., 1994). Functionally, OPN has been implicated in cell attachment and chemotaxis for macrophages, smooth muscle cells, and endothelial cells (Singh et al., 1990; Liaw et al., 1994; Giachelli et al., 1995), which may bear on vascular remodeling processes in restenosis and atherosclerosis.
Cerebral ischemia is another condition characterized by significant leukocyte infiltration and tissue remodeling (Hallenbeck et al., 1986;Clark et al., 1993, 1994; Garcia et al., 1994; Barone et al., 1995). The inflammatory reaction after focal stroke is characterized by early neutrophil adherence to blood vessels and infiltration into brain tissue, followed by significant accumulation of monocytes and activated macrophages in the lesion. These activated macrophages are associated with phagocytosis, connective tissue matrix formation, and resolution of the injured brain tissue. Although the expression of mRNA and protein for several inflammatory cytokines, chemokines, and cell adhesion molecules has been described previously (Feuerstein et al., 1997), little is known about matrix remodeling processes in brain ischemia and especially RGD–matrix proteins that enable inflammatory cell adhesion and migration into the injury zone.
Because OPN expression has been found in a number of inflammatory reactions in peripheral organs (Giachelli et al., 1993, 1994; Murry et al., 1994; O’Brien et al., 1994; Pichler et al., 1994; Wang et al., 1996), with spatial and temporal association to macrophage migration and accumulation (Singh et al., 1990), we speculated that OPN also might be involved in a similar reaction elicited by ischemic brain injury. Therefore, we examined the spatial and temporal expression of OPN mRNA in two well characterized models of focal brain ischemia (permanent or temporary occlusion of the middle cerebral artery). Usingin situ hybridization and immunohistochemistry methods, we investigated the temporal and spatial distribution of OPN in the ischemic cortex. In addition, because gliosis is one of the critical remodeling events in response to focal stroke, we investigated the chemotactic activity of OPN on glial cell migration in vitroand demonstrated a potential role of OPN in glial cell activation and migration in addition to the actions postulated for OPN on macrophage recruitment and matrix remodeling after ischemic injury.
MATERIALS AND METHODS
Focal brain ischemia. Permanent or temporary cerebral focal ischemia or sham surgery was performed under stereotaxic control in male spontaneously hypertensive rats (SHR) or in a normotensive rat strain [Wistar-Kyoto (WKY)], at 18 weeks of age and weighing 250–330 gm, by permanent or temporary occlusion of the middle cerebral artery (MCAO), as described in detail previously (Barone et al., 1992a,b,1995, 1997a). These models are similar to those established and used by others (Brint et al., 1988; Duverger and MacKenzie, 1988; Buchan et al., 1992) and have been characterized extensively over time, indicating extended time course evaluations, for the consistency of ischemic cortical blood flow effects and infarction (Barone et al., 1992a,b), neurological deficits (Barone et al., 1993), increased cytokine expression and influence on tissue injury (Barone et al., 1997a; Wang et al., 1997), cellular infiltration, inflammation, tissue changes, and resolution of injury (Barone et al., 1991, 1992b, 1995;Clark et al., 1993, 1994), and the influence of temperature on injury (Barone et al., 1997b). Complete tissue injury/necrosis was observed by 24 hr of permanent MCAO or within 24 hr of reperfusion in these models (Clark et al., 1993, 1994). Briefly, for permanent MCAO, the middle cerebral artery was occluded permanently and cut dorsal to the lateral olfactory tract at the level of the inferior cerebral vein by using electrocoagulation (Force 2 Electrosurgical Generator, Valley Lab). For temporary MCAO with reperfusion, the MCA was lifted from the brain surface to occlude blood flow for 160 min and then reperfused as described in detail previously (Barone et al., 1992a,b, 1995, 1997a). Blood flow determinations have demonstrated the validity of these techniques, i.e., flow is permanently decreased <25% of baseline after permanent MCAO and recovers to 100% of blood flow after reperfusion of the temporary MCAO (Barone et al., 1992a). SHR were selected as the focus of study in the present experiments because of their consistent response to permanent or transient focal ischemic injury (i.e., consistent infarctions with low variability) (Duverger and MacKenzie, 1988; Ginsberg and Busto; 1989; Barone et al., 1992a). Body temperature was maintained at 37°C with a heating pad during all surgical procedures and continued to be regulated at 37°C after head closure (i.e., during recovery of anesthesia) for several hours until normal motor activity was resumed by individual animals. In sham-operated rats the dura was opened over the MCA, but the artery was not occluded. Rats then were overdosed with pentobarbital, and forebrains were removed at 1, 3, 6, and 12 hr and 1, 2, 5, 10, and 15 d after permanent MCAO or after reperfusion that followed temporary MCAO and at 12 hr and 5 d after sham surgery. The ischemic cortex (i.e., the cortex ipsilateral to surgery) was dissected from the ipsilateral hemisphere; the contralateral (control) cortex was dissected from the nonischemic contralateral hemisphere from the same rat (Barone et al., 1991, 1992b, 1995; Wang et al., 1997). The cortical samples were frozen immediately in liquid nitrogen and stored at −80°C.
Northern hybridization analysis. For RNA preparation, cortical samples were homogenized in an acid guanidinium thiocyanate solution and extracted with phenol and chloroform as described previously (Chomczynski and Sacchi, 1987; Wang et al., 1994a). RNA samples (40 μg/lane) were electrophoresed through formaldehyde–agarose slab gels and transferred to GeneScreen Plus membranes (NEN Life Science Products, Boston, MA). For Northern blot analysis, an OPN or ribosomal protein L32 cDNA was prepared as described previously (Wang et al., 1996) and was labeled uniformly with [α-32P]dATP (3000 Ci/mmol, Amersham, Arlington Heights, IL), using a random-priming DNA labeling kit (Boehringer Mannheim, Indianapolis, IN). Hybridization of each probe was performed overnight with 1 × 106 cpm/ml of probe at 42°C in 5× SSPE (750 mm NaCl, 50 mmNaH2PO4, pH 7.6, and 5 mmEDTA), 50% formamide, 5× Denhardt’s solution, 2% SDS, 100 μg/ml polyA, and 200 μg/ml boiled salmon sperm DNA. The membranes were washed in 2× SSPE/2% SDS at 65°C for 1–2 hr, with a change every 30 min, and then autoradiographed at −70°C with a Cronex Lightning-Plus intensifying screen for various times, depending on the signal intensity. The relative band intensities were measured by a PhosphorImager with an ImageQuant software package (Molecular Dynamics, Sunnyvale, CA). A probe was stripped from the membranes in 10 mm Tris, pH 7.5, 1 mm EDTA, pH 8.0, and 1% SDS for 20 min at 95°C and then washed in 2× SSPE for 10 min before rehybridization with the other probe. The expression of rpL32 gene is relatively constant in the present experimental conditions (Wang et al., 1994a,b) and therefore was used to normalize the differences of the samples loaded in each lane.
In situ hybridization. Tissue preparation, in vitro transcription, and combined in situ hybridization and immunohistochemistry were performed as reported previously (Ellison et al., 1996), with slight modifications to the in situhybridization protocol as indicated. Tissue sections (12 μm) were incubated with the ED-1 antibody (BioSource International, Camarillo, CA) overnight at 4°C. Detection of the antibody was performed with the Vector Labs ABC kit (Burlingame, CA) according to the manufacturer’s instructions, with diaminobenzidine as the substrate. Immediately after antibody detection, the tissue was deproteinated in 0.2 m HCl and then acetylated with 0.1 mtriethanolamine, pH 8, with 0.25% acetic anhydride. Antisense or sense [33P]uridine triphosphate-labeled OPN probes (1 × 106 cpm/ml) were applied to tissue and hybridized overnight at 60°C. Posthybridization washes were modified as follows: 4× SSC twice for 10 min each at 50°C; 20 μg/ml RNase for 30 min at 37°C; 5 min each of 2× SSC, 1× SSC, and 0.5× SSC at room temperature; 0.1× SSC for 20 min at 55°C; and 0.1× SSC for 5 min at room temperature. Slides were dehydrated and dried and then exposed to Hyperfilm βmax (Amersham) overnight. For emulsion autoradiography the slides were dipped in NTB2 emulsion (Kodak, Rochester, NY) and exposed for 7 d at 4°C.
Immunohistochemistry. For immunohistochemical studies, rats were killed at 6, 12, and 24 hr and 5, 10, and 15 d after permanent MCAO. After whole-body perfusion with 10% phosphate-buffered formalin, the brain from three rats (n = 3) at each time point was excised and stored in formalin for 24 hr, after which time the samples were transferred to 70% ethanol and then subjected to standard histological processing by using a Vacuum Infiltration Processor (Miles, Elkhart, IN). After paraffin embedding, 5 μm sections were cut, stained with hematoxylin and eosin, and evaluated microscopically. Additional sections were placed on Capillary Gap Plus Microscope Slides (Bio-Tek Instruments, Burlingame, CA) for immunohistochemical evaluation of OPN, ED-1 (monocyte/macrophage marker), glial fibrillary acidic protein (GFAP; marker for activated glia), and S-100 (neurofilament marker) expression. Immunohistochemistry was performed as described previously (Wang et al., 1996) with mouse anti-rat OPN antibody, MPIIIB10 (at 1:50 dilution; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), monoclonal anti-ED-1 (at 1:100 dilution; Harlan BioProducts for Science, Indianapolis, IN), rabbit anti-cow GFAP (at a dilution of 1:750 or 1:20,000; Dako, Carpinteria, CA), and rabbit anti-cow S-100 (at 1:20,000 dilution; DAKO), respectively.
Cell culture and migration assay. Rat C6 glial cells (ATCC CCL107) were obtained from American Type Cultured Collection (ATCC, Rockville, MD) and cultured in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 5% fetal bovine serum. Normal human astrocytes were purchased from Clonetics and cultured in an astrocyte basal medium (Clonetics, San Diego, CA) containing 5% fetal bovine serum, 20 ng/ml human epidermal growth factor (hEGF), 25 μg/ml insulin, 25 ng/ml progesterone, 50 μg/ml transferrin, 50 μg/ml gentamicin, and 50 ng/ml amphotericin-B. The human astrocytes were maintained and subcultured according to the manufacturer’s specifications and were applied for the cell migration assays before the passage 3.
Cell migration assays were performed in a Transwell cell culture chamber, using a polycarbonate membrane with 8 μm pores (Costar, Cambridge, MA) as reported previously (Hidaka et al., 1992). The lower surface of the membrane was precoated with a different concentration of recombinant rat OPN (Yue et al., 1994). C6 cells were suspended in DMEM, and human astrocytes were resuspended in astrocyte basal medium supplemented with 0.2% bovine serum albumin at a concentration of 3 × 106 and 2 × 105 cells per milliliter, respectively. As a standard assay, 0.2 ml of cell suspension was placed in the upper compartment of the chamber, and the lower compartment contained 0.6 ml of DMEM or astrocyte basal medium supplemented with 0.2% bovine albumin before use. Incubation was performed at 37°C in 5% CO2 for 24 hr. After incubation, nonmigrated cells on the upper surface were scraped gently, and the filters were fixed in methanol and stained with 10% Giemsa stain. The number of cells that migrated to the lower surface of the filters either was measured by optical density at 640 nm (C6 cells) or counted in four high-power fields (100×) per filter (Hidaka et al., 1992; Yue et al., 1994). The relative number of cells for optical density measurement was evaluated in parallel with counting the cells under microscope, and the percentage of cells migrated was determined (see Figure 7 legend for detail). Experiments were performed in triplicate.
Fig. 7.
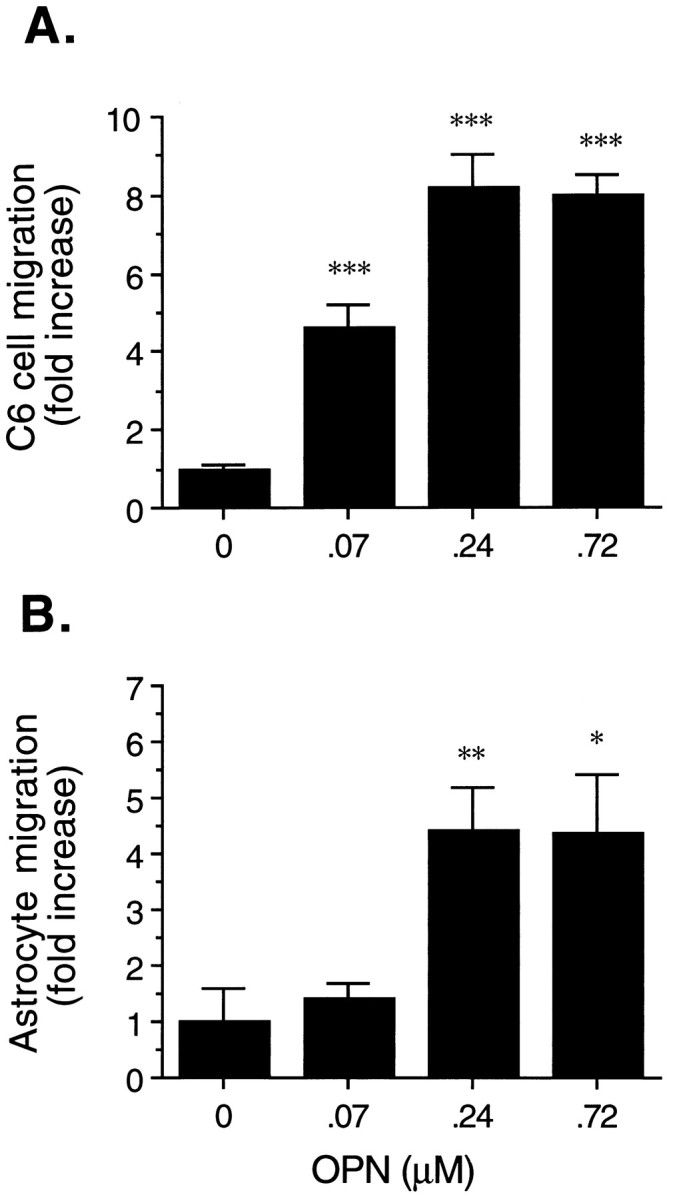
Effects of OPN on C6 astroglia (A) and normal human astrocyte (B) cell migration. The migration assay was performed in the Transwell cell culture chamber, as described in Materials and Methods. The indicated concentration of OPN was coated on the lower surface of the membrane. The cell suspension (containing 6 × 105 C6 cells or 4 × 104 human astrocytes in a vol of 0.2 μl) was added in the upper compartment and incubated for 24 hr. The relative number of migrated cells (on the lower surface of the filters) was measured by optical density at 640 nm for C6 cells or determined microscopically by counting four high-power fields per filter for human astrocytes. In the concentration range of 0–0.24 μm OPN, ∼0.5–4% of C6 cells or 0.7–3% of human astrocytes were migrated. The data are the mean ± SE of three experiments performed in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001, as compared with the control.
Statistical analysis. Statistical comparisons were made by ANOVA (Fisher’s protected least-squares difference); values were considered to be significant when p < 0.05.
RESULTS
Temporal expression of OPN mRNA in rat ischemic cortex after MCAO
Figure 1Aillustrates a representative Northern blot for the OPN mRNA in the focal ischemic and nonischemic cortex and in the sham-operated sample. Quantitative Northern blot signals for OPN (n = 4), after being normalized to a rpL32 probe, are illustrated in Figure1B. OPN mRNA in the sham-operated animals or in the contralateral (nonischemic) cortex was not detected consistently. The expression of OPN mRNA was induced in the ipsilateral (ischemic) cortex 12 hr after ischemic injury (3.5-fold increase over control), significantly upregulated at 2 d (12.1-fold; p < 0.05), peaked at 5 d (49.5-fold; p < 0.001), and then gradually decreased after 10 d (11.0-fold; p< 0.05) following MCAO (Fig. 1).
Fig. 1.

Northern blot analysis of OPN mRNA expression in rat ischemic cortex after permanent MCAO. A, Represent Northern blot for OPN and rpL32 cDNA probes to the samples from spontaneously hypertensive rats (SHR) after permanent MCAO. Total cellular RNA (40 μg/lane) was resolved by electrophoresis, transferred to a nylon membrane, and hybridized to the indicated cDNA probe. Ipsilateral and contralateral cortex samples (denoted by +) from individual rats of sham surgery (S; 5 d) or after 1, 3, 6, 12, and 24 hr and 2, 5, 10, and 15 d of permanent MCAO are depicted. B, Quantitative Northern blot data (n = 4) for OPN mRNA expression after focal stroke. The data were generated by PhosphorImager analysis and were displayed graphically after being normalized with rpL32 mRNA signals. *p < 0.05; ***p < 0.001, as compared with sham-operated animals.
A similar induction profile of OPN mRNA expression also was observed in a rat model of temporary MCAO (160 min) with reperfusion (Fig.2), except that the upregulation of OPN mRNA at early time points (12 and 24 hr after reperfusion) revealed statistical significance (14.8- and 15.7-fold increase, respectively;p < 0.01), which may reflect the early infiltration of macrophages into the ischemic lesions after temporary MCAO (Clark et al., 1994). The peak expression of OPN mRNA was observed at 5 d after reperfusion (40.0-fold over control; p < 0.001) (Fig. 2).
Fig. 2.

Northern analysis of OPN mRNA expression in rat ischemic cortex after temporary MCAO with reperfusion. This figure is illustrated as described in the Figure 1 legend except that temporary occlusion (160 min) of the middle cerebral artery was used. The indicated time points refer to the time of reperfusion. The hybridization was performed in the presence of OPN and rpL32 probes simultaneously.
To evaluate whether the elevated levels of OPN mRNA in the ischemic cortex are particular to the specific SHR strain that carries the stroke risk factor of hypertension, we compared the hypertensive strain (SHR) with the normotensive rat strain (Wistar-Kyoto) at 12 hr and 5 d after permanent MCAO (Fig. 3). Northern blot analysis revealed that MCAO induced a higher level of OPN mRNA expression in the ipsilateral (ischemic) cortex over controls of both rat strains (at 5 d; p < 0.01) and in particular the SHR. The elevated OPN mRNA level was significantly higher in SHR (8.7-fold; p < 0.001) than in WKY at 5 d after MCAO, whereas no difference was noted at 12 hr (Fig. 3). This difference is in accord with the previous observation (Clark et al., 1993) of maximal accumulation of macrophages in the ischemic cortex 5 d after MCAO in SHR, as compared with WKY, but not at the early time point (12 hr).
Fig. 3.

Northern blot analysis of OPN mRNA expression in SHR and WKY at 12 hr and 5 d after permanent MCAO.A, Total cellular RNA was isolated from the ischemic (Ipsilateral) and nonischemic (Contralateral) cortex of hypertensive (SHR) and normotensive (WKY) rats at 12 hr and 5 d after permanent MCAO. This representative Northern blot was performed by using an OPN cDNA probe, as described in Materials and Methods and in the legend to Figure 1A. B, Quantitative analysis of OPN mRNA expression in hypertensive (SHR) and normotensive (WKY) rats after permanent MCAO, as described in the legend to Figure 1B. The data are presented as the mean values of four animals (n= 4) from each strain after the samples loaded in each lane with rpL32 were normalized. ***p < 0.001, as compared with WKY at the same time point.
In situ localization of OPN mRNA in ischemic cortex
Because maximal expression of OPN mRNA was observed at 5 d (n = 3) after MCAO (see Fig. 1), in situhybridization was performed at this time point to identify the cells that were expressing OPN mRNA. As illustrated in Figure4A, the expression of OPN mRNA detected by in situ hybridization was seen in the ischemic cortex of the entire infarcted region (Fig.4A). To identify phenotypically the cells expressing OPN mRNA, we used a technique of combined in situhybridization and immunohistochemistry. All of the cells expressing OPN mRNA in the infarct could be identified phenotypically as macrophages by the colocalization of ED-1 and OPN mRNA (Fig. 4B). On the other hand, not all of the ED-1-positive macrophages expressed OPN mRNA. Instead, only those ED-1-positive macrophages with an ameboid morphology characteristic of cells that were actively phagocytosing expressed OPN mRNA (Fig. 4B, inset). No macrophage was observed in the surgery cortex of sham-operated rats or in the contralateral (nonischemic) cortex of MCAO rats. In addition, the sense probe gave no signal (data not shown). Therefore, macrophage infiltration was observed only because of ischemia.
Fig. 4.
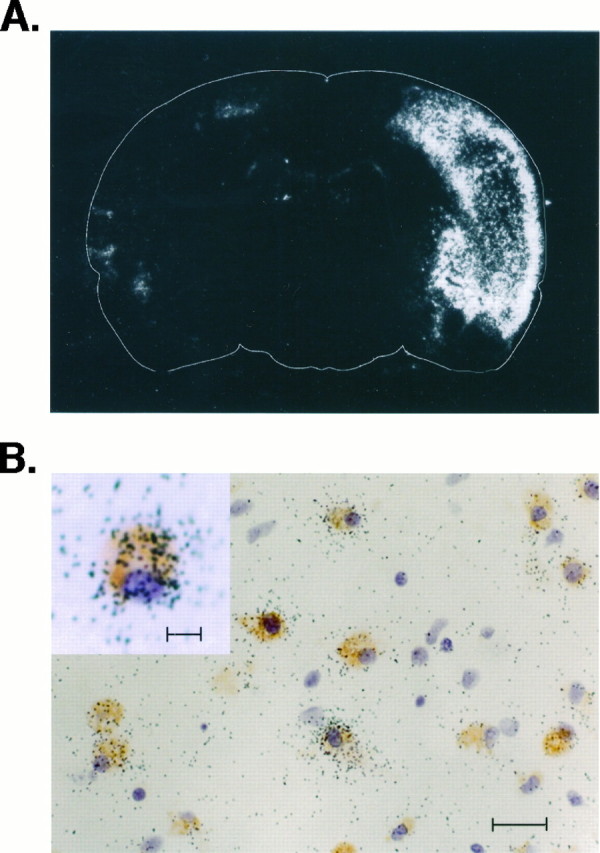
In situ localization of OPN mRNA expression in the ischemic cortex. Shown is film autoradiography of OPN mRNA expression 5 d after permanent MCAO (A). OPN mRNA-expressing cells are located abundantly throughout the entire infarcted region. The ED-1 antibody and OPN mRNA colocalized in many, but not all, of the macrophages in the infarct (B). Morphologically, the ED-1-positive and OPN-positive cells (B,inset) have an ameboid appearance indicative of their activated phagocytic state. Scale bars: in B, 20 μm; in inset, 10 μm.
Immunohistochemical analysis of OPN expression in ischemic cortex
To define further the cellular components and the upregulation of OPN peptide in focal stroke, immunohistochemical techniques were applied by using a mouse anti-rat OPN antibody, MPIIIB10(1), to examine OPN expression in normal (sham) or ischemic brain tissues at 6, 12, and 24 hr and 5, 10, and 15 d after permanent MCAO (n= 3). Selected slides for immunohistochemical staining and hematoxylin and eosin staining are illustrated in Figures5 and 6. Control sections stained with preimmune sera or the exclusion of the primary antibody did not demonstrate detectable staining for OPN (data not shown). In accord with the OPN mRNA expression data, OPN peptide expression was not detected in uninjured or sham-operated brain sections. A very weak positive immunoreactive signal was observed in some cells (GFAP-positive and ED-1-positive) in the ischemic cortex 6–24 hr after permanent MCAO (data not shown). In contrast, there was strong positive immunoreactivity in all of the animals 5 d after MCAO (Fig. 5D at low magnification and Fig. 6C at high magnification), indicating abundant OPN expression in the focal ischemic brains.
Fig. 5.
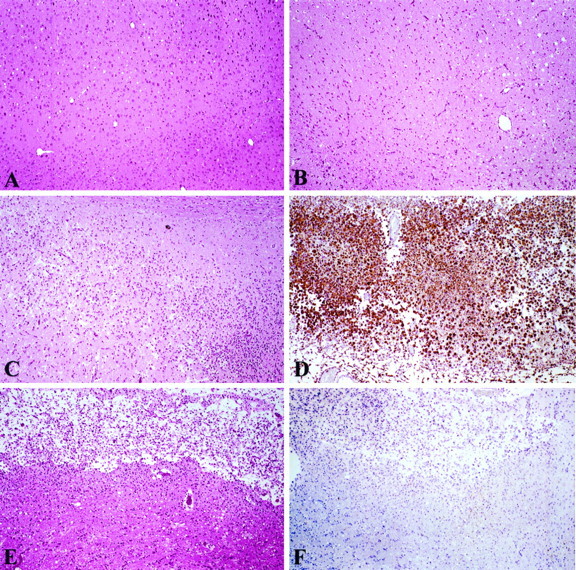
Histological and immunohistochemical staining of OPN expression in ischemic cortex. A, Hematoxylin and eosin-stained (H&E) section of normal cerebral cortex (sham-operated animals at day 5). B, H&E-stained focal ischemic zone at 24 hr after permanent MCAO. C, Focal ischemic zone at 5 d (H&E). D, Immunohistochemistry showing numerous OPN-positive macrophages in this 5 d lesion. E, Focal ischemic cortex at 15 d (H&E). F, Immunohistochemistry for OPN in focal ischemic lesion at 15 d.
Fig. 6.
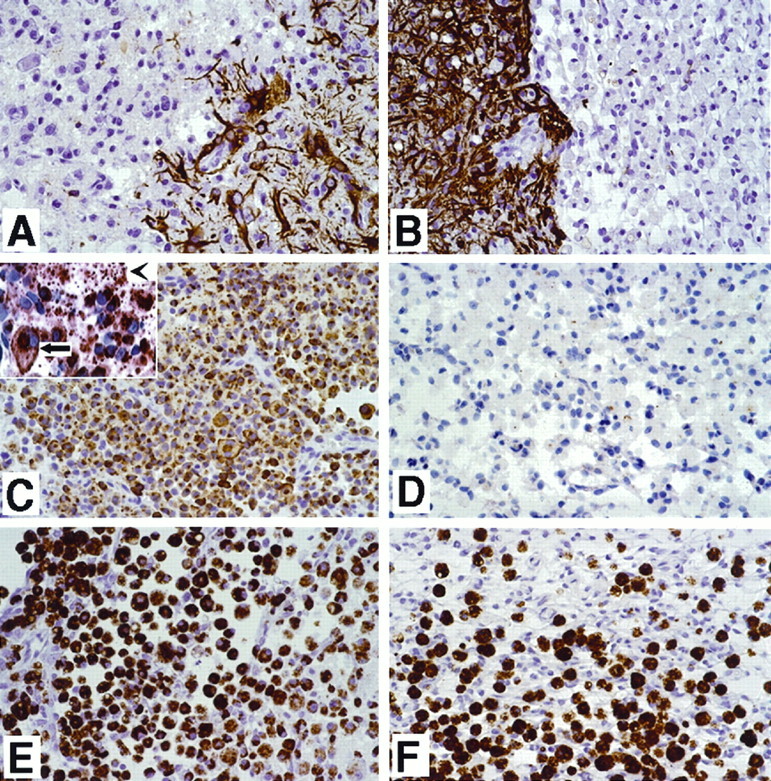
Immunohistochemical detection of temporal OPN expression in ischemic cortex after permanent MCAO. A, Immunostaining for GFAP at the boundary of the ischemic lesion 5 d after permanent MCAO. Note the strong immunostaining of GFAP in astrocytes of the nonischemic lesion. B, Immunostaining for GFAP 15 d after MCAO. The GFAP expression pattern is similar to day 5, but the signal is stronger. C, Immunostaining for OPN 5 d after MCAO. Note the diffuse cellular staining and the large number of positive-staining cells in the middle of the lesion. See the inset for a detailed view of OPN in the cells (arrow) and extracellular matrix (arrowhead). D, Immunostaining for OPN in the ischemic lesion 15 d after MCAO. Most of the cells are negative for OPN. E, Ischemic lesion 5 d after MCAO, filled with a large number of ED-1-positive cells that indicate macrophage/monocyte lineage. F, Ischemic 15 d lesion with ED-1-positive cells. Note the organization of this lesion.
The immunostaining demonstrated OPN expression in numerous cells in the necrotic area. Furthermore, the necrotic zone was demarcated by a dense band of OPN-positive cells at the interface of necrotic and viable tissue. OPN staining (Fig. 6C) was primarily cytoplasmic, with prominent staining around the perinuclear region that suggested localization to the Golgi apparatus. In addition, there was extracellular staining for OPN in the necrotic tissue, which appeared as tiny granules in the extracellular space (Fig. 6C, inset). The OPN-positive staining cells had histological features of macrophages; to confirm this observation, we stained serial sections for ED-1 expression (Fig. 6E), a marker that is specific for cells in the monocytes/macrophage lineage (Dijkstra et al., 1985). In general, similar to our in situ hybridization data, all cells expressing OPN also expressed ED-1, whereas a few ED-1-positive staining cells did not express OPN.
It was interesting to observe that at day 15 after ischemia there was a remarkable decrease in the number of OPN-positive cells in the lesions (Figs. 5F, 6D) despite the fact that ED-1-positive macrophages were still abundant in the granulation tissue (Fig. 6F). At this late time point after injury, only a few OPN-positive cells were noted, which were located primarily in the peripheral region of the lesion, in the perivascular region of the necrotic tissue, and in some smooth muscle and endothelial cells of repairing vessels.
Effects of OPN on astrocyte cell migration
On the basis of the chemotactic feature of OPN for macrophages, smooth muscle cells, and endothelial cells (Singh et al., 1990; Liaw et al., 1994; Giachelli et al., 1995) and the induction of glial cell activation and apparent mobilization in ischemic stroke (Clark et al., 1993), we speculated that elevated expression of OPN after focal stroke might serve for glial cell activation and migration, a critical feature related to gliosis after focal stroke. To explore this potential role of OPN in stroke, we evaluated the effect of OPN on C6 glial cell migration in an in vitro assay. As shown in Figure7A, OPN caused a concentration-dependent stimulation of C6 glial cell migration after a 24 hr incubation. A marked induction was observed at 0.07 μm (4.6-fold induction over control; p < 0.001) and 0.24 μm (8.2-fold increase; p< 0.001). Similar dose-dependent effects of OPN on normal human astrocytes were observed (Fig. 7B), showing 4.4-fold induction at 0.24 and 0.72 μm (p< 0.05) over controls. Approximately 4% C6 cells or 3% human astrocytes migrated in response to 0.24 μm OPN stimulation.
DISCUSSION
The key novel findings of the present study are the de novo induction of OPN mRNA and peptide after focal stroke, its cellular localization, and putative function. OPN is a secreted glycoprotein present in the extracellular matrix. The conserved RGD sequence in OPN was demonstrated for its adhesive activity (Xuan et al., 1995) to cells via the cell surface receptor, αvβ3 integrin (Liaw et al., 1995). Various extracellular matrix proteins such as fibronectin and collagen or plasma proteins such as vitronectin and fibrinogen also have been shown to contain an RGD motif and are associated with specific cellular adhesion and migration (Yamada, 1991). However, little is known about the involvement of the RGD-containing adhesive molecules in focal stroke and/or CNS injuries. Nevertheless, enhanced expression of tenasin, a developmentally regulated extracellular molecule, has been reported in a discrete region of astrocytes (around the lesion) after stab brain injury (Laywell et al., 1992). Very recently, the induced expression of SC1, a brain extracellular matrix glycoprotein related to secreted protein acidic and rich in cysteine (SPARC), also has been demonstrated in the active astrocytes after surgical wound (Mendis et al., 1996a), suggesting a potential role of SC1 in glial scar formation. The SPARC family of extracellular matrix-associated glycoproteins has been shown to have anti-adhesive properties (Sage and Bornstein, 1991; Lane and Sage, 1994; Girard and Springer, 1996), which lack an RGD motif. It is interesting to note that cellular expression of OPN (primarily in macrophages; the present report) is distinct from those of tenascin and SC1 (mainly in astrocytes; Laywell et al., 1992; Mendis et al., 1996a) after brain injury, although all of these molecules may be involved in matrix remodeling after injury. Similarly, differential expression of OPN with tenascin, SC1, or SPARC has been reported during development (Nomura et al., 1988; Laywell et al., 1992; Mendis et al., 1996b).
The biological or pathophysiological significance of OPN expression in cerebral ischemia is not yet clear, but several potential roles can be inferred on the basis of previous reports on OPN expression in peripheral organs in response to injury. First, the induced expression of OPN in macrophages in the ischemic lesion may contribute to the recruitment of more macrophages into the injured tissue, based on its adhesive and chemotactic properties (Giachelli et al., 1995). In particular, OPN has been shown to bind to macrophages in vitro and induce macrophage infiltration in mice (Singh et al., 1990). The temporal expression of OPN in the ischemic cortex is remarkably parallel to the accumulation of macrophages after focal stroke, suggesting that the induced expression of OPN may recruit additional macrophages in the ischemic brain.
Second, OPN-expressing macrophages in the ischemic lesion may play an active role in tissue remodeling after ischemic stroke. OPN-expressing macrophages with the ameboid morphology characteristic of active cell phagocytosis were clustered mainly in the necrotic lesion. The spatial expression of OPN after focal stroke confirms the previous observation concerning myeloperoxidase activity (Barone et al., 1995), suggesting that these macrophages are active and play a role in removing necrotic tissue. Interestingly, strong OPN-expressing macrophages also were localized in the necrotic tissues after myocardial ischemia (Murry et al., 1994) or clustered around tubules of tubulointerstitial nephritis (Giachelli et al., 1994), supporting a common pattern of OPN function in acute tissue injury. Additional parallelism can be drawn from the diminished expression of OPN in macrophages right after the removal of the necrotic tissue in both myocardial ischemia (Murry et al., 1994) and focal brain ischemia (the present report), although a large number of macrophages were still present in the injury site, suggesting that OPN expression marks a functional (phagocytosing) state of the macrophage.
The induced expression of OPN after focal stroke may involve general tissue repair and remodeling processes after ischemia. Time course studies reveal that ischemia-induced expression of OPN parallels not only massive macrophage infiltration but also significant extracellular matrix formation, neovascularization, and gliosis (Clark et al., 1993,1994). Induced expression of αvβ3 integrin recently has been demonstrated in selected microvessels of ischemic lesion after focal cerebral ischemia (Okada et al., 1996), suggesting that the concomitant upregulation of OPN and its receptor may contribute to angiogenesis induced by focal ischemia. In addition, the dose-dependent effects of OPN on astroglial cell migration were demonstrated for the first time, suggesting a role of OPN in glial cell activation/migration, a critical element of gliosis/remodeling in focal stroke. It is interesting to speculate that the change of glial cells from being scattered adjacent to the infarct to a focused glial scar and a walling off of the resolved infarct after 15 d [see Fig. 6A,B for the GFAP-positive astrocyte staining and Clark et al. (1993)] may be associated with the migratory activity of OPN. Further studies must be explored when specific antagonists against αvβ3 integrin or neutralizing antibodies against OPN become available.
Additional functions for ischemia-induced OPN in brain may include the inhibition of nitric oxide production (Rollo et al., 1996). Rollo et al. (1996) demonstrated that OPN could dose-dependently suppress inducible nitric oxide synthase (iNOS) expression in activated RAW264.7 macrophages. The temporal expression of iNOS (induced at 48 hr and returned to basal level at 7 d after ischemia) (Iadecola et al., 1995, 1996) and OPN after focal stroke also may suggest such an inhibitory role.
The factors that induce OPN expression after focal stroke are not yet known. However, time course studies suggest that inflammatory cytokines, including TNF-α and IL-1β that are induced earlier (onset 1–3 hr and peak at 12 hr) (Liu et al., 1993, 1994; Wang et al., 1994b), or factors associated with tissue remodeling processes that are upregulated late after ischemic injury, such as TGF-β1 (upregulated until 2–15 d after ischemic injury) (Wang et al., 1995), may be involved in the regulation of OPN gene expression in focal stroke. The effect of these factors on OPN expression has been demonstrated previously in various cultured cells (Butler, 1989; Patarca et al., 1989; Singh et al., 1990; Denhardt and Guo, 1993; Giachelli et al., 1995; Wang et al., 1996). The present model of focal ischemia (i.e., with infarct restricted to cortex) provides long-term histopathological and neurobehavioral evaluation (i.e., 100% survival beyond 30 d;Barone et al., 1993; Clark et al., 1993) and is ideal for full time course gene expression studies, including OPN, as demonstrated in the present work.
In conclusion, the present report demonstrates the delayed expression of OPN and its robust induction in macrophages after ischemic brain injury. The temporal and spatial distribution of OPN suggests that the induced OPN in the ischemic cortex may play a role in macrophage recruitment, matrix repair, gliosis, and remodeling processes that occur after focal ischemia, although direct evidence for these potential functions still needs to be explored further.
Footnotes
We extend our appreciation to Ray White, Juan-Li Gu, and Christine Webb for excellent technical assistance.
Correspondence should be addressed to Dr. Xinkang Wang, Department of Cardiovascular Pharmacology, SmithKline Beecham Pharmaceuticals, 709 Swedeland Road, P.O. Box 1539, UW 2511, King of Prussia, PA 19406.
REFERENCES
- 1.Barone FC, Hillegass LM, Price WJ, White RF, Feuerstein GZ, Sarau HM, Clark RK, Griswold DE. Polymorphonuclear leukocyte infiltration into cerebral focal ischemic tissue: myeloperoxidase activity assay and histologic verification. J Neurosci Res. 1991;29:336–348. doi: 10.1002/jnr.490290309. [DOI] [PubMed] [Google Scholar]
- 2.Barone FC, Price WJ, White RF, Willette RN, Feuerstein GZ. Genetic hypertension and increased susceptibility to cerebral ischemia. Neurosci Biobehav Rev. 1992a;16:219–233. doi: 10.1016/s0149-7634(05)80182-4. [DOI] [PubMed] [Google Scholar]
- 3.Barone FC, Schmidt DB, Hillegass LM, Price WJ, White RF, Feuerstein GZ, Clark RK, Griswold DE, Sarau HM. Reperfusion increases neutrophil and LTB4 receptor binding in focal ischemia. Stroke. 1992b;23:1337–1348. doi: 10.1161/01.str.23.9.1337. [DOI] [PubMed] [Google Scholar]
- 4.Barone FC, Clark RK, Price WJ, White RF, Storer BL, Feuerstein GZ, Ohlstein EH. Neuron specific enolase increases in cerebral and systemic circulation following focal ischemia. Brain Res. 1993;623:77–82. doi: 10.1016/0006-8993(93)90012-c. [DOI] [PubMed] [Google Scholar]
- 5.Barone FC, Hillegass LM, Tzimas MN, Schmidt DB, Foley JJ, White RF, Price WJ, Feuerstein GZ, Clark RK, Griswold DE, Sarau HM. Time-related changes in myeloperoxidase activity and leukotriene B4 receptor binding reflect leukocyte influx in cerebral focal stroke. Mol Chem Neuropathol. 1995;24:13–30. doi: 10.1007/BF03160109. [DOI] [PubMed] [Google Scholar]
- 6.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor α: a mediator of focal ischemic brain injury. Stroke. 1997a;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 7.Barone FC, Feuerstein GZ, White RF. Reduced brain temperature during transient focal ischemia provides complete neuroprotection. Neurosci Biobehav Rev. 1997b;21:31–44. doi: 10.1016/0149-7634(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 8.Brint S, Jacewicz M, Kiessling M, Tanable J, Pulsinelli W. Focal brain ischemia in the rat: methods for reproducible neocortical infarction using tandem occlusion of the distal middle cerebral artery and ipsilateral common carotid arteries. J Cereb Blood Flow Metab. 1988;8:474–485. doi: 10.1038/jcbfm.1988.88. [DOI] [PubMed] [Google Scholar]
- 9.Buchan AM, Xue D, Slivka A. A new model of temporary focal neocortical ischemia in the rat. Stroke. 1992;23:273–279. doi: 10.1161/01.str.23.2.273. [DOI] [PubMed] [Google Scholar]
- 10.Butler WT. The nature and significance of osteopontin. Connect Tissue Res. 1989;23:123–136. doi: 10.3109/03008208909002412. [DOI] [PubMed] [Google Scholar]
- 11.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinum thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 12.Clark RK, Lee EV, Fish CJ, White RF, Price WJ, Jonak ZL, Feuerstein GZ, Barone FC. Progression of cerebral changes following middle cerebral artery occlusion in the rat: a quantitative planimetric, histologic, and immunohistochemical study. Brain Res Bull. 1993;31:565–572. doi: 10.1016/0361-9230(93)90124-t. [DOI] [PubMed] [Google Scholar]
- 13.Clark RK, Lee EV, White RF, Jonak ZL, Feuerstein GZ, Barone FC. Reperfusion following focal stroke hastens inflammation and resolution of ischemic injured tissue. Brain Res Bull. 1994;35:387–391. doi: 10.1016/0361-9230(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 14.Denhardt DT, Guo X. Osteopontin: a protein with diverse functions. FASEB J. 1993;7:1475–1482. [PubMed] [Google Scholar]
- 15.Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in rat recognized by monoclonal antibodies ED1, ED2, and ED3. Adv Exp Med Biol. 1985;186:409–419. doi: 10.1007/978-1-4613-2463-8_50. [DOI] [PubMed] [Google Scholar]
- 16.Duverger D, MacKenzie T. The quantification of cerebral infarction following focal ischemia in the rat: influence of strain, arterial pressure, blood glucose concentration, age. J Cereb Blood Flow Metab. 1988;8:449–461. doi: 10.1038/jcbfm.1988.86. [DOI] [PubMed] [Google Scholar]
- 17.Ellison JA, Scully SA, de Vellis J. Evidence for neuronal regulation of oligodendrocyte development: cellular localization of platelet-derived growth factor α receptor and A-chain mRNA during rat cerebral cortex development. J Neurosci Res. 1996;45:28–39. doi: 10.1002/(SICI)1097-4547(19960701)45:1<28::AID-JNR3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Feuerstein GZ, Wang XK, Barone FC. Inflammatory mediator of ischemic injury: cytokine gene regulation in stroke. In: Ginsberg MD, Bogousslavsky J, editors. Cerebrovascular disease. Blackwell Scientific; Cambridge, MA: 1998. pp. 507–531. [Google Scholar]
- 19.Garcia JH, Liu KF, Yoshida Y, Lian J, Chen S, del Zoppo GJ. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am J Pathol. 1994;144:188–199. [PMC free article] [PubMed] [Google Scholar]
- 20.Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92:1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giachelli CM, Pichler R, Lombardi D, Denhardt DT, Alpers CE, Schwartz SM, Johnson RJ. Osteopontin expression in angiotensin II-induced tubulointerstitial nephritis. Kidney Int. 1994;45:515–524. doi: 10.1038/ki.1994.67. [DOI] [PubMed] [Google Scholar]
- 22.Giachelli CM, Schwartz SM, Liaw L. Molecular and cellular biology of osteopontin: potential role in cardiovascular disease. Trends Cardiovasc Med. 1995;5:88–95. doi: 10.1016/1050-1738(95)00005-T. [DOI] [PubMed] [Google Scholar]
- 23.Ginsberg DM, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- 24.Girard JP, Springer TA. Modulation of endothelial cell adhesion by hevin, an acidic protein associated with high endothelial venules. J Biol Chem. 1996;271:4511–4517. doi: 10.1074/jbc.271.8.4511. [DOI] [PubMed] [Google Scholar]
- 25.Hallenbeck JM, Dutka AJ, Tanishima T, Kochanek PM, Kumaroo KK, Thompson CB, Obrenovitch TP, Contreras TJ. Polymorphonuclear leukocyte accumulation in brain region with low blood flow during the early postischemic period. Stroke. 1986;17:246–253. doi: 10.1161/01.str.17.2.246. [DOI] [PubMed] [Google Scholar]
- 26.Hidaka YT, Eda T, Yonemoto M, Kamei T. Inhibition of cultured vascular smooth muscle cell migration by simvastatin (MK-7333). Atherosclerosis. 1992;95:87–94. doi: 10.1016/0021-9150(92)90179-k. [DOI] [PubMed] [Google Scholar]
- 27.Iadecola C, Zhang F, Xu S, Casey R, Ross ME. Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab. 1995;15:378–384. doi: 10.1038/jcbfm.1995.47. [DOI] [PubMed] [Google Scholar]
- 28.Iadecola C, Zhang F, Xu S, Casey R, Clark HB, Ross ME. Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke. 1996;27:1373–1380. doi: 10.1161/01.str.27.8.1373. [DOI] [PubMed] [Google Scholar]
- 29.Lampe MA, Patarca R, Ireguli MV, Cantor H. Polyclonal B cell activation by the Eta-1 cytokine and the development of systemic autoimmune disease. J Immunol. 1991;147:2902–2906. [PubMed] [Google Scholar]
- 30.Lane TF, Sage EH. The biology of SPARC, a protein that modulates cell–matrix interactions. FASEB J. 1994;8:163–173. [PubMed] [Google Scholar]
- 31.Laywell ED, Dorries U, Bartsch U, Faissner A, Schachner M, Steindler DA. Enhanced expression of the developmentally regulated extracellular matrix molecule tenascin following adult brain injury. Proc Natl Acad Sci USA. 1992;89:2634–2638. doi: 10.1073/pnas.89.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liaw L, Almeida M, Hart CH, Schwartz SM, Giachelli CM. Osteopontin promotes vascular cell adhesion and spreading and is chemotactic for smooth muscle cells in vitro. Circ Res. 1994;74:214–224. doi: 10.1161/01.res.74.2.214. [DOI] [PubMed] [Google Scholar]
- 33.Liaw L, Skinner MP, Raines EW, Ross R, Cheresh DA, Schwartz SM, Giachelli CM. The adhesive and migratory effects of osteopontin are mediated via distinct cell surface integrins: role of αvβ3 in smooth muscle cell migration to osteopontin in vitro. J Clin Invest. 1995;95:713–724. doi: 10.1172/JCI117718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T, McDonnell PC, Young PR, White RF, Siren AL, Hallenbeck JM, Barone FC, Feuerstein GZ. Interleukin-1β mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1751. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- 35.Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor α expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- 36.Mendis DB, Ivy GO, Brown IR. SC1, a brain extracellular matrix glycoprotein related to SPARC and follistin, is expressed by rat cerebellar astrocytes following injury and during development. Brain Res. 1996a;730:95–106. doi: 10.1016/0006-8993(96)00440-4. [DOI] [PubMed] [Google Scholar]
- 37.Mendis DB, Shahin S, Gurd JW, Brown IR. SC1, a SPARC-related glycoprotein, exhibits features of an ECM component in the developing and adult brain. Brain Res. 1996b;713:53–63. doi: 10.1016/0006-8993(95)01472-1. [DOI] [PubMed] [Google Scholar]
- 38.Murry CE, Giachelli CM, Schwartz SM, Vracko R. Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol. 1994;145:1450–1462. [PMC free article] [PubMed] [Google Scholar]
- 39.Nomura S, Wills AJ, Edwards DR, Heath JK, Hogan BLM. Developmental expression of 2ar (osteopontin) and SPARC (osteonectin) RNA as revealed by in situ hybridization. J Cell Biol. 1988;106:441–450. doi: 10.1083/jcb.106.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Brien ER, Garvin MR, Stewart DK, Hinohara T, Simpson JB, Schwartz SM, Giachelli CM. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler Thromb. 1994;14:1648–1656. doi: 10.1161/01.atv.14.10.1648. [DOI] [PubMed] [Google Scholar]
- 41.Okada Y, Copeland BR, Hamann GF, Koziol JA, Cheresh DA, del Zoppo GJ. Integrin avb3 is expressed in selected microvessels after focal cerebral ischemia. Am J Pathol. 1996;149:37–44. [PMC free article] [PubMed] [Google Scholar]
- 42.Patarca R, Freeman GJ, Singh P, Wei FY, Durfee T, Blattner F, Regnier DC, Kozak CA, Mock BA, Morse HC, III, Jerrells TR, Cantor DC. Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. J Exp Med. 1989;170:145–161. doi: 10.1084/jem.170.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patarca R, Wei FY, Singh P, Morasso MI, Cantor H. Dysregulated expression of the T cell cytokine Eta-1 in CD4−8− lymphocytes during the development of murine autoimmune disease. J Exp Med. 1990;172:1177–1183. doi: 10.1084/jem.172.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pichler R, Giachelli CM, Lombardi D, Pippin J, Gordon K, Aplers CE, Schwartz SM, Johnson RJ. Tubulointerstitial disease in glomerulonephritis: potential role of osteopontin (uropontin). Am J Pathol. 1994;144:915–926. [PMC free article] [PubMed] [Google Scholar]
- 45.Rollo EE, Laskin DL, Denhardt DT. Osteopontin inhibits nitric oxide production and cytotoxicity by activated RAW264.7 macrophages. J Leukoc Biol. 1996;60:397–404. doi: 10.1002/jlb.60.3.397. [DOI] [PubMed] [Google Scholar]
- 46.Sage EH, Bornstein P. Extracellular proteins that modulate cell–matrix interactions: SPARC, tenascin, and thrombospondin. J Biol Chem. 1991;266:14831–14834. [PubMed] [Google Scholar]
- 47.Singh RP, Patarca R, Schwartz J, Singh P, Cantor H. Definition of a specific interaction between the early T lymphocyte activation 1 (Eta-1) protein and murine macrophages in vitro and its effect upon macrophages in vivo. J Exp Med. 1990;171:1931–1942. doi: 10.1084/jem.171.6.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang XK, Siren A-L, Liu Y, Yue T-L, Barone FC, Feuerstein GZ. Upregulation of intercellular adhesion molecule 1 (ICAM-1) on brain microvascular endothelial cells in rat ischemic cortex. Mol Brain Res. 1994a;26:61–68. doi: 10.1016/0169-328x(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 49.Wang XK, Yue T-L, Barone FR, White RF, Feuerstein GZ. Concomitant cortical expression of TNF-α and IL-1β mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994b;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- 50.Wang XK, Yue TL, White RF, Barone FC, Feuerstein GZ. Transforming growth factor-β1 exhibits delayed gene expression following focal cerebral ischemia. Brain Res Bull. 1995;36:607–609. doi: 10.1016/0361-9230(94)00243-t. [DOI] [PubMed] [Google Scholar]
- 51.Wang XK, Louden C, Ohlstein EH, Stadel JM, Gu JL, Yue TL. Osteopontin expression in platelet-derived growth factor-stimulated vascular smooth muscle cells and carotid artery after balloon angioplasty. Arterioscler Thromb Vasc Biol. 1996;16:1365–1372. doi: 10.1161/01.atv.16.11.1365. [DOI] [PubMed] [Google Scholar]
- 52.Wang XK, Barone FC, Aiyar NV, Feuerstein GZ. Increased interleukin-1 receptor and interleukin-1 receptor antagonist gene expression after focal stroke. Stroke. 1997;28:155–162. doi: 10.1161/01.str.28.1.155. [DOI] [PubMed] [Google Scholar]
- 53.Xuan JW, Hota C, Shigeyama Y, D’Errico JA, Somerman MJ, Chambers AF. Site-directed mutagenesis of the arginine-glycine-aspartic acid sequence in osteopontin destroys cell adhesion and migration functions. J Cell Biochem. 1995;57:680–690. doi: 10.1002/jcb.240570413. [DOI] [PubMed] [Google Scholar]
- 54.Yamada KM. Adhesive recognition sequences. J Biol Chem. 1991;266:12809–12812. [PubMed] [Google Scholar]
- 55.Yue TL, McKenna PJ, Ohlstein EH, Farach-Carson MC, Butler WT, Johanson K, McDevitt P, Feuerstein GZ, Stadel JM. Osteopontin-stimulated vascular smooth muscle cell migration is mediated by β3 integrin. Exp Cell Res. 1994;214:459–464. doi: 10.1006/excr.1994.1282. [DOI] [PubMed] [Google Scholar]