

Abstract
Protein l-isoaspartyl methyltransferase (PIMT) is suggested to play a role in the repair of aged protein spontaneously incorporated with isoaspartyl residues. We generated PIMT-deficient mice by targeted disruption of the PIMT gene to elucidate the biological role of the gene in vivo. PIMT-deficient mice died from progressive epileptic seizures with grand mal and myoclonus between 4 and 12 weeks of age. An anticonvulsive drug, dipropylacetic acid (DPA), improved their survival but failed to cure the fatal outcome. l-Isoaspartatate, the putative substrate for PIMT, was increased ninefold in the brains of PIMT-deficient mice. The brains of PIMT-deficient mice started to enlarge after 4 weeks of age when the apical dendrites of pyramidal neurons in cerebral cortices showed aberrant arborizations with disorganized microtubules. We conclude that methylation of modified proteins with isoaspartyl residues is essential for the maintenance of a mature CNS and that a deficiency in PIMT results in fatal progressive epilepsy in mice.
Keywords: protein l-isoaspartyl methyltransferase, gene targeting, isoaspartate, epilepsy, disorganized microtubules, aberrant arborizations
Asparaginyl and aspartyl residues are the most labile amino acids subject to spontaneous degradation reactions that modify aging proteins. This is attributable, in part, to their susceptibility to the intramolecular attack of the side chain carbonyl carbon by the adjacent backbone nitrogen, resulting in the formation of a succinimide intermediate (Geiger and Clarke, 1987). This intermediate ring can hydrolyze at either of its two carbonyl groups, yielding both isoaspartyl (isomerization) and normal aspartyl residues (McFadden and Clarke, 1987). The methyltransferase activity has been widely detected in various organisms, which transfer the methyl residue to the side chain carboxyl group of l-isoaspartyl andd-aspartyl residues but not to normall-aspartyl residues (McFadden and Clarke, 1982; Aswad, 1984; Clarke, 1985; Ingrosso et al., 1989), and has been designated protein l-isoaspartyl methyltransferase (PIMT) (Clarke, 1985). Experiments performed in vitro have demonstrated that the incubation of synthetic l-isoaspartyl-containing peptides with PIMT results in the conversion of at least 50% of the peptide to the normal l-aspartyl-containing form (Johnson et al., 1987; McFadden and Clarke, 1987). This finding led to the hypothesis that the function of PIMT in vivo is to minimize the accumulation of potential l-isoaspartyl residues in long-lived proteins as proteins age. However, this hypothesis is only based on data obtained by in vitro aging experiments, whereas the in vivo function of PIMT is still unknown. To clarify the biological role of PIMT, we disrupted the PIMTgene by gene targeting and found that protein methylation is essential for the maintenance of the CNS and that a deficiency of PIMT results in fatal progressive epilepsy in mice.
MATERIALS AND METHODS
Construction of a targeting vector and production of gene-disrupted mice. The C57BL/6 mouse genomic library in λDASH II was screened with human PIMT cDNA (Shirasawa et al., 1995) as a probe. Out of 1 × 106 phages screened, a recombinant phage containing genomic DNA of PIMTlocus was isolated. The targeting vector pPNT-PIMT-neo was constructed by inserting a 0.79 kb PCR-amplified 3′-homologous fragment (short arm) into aXhoI/NotI-restricted pPNT vector (Tybulewicz et al., 1991). The short arm was amplified from a genomic clone by PCR with a XhoI-anchored sense primer (5′-ATCTCGAGTGCTGCACAAACAGGGCACATG-3′) and aNotI-anchored complementary primer (5′-AAGCGGCCGCTCACATATCTTTCCACCTGC-3′). The 3′-NsiI site of the 10.4 kb EcoRI–NsiI-restricted fragment was converted to the BamHI site by ligating adapters (5′-AGCTT-3′ and 5′-GATCAAGCTTGGAT-3′) and subcloned into theEcoRI/BamHI-restricted targeting vector.
The ES cell line E14 (Hooper et al., 1987) was cultured, transfected, and screened as described previously (Kitamura et al., 1991). After transfection of the NotI-linearized pPNT-PIMT-neor construct, 250 colonies resistant to both G418 and GANC were screened for the homologous recombination event by PCR with a primer located downstream of the short arm (5′-GGCACACAGAGACACCAAG-3′) and a primer complimentary to the PGK-neor gene sequence (5′-ACTTGTGTACGCCAAGTGC-3′) at an annealing temperature of 65°C. The precise homologous recombination of PCR-positive clones was confirmed by Southern blot analysis. Genomic DNA from a PCR-positive clone was digested with BamHI andXhoI and hybridized with a 1.2 kb SacI fragment as a 5′-probe or digested with BamHI and EcoRI and hybridized with a 0.73 kb BspHI/SspI fragment as a 3′-probe (Fig.1A). One ES clone designated ES84 contained the expected PIMT-targeted allele. Hybridization with the neor probe verified the presence of a single copy ofneor gene for ES84. ES84 was then injected into the blastocysts of C57BL/6 mice as described previously (Bradly, 1987). Resulting male chimeras were mated to C57BL/6 females, and their agouti offspring were screened by PCR and Southern blot analysis to detect the heterozygous mutation in the genome using DNA prepared from their tails. ES84 was able to transmit thePIMT-targeted allele in the germline. The heterozygous mice were intercrossed to produce homozygous offspring.
Fig. 1.

Targeted disruption of the PIMT gene.A, top, Genomic organization of thePIMT gene containing exons I–VII (closed boxes). Middle, The targeted vector pPNT in which the 10 kb EcoRI/NsiI and the 0.8 kbHpaI/NsiI fragments of thePIMT genomic clone were inserted into theEcoRI/XbaI andXhoI/NotI sites, respectively.Arrows indicate the direction of transcription. Theshaded boxes of tK and neogenes represent the PGK-1 promoter. Bottom, The predicted structure of the PIMT locus after a targeting event. Exon II, intron II, and exon III are replaced with theneo gene. The fragments used for Southern blot and PCR amplification are E, EcoRI;B, BamHI; S,SacI; Xb, XbaI;Ns, NsiI; Hp,HpaI; Xh, XhoI; andN, NotI. B,C, Southern blot analysis of genomic DNA extracted from mouse tails. DNA was digested with BamHI andXhoI and hybridized with the 5′-probe (B). DNA was digested with BamHI and EcoRI and hybridized with the 3′-probe (C). The sizes of wild-type (3.4 kb) and disrupted (11.4 kb) alleles are shown (C). The genotypes of mice are presented above the lanes.D, RNase protection analysis of PIMT+/+,PIMT+/−, and PIMT−/− mouse brain and testis. Ten micrograms of total RNA prepared from the indicated mouse genotypes were subjected to RNase protection analysis probed by a35S-labeled cRNA probe. Protected wild-type (365 bp) and targeted (256 bp) transcripts were separated by a denaturing PAGE gel.E, Immunoblot analysis of PIMT+/+,PIMT+/−, and PIMT−/− mouse brain and testis. Upper, Lysates of homogenized tissues derived from the indicated mouse genotypes were subjected to SDS-PAGE and blotted to a nitrocellulose membrane. The blot was probed with anti-PIMT antibody. Lower, The bands (26 kDa) corresponding to PIMT were measured by densitometry, and their intensities were presented in a graph.
RNase protection analysis and immunoblot analysis. Total RNA was prepared from brains or testes from PIMT−/− and control mice by the guanidine-CsCl method as described (Chirgwin et al., 1979). Ten micrograms of total RNA were hybridized with32P-labeled cRNA antisense probe at 50°C overnight and were digested with 40 μg/ml of RNase A and 2 μg/ml of RNase T1 at 37°C for 30 min using the PRAII kit (Ambion, Austin, TX). Protected fragments were resolved on denaturing PAGE gels.32P-labeled cRNA probe was prepared from the pGEM7Z vector (Promega, Madison, WI) carrying the 365 bp mouse PIMT coding sequence [nucleotides (nts) 203–567; D38023] by T7 RNA polymerase according to the protocol of the manufacturer (Promega).
For Western blot analysis, brain tissues were homogenized with Tris-saline (TS) buffer (50 mm Tris-HCl, pH 7.6, and 150 mm NaCl), treated with SDS sample buffer, and resolved on 15–22% gradient SDS-acrylamide gels. Proteins were electrophoresed to a polyvinylidene fluoride membrane (Millipore, Bedford, MA). The blot was blocked in TS buffer with 3% gelatin, incubated with anti-PIMT antiserum diluted 1:500 in TS buffer, washed briefly in TS buffer, and incubated with biotinylated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:500 in TS buffer, followed by the avidin–biotin complex. Immunoreactions were visualized by color development using 4-chloro-1-naphthol, and optical densities were measured to determine the relative amount of PIMT in the immunoblot.
Histopathological examinations, immunohistochemical analysis, and electron microscopy. For routine morphological evaluations, tissues were dissected, weighed, and fixed in 4% paraformaldehyde in PBS, pH 7.0. Paraffin sections were prepared by standard procedures and stained with hematoxylin and eosin (H&E) or Nissl stain. For immunohistochemical staining of neurofilament (NF), the animals were perfused with ice-cold acid and alcohol solution (95% ethanol and 5% acetic acid), and extracted brain tissues were fixed in the same solution. Eight micrometer paraffin sections were incubated with anti-NF antibody (1:500 dilution) (Fukuda et al., 1997) at 4°C overnight. After being washed with PBS, sections were incubated with peroxidase-conjugated goat anti-rabbit IgG Fab fragment (1:100 dilution; Medical and Biological Laboratories, Nagoya, Japan). Immunoreactivity was developed in 50 mm Tris, pH 7.4, containing 0.01% DAB and 0.01% hydrogen peroxide. For immunohistochemical staining of MAP-2, tissues were fixed in a 4% paraformaldehyde and 30% sucrose mixture in PBS, pH 7.0, mounted in OCT compound (Miles, Elkhart, IN) and were snap frozen in dry ice. Ten micrometer cryostat sections were incubated with anti-MAP-2 antibody (1:200 dilution; Amersham, Arlington Heights, IL). After washing with PBS, sections were incubated with FITC-conjugated anti-mouse immunoglobulins (1:500 dilution; Amersham). Labeled sections were sealed in mounting medium containing antifader (IMMUNON). Immunoreactivity was visualized by confocal laser microscopy (MRC1000; Bio-Rad, Hercules, CA). For electron microscopic analysis, mice were perfused with a 2.5% glutaraldehyde and 1% paraformaldehyde mixture in 0.1 m phosphate buffer, pH 7.0. Extracted tissues were post-fixed with 0.1% OsO4 in 0.1 m phosphate buffer for 1 hr, stained en bloc in 1% uranyl acetate, and embedded in Epon 812 after dehydration with ethanol. Serial sections of 0.5 or 1 μm thickness from cerebral cortices from PIMT−/− or control mice were cut by a Reichert microtome, placed on a hormbar membrane of single hole mesh (Synaptec), stained conventionally, and observed with a JEOL 4000EX intermediate voltage electron microscope at 350 kV.
Methyltransferase activity and determination of isoaspartate andd-amino acids. PIMT activity was measured essentially as described (Lowenson and Clarke, 1991) with ovalbumin as a substrate. Tissues extracted from PIMT−/− and control mice were homogenized in 50 mm phosphate buffer, pH 7.0, containing 2 mm EDTA and 15 mm β-mercaptoethanol, and the protein concentration was determined by BCA assay. In assays, 100 μg of ovalbumin was incubated in 30 μl of 0.2 msodium–citrate buffer, pH 6.0, with 10 μmS-adenosyl-l-[14C]methionine (2.15 GBq/mmol; CFA.360; Amersham) and in 20 μg of tissue homogenate at 37°C for 30 min. Each reaction was quenched with 50 μl of 0.2m NaOH in 1% SDS, and immediately a 60 μl aliquot was spotted on a piece of filter paper. The paper was wedged into the neck of a 10 ml scintillation vial containing ACSII (Amersham), which was then capped and allowed to equilibrate at room temperature for 3 hr. Specific activity was presented as picomoles of 14C per minute per milligram of protein. Data were presented as an average ± SD of six experiments.
Isoaspartic acid residues were quantified using the ISOQUANT protein deamidation kit (Promega) with isoaspartyl-DSIP as a standard substrate as described (Paranandi et al., 1994). Brain homogenates were prepared as described above. Isoaspartic acid residues was presented as pmol/μg of homogenate. Data were presented as an average ± SD of triplicate experiments.
d-Amino acids were quantified essentially as described (Hayashi and Sasagawa, 1993). Brain tissues from PIMT−/−and control mice were hydrolyzed by adding 300 μl of 6N HCl and incubating at 110°C for 6 hr. Samples were then dried, dissolved in 500 μl of DDW, and derivatized with (+)-1-(9-fluorenyl)-ethylcholoroformate (Fluka, Neu-Ulm, Germany). Derivatized samples were analyzed by HPLC (Hitachi, 655A12). The amounts of d-amino acids were presented as the ratio of the amount of d-amino acid to the combined amount ofd- and l-amino acid.
Video monitor of epileptic attack. Behavior and epileptic attacks of PIMT−/− mice were monitored and recorded for 24 hr by home video apparatus. The type and frequency of epileptic seizures were visually diagnosed using the recorded video. Dipropylacetic acid (DPA) sodium was orally administered by dissolving 3 mg of DPA in 3 ml of drinking water.
Electroencephalograph. When animals were under anesthesia with Ketamin (20 mg/ml) and Xylazine (9 mg/ml), four epidural silver ball electrodes, 0.1 mm in diameter, were implanted in the left frontal, right frontal, left occipital, and right occipital epidural space via holes in the skull by the implantation method described previously (Nakano et al., 1993, 1994). Five hundred microliters of 5% glucose in physiological saline were intraperitoneally administered to aid rapid postoperative recovery from anesthesia, and all examined mice survived the operation. A stainless electrode was placed subcutaneously near the nose as a reference for monopolar recordings. A cassette connector was placed on the back with the outlet leads connected to the preamplifier of recording apparatus as described previously (Nakano et al., 1994). Electroencephalographs (EEGs) including four monopolar recordings and four bipolar recordings were recorded everyday for 2 hr between 10 A.M. and 5 P.M. from the second day after the operation. All epileptic spikes successfully recorded were spontaneous cortical discharges without any evocation.
RESULTS
Generation of PIMT knock-out mice
A 15 kb PIMT genomic clone obtained from a mouse C57BL/6 genomic library was used to construct a targeting vector (pPNT-PIMT) for homologous recombination by positive–negative selection (Mansour et al., 1988), as shown in Figure1A. Exons II and III of the PIMT gene (nt 75–311; GenBank/EMBL accession number D38023) were replaced by the PGK-neor cassette in inverse orientation, which disrupted the gene by deleting a segment encompassing from Lys19 to Met64 of thePIMT transcript. The disrupted gene failed to be productively spliced from exons I to IV in frame (Romanik et al., 1992). NotI-linearized pPNT-PIMT was electroporated into E14 ES cells, and neomycin-resistant clones were selected using G418. After selection, 250 ES clones were isolated, of which one clone carried the expected targeted recombination event as determined using the 3′-probe outside of the targeted construct (Fig.1A).
Targeted ES cells were injected into C57BL/6 blastocysts, and three chimeric animals were generated. One male transmitted the targeted allele to germline cells. Heterozygous F1 animals were interbred to generate homozygous PIMT-deficient mice. The genotype was determined using PCR (Fig. 1A) and confirmed by Southern blot analysis (Fig. 1B,C). The 5′-probe detected a 7.4 kbBamHI fragment of the wild-type allele and a 8.9 kbBamHI/XhoI fragment of the knock-out allele in the BamHI/XhoI double-digested genome (Fig.1B). The 3′-probe detected a 3.4 kbBamHI/EcoRI fragment in a wild-type allele and a 11.4 kb in a knock-out allele (Fig. 1C). These results are consistent with the predicted replacement by the PGK-neor cassette (Fig.1A). In addition, RNase protection analysis of brain and testis RNA probing the 365 bp fragment (nt 203–567) of mousePIMT (exons II-VI) showed the 365 bp fully protected transcript in RNAs prepared from brains and testes ofPIMT+/+ and PIMT+/− mice, whereas the 256 bp partially protected transcript (nt 312–567) was found inPIMT+/− and PIMT−/− mice (Fig.1D). Finally, immunoblot analysis of brain and testis lysates using anti-PIMT antibody (Shirasawa et al., 1995) showed the strong 26 kDa band in PIMT+/+ mice and a less intense band in PIMT+/− mice, but no corresponding band was detected in the lysates from PIMT−/− mice (Fig. 1E,upper panel). The intensity of immunoreactivity in PIMT+/− mice was approximately half of the intensity detected in PIMT+/+ mice (Fig. 1E,lower panel). The generation ofPIMT−/− animals followed Mendelian segregation, indicating that the homozygous genotype was not embryonically lethal.
The l-isoaspartate but not the d-aspartate residue is increased in the brain of PIMT-deficient mice
To confirm the deficiency of enzymatic activity inPIMT−/− mice, we assayed the lysates prepared from brain, kidney, testis, and eye of PIMT+/+, PIMT+/−, andPIMT−/− mice for methyltransferase activity by assessing the incorporation with [14C]methyl residues ofS-adenosyl-l-methionine (SAM) into ovalbumin (Fig. 2A). The lysate prepared from PIMT+/+ mice showed a specific activity of 14.6 ± 2.5 (pmol/min/mg of protein, average ± SD;n = 3) in testis, 12.2 ± 0.99 in the brain (n = 3), 3.60 ± 0.58 in the kidney (n = 3), and 1.46 ± 0.16 in the eye (n = 3; Fig. 2A). The lysate fromPIMT−/− mice showed no enzymatic activities in the testis, brain, and eye but reduced activity in the kidney. The results suggested that major enzymatic activities for methyltransferase were completely deficient in examined tissues from PIMT−/−mice, except in the kidney where other minor methyltransferase activity may exist. The results also showed that PIMT+/− mice exhibited approximately half of enzymatic activity ofPIMT+/+ mice (Fig. 2A), which was consistent with the expression of PIMT in immunoblot analysis (Fig.1E).
Fig. 2.

PIMT specific activity was deficient inPIMT−/− mice, and the amount of l-isoaspartyl residues but not that of d-aspartyl residues was increased in the brains of PIMT−/− mice. A, Lysates prepared from brains, kidneys, testes, and eyes of the indicated mouse genotypes were assayed for PIMT-specific activity. Eachbar represents the mean ± SD for three 5-week-old mice. B, l-Isoaspartyl residues were measured in the lysates prepared from brains of the indicated genotypes by the incorporation of [3H]methyl residues by recombinant PIMT. Each bar represents the mean ± SD for three 5-week-old mice. The significance of differences (*p < 0.001) was determined by a Student’st test. C, The ratio ofd-amino acids to l-amino acids was measured in the brain (gray matter) of the indicated genotypes. No significant differences were found between PIMT+/− andPIMT−/− mice in the amount of d-amino acid divided by the total amount of d-amino acid plusl-amino acid (D/D + L ratios) for total d-amino acids, d-aspartate, d-methionine,d-tyrosine, d-phenylalanine,d-histidine, and d-arginine.
We next investigated whether the substrate molecules were accumulated in PIMT-deficient mice. In previous studies in vitro, PIMT reacted in a stoichiometric manner to l-isoaspartate residues in a synthetic peptide but in a substoichiometric manner tod-aspartate residues (O’Connor et al., 1984). However, still undefined is whether d-aspartate andl-isoaspartate residues are bona fide substrates for PIMTin vivo. Therefore, we investigated the accumulation of substrates with l-isoaspartate and d-aspartate residues in the brain tissues from PIMT−/− mice. The brain lysates prepared from PIMT+/+, PIMT+/−, orPIMT−/− mice were assayed for the capacity of methyl incorporation by recombinant PIMT (Fig. 2B). The results showed that the amount of isoaspartyl residues was increased ninefold in PIMT−/− mice (0.703 ± 0.145 pmol/mg of lysate, average ± SD; n = 3) compared withPIMT+/+ mice (0.075 ± 0.018; n = 3) orPIMT+/− mice (0.079 ± 0.002; n = 3). On the other hand, the ratio of d-aspartate tod-aspartate plus l-aspartate was not increased in the brain from PIMT−/− mice compared withPIMT+/− mice, as well as the ratios of otherd-amino acids including d-methionine,d-tyrosine, d-phenylalanine,d-histidine, and d-arginine (Fig.2C). These results suggested that the proteins carryingl-isoaspartate but not d-aspartate residues are the in vivo substrates for PIMT.
Clinicopathological phenotype of PIMT−/− mice
PIMT−/− mice were born at the normal Mendelian frequency from heterozygote crosses in 129 × the C57BL/6 background. PIMT−/− neonates developed normally and were viable, but their weights were ∼35% smaller than PIMT+/−or PIMT+/+ mice until 7 weeks after birth (data not shown). However, PIMT−/− mice all died between 4 and 12 weeks of age (Fig. 3A, solid line with closed circles). A macroscopic study on postmortem examinations has revealed that the brains ofPIMT−/− mice were 15% heavier than were those ofPIMT+/− mice (509.6 ± 37.9 vs 459.2 ± 28.5 mg;p < 0.0001; Figs. 3B,4B), and the spleens ofPIMT−/− mice were 28% smaller than were those ofPIMT+/− mice (66.5 ± 16.8 vs 91.3 ± 38.8 mg;p < 0.05). Nevertheless, a routine histological examination failed to reveal any apparent pathological abnormality causing death, excluding the possibility that PIMT−/− mice had died from a chronic, progressive pathological process of the heart, lung, or other structures examined. Because gross appearances of somePIMT−/− mice (∼20%) showed a kyphosis of the spine (Fig. 4A), we checked their bones by x ray. Radiological studies, however, failed to show any abnormality in the bone of the vertebrae, skulls, and limbs, suggesting that the kyphosis of the spine may not result from a bone abnormality but rather from the weakness of the muscle providing a support for the vertebral columns, as seen in muscular dystrophies (Wilkins and Gibson, 1976).
Fig. 3.

Survival curve and brain weight ofPIMT−/− mice. A, Kaplan–Meier survival curves of PIMT−/− (closed circles) andPIMT+/− (open circles) mice. The survival of PIMT−/− mice was improved by antiepileptic medication with DPA (closed diamonds). B, Brain weights of 24 PIMT−/− mice (closed circles) and of 24 littermate control mice (PIMT+/−, open circles) plotted versus their age. An upper regression line represents the weights of brains from PIMT−/− mice with a slope of 17.959, and a lower regression line represents those ofPIMT+/− mice with a slope of 4.375.
Fig. 4.

Gross appearance and brain pathology ofPIMT−/− mice. A, Picture of 5-week-old mice derived from intercross-breeding of PIMT heterozygous mice.PIMT−/− (left) andPIMT+/− (right). B, Gross appearance of brains of 5-week-old PIMT−/−(left) and PIMT+/+ (right) mice. C, D, Nissl staining of neurons in layer 5 of the precentral cortex from PIMT−/−(C) and PIMT+/+(D) mice. Magnification: C,D, 400×.
A routine microscopic examination of the brain sections fromPIMT−/− mice using hematoxylin–eosin staining showed the proportional enlargement in all examined areas including gray matter and white matter of the cerebral cortex, hippocampus, striatum, thalamus, and cerebellum. We then investigated the number of neurons or astrocytes in the dentate gyrus of the hippocampus or cerebral cortices (Fig. 4C,D, PIMT−/− and PIMT+/+, respectively) and the number of oligodendrocytes in given microscopic fields of the white matter, revealing no significant increase in the number of neurons or astrocytes (Fig. 4C,D) or of oligodendrocytes in PIMT−/− mice (data not shown); these observations imply that enlargement of the brain was not because of the increased number of neurons as described in CPP-32-deficient mice (Kuida et al., 1996) or because of the gliosis often observed after brain injuries or inflammations but may be because of the increased volume of extracellular matrix or the increased extrasomatal structures such as neural fibers in the brain of PIMT−/− mice. In Nissl sections, cortical pyramidal neurons of bizarre morphology with an abnormally ballooned nucleus and clearly demarcated nucleolus were found in layer V of PIMT−/− mice (Fig. 4C) but not in PIMT+/+ mice (Fig. 4D). However, the appearance of these morphologically abnormal neurons in the precentral cortex of PIMT−/− mice suggested that this abnormality would not be attributable to the generalized enlargement of the brain but may rather be related to myoclonic seizures observed in this mice (see below).
We then investigated blood chemistry, lipid metabolism, electrolyte balance, and hormones in pooled sera from PIMT−/− as well as PIMT+/+ mice. In blood chemistry, PIMT−/−mice showed normal levels for total protein, albumin, glutamic-oxaloacetic transaminase, glutamic–pyruvic transaminase, alkaline phosphatase, and cholinesterase but decreased levels for lactate dehydrogenase (996 vs 1306 IU/l in PIMT−/− vsPIMT+/+), creatine phosphokinase (1138 vs 2135 IU/l), and ferritin (2.5 vs 5.2 ng/ml). In lipid metabolism, PIMT−/−mice showed normal levels for phospholipid and free fatty acid but an increased level for triglyceride (70 vs 52 mg/dl) and decreased levels for total cholesterol (86 vs 116 mg/dl) and esterified cholesterol (75 vs 105 mg/dl). In electrolytes, PIMT−/− mice showed normal levels for Na, K, Cl, Ca, and creatinin but showed a decreased phosphate level (9.8 vs 12.1 mg/dl) and an increased blood urea nitrogen level (30.3 vs 19.7 mg/dl). In hormones examined so far,PIMT−/− mice showed normal levels for free T3 and T4 but increased levels for aldosterone (830 vs 430 pg/ml) and corticosterone (0.24 vs 0.12 mg/ml). All these data may suggest a catabolic state ofPIMT−/− mice, whereas a decrease in CPK may suggest a decreased amount of body muscle.
PIMT-deficient mice die from generalized tonic–clonic seizures
PIMT-deficient mice showed a shortened survival (Fig.3A). Approximately half of PIMT−/− mice spontaneously died from 4 to 5 weeks of age, and the remaining half died at a rather constant rate until 13 weeks of age (Fig.3A, left solid line with closed circles). The fact that few PIMT−/− mice die before 4 weeks of age in spite of abnormalities such as small body size suggests that the abnormality causing a sudden death may be related to maturation defects or a failure in the maintenance of matured organ systems rather than to developmental defects. We then monitored the clinical features of PIMT−/− mice for 24 hr by video camera to catch the clinical feature at the last moment of death. To our surprise, PIMT−/− mice died from grand mal-type epileptic attacks with jumping fits or refractory status epilepticus. In their video recording, PIMT−/− mice showed various types of epileptic seizures including jumping fits, forelimb clonus, running fit, tonic–clonic (GTC) seizures, and myoclonic jerks of the limbs and the trunk. These clinical features, taken together with the following electroencephalographic data, implied that the underlying basis for the epilepsy may be attributable to an increased neuronal excitability in cerebral cortices, hippocampi, the limbic system, and the brainstem.
We next recorded EEGs of PIMT−/− mice to determine the type of epilepsy. EEGs from three PIMT−/− mice were successfully recorded and compared with that of control mice. ThreePIMT−/− mice manifested clinical convulsions with EEG recordings of paroxysmal cortical discharges with spikes and waves as shown in Figure 5, whereas controlPIMT+/+ mice had no clinical convulsions or abnormal cortical discharges. A typical EEG recording of grand mal seizure was presented in Figure 5 in which PIMT−/− mice clinically showed static tonic phase (Fig. 5, phase A), dynamic tonic phase (phase B), long clonic phase (phase C), and a jumping fit (phase D) followed by the posticteric cortical suppression with long duration. Static tonic seizure started abruptly in all bipolar and monopolar leads as presented in Figure 5. After the generalized spikes in tonic phase, typical spikes and waves were recorded in the left frontal lead (Fig. 5, phase C) for >20 sec when the mouse showed a forelimb clonus in the standing position on hindlimbs. Although these spikes and waves failed to spread to the other parts of the brain during clonic phase, the spikes became generalized again in phase D when the mouse showed an abrupt jumping fit (Fig. 5, phase D), followed by a deep coma of long duration. The epileptic animal presented characteristic clinical features in its long-lasting posticteric period when respiratory arrests were often observed in PIMT-deficient mice. As shown in Figure5, only muscle activity corresponding to respiratory movement was recorded in all EEG leads, suggesting that electrical activity in the brains of PIMT−/− mice was severely suppressed in this period. The fact that PIMT−/− mice showed the respiratory arrests after grand mal attacks on the recorded video monitor was well explained by this EEG record. We suggest based on these data that the respiratory suppression after the epileptic seizure may be the direct cause of death in PIMT−/− mice, although alternatively fatal accidents such as arrhythmia could arise in this period.
Fig. 5.

EEG recordings of PIMT−/− mice. A GTC seizure of a PIMT−/− mouse. The beginning of GTC seizure is indicated as Onset of GTC. During the initial static tonic phase (A), the mouse stopped moving and stayed in a prone posture with strong lordosis of the spine and progressive elevation of the tail. During the following dynamic tonic phase (B), the mouse squeaked once and fell over on each side with dynamic torsion of the trunk and tonic elevation of the tail, followed by an opistotonic posture. During the clonic phase (C), the mouse resumed a prone posture, immediately stood on the hindlimbs, and continued the periodic or clonic movements of the forelimbs. The EEG showed a continuous spike-and-wave complex on left frontal recordings, whereas the other monopolar recordings showed continuous rapid rhythm. During a jumping fit (D), the mouse began to walk and jumped several times. When the GTC seizure terminated, the mouse become immobilized, accompanied by a deep respiration. During this phase, the baseline fluctuation in monopolar recordings reflected respiratory movements (up arrowhead), whereas the bipolar recordings showed strong postictal cortical suppression. L, Left; R, right; F, frontal; O, occipital.
We next explored the possibility that DPA sodium (valproate), a widely used antiepileptic drug, could prevent the seizure ofPIMT−/− mice and thus elongate the survival ofPIMT−/− mice. A therapeutic dose of DPA (3 mg/ml in drinking water) was administered orally to PIMT−/− as well as to PIMT+/+ mice, and the frequency of epileptic seizures was checked on the video monitor. Serum concentrations of DPA checked in several treated mice were 20–50 μg/ml. The recordings on the video monitor showed that the frequency of epileptic seizures diagnosed on the video monitor was significantly reduced (minimally seen) inPIMT−/− mice until 7–8 weeks, and the survival ofPIMT−/− mice was significantly elongated by an average of 4 weeks (Fig. 3A, solid line withclosed squares). However, after 8 weeks of age,PIMT−/− mice showed the reluctant epileptic attacks in spite of the therapeutic dose of DPA and finally died either from status epileptics or the toxic suppression of DPA before 12 weeks of age (Fig. 3A). Based on the clinical effects of DPA in the survival of PIMT−/− mice, we reconfirmed that the direct cause of death in PIMT-deficient mice was attributable to epileptic attacks that were initially controlled by DPA but became uncontrollable by 12 weeks of age.
Abnormal dendritic arborizations in pyramidal neurons of PIMT-deficient mice
To define the pathological basis for the abnormal neuronal excitability, we examined the brain morphology of PIMT−/−mice. As noted earlier, there was a 15% increase in brain size inPIMT−/− mice compared with wild-type littermates (Fig.4B) without significant macroscopic abnormalities such as hydrocephalus, brain edema, or brain tumor. We next investigated the brain size in various stages of PIMT−/−mice and compared the sizes with those of PIMT+/− mice (Fig. 3B). Surprisingly, the brains of PIMT-deficient mice began to increase in volume after 4 weeks of age when the basic structure of the brain was, in general, supposed to have been formed. The brain size of PIMT−/− mice increased continuously with age until it finally showed approximately a 25% increase on average at 10 weeks of age (Fig. 3B). The abnormal increase in the brain size seems to coincide with the occurrence of epileptic attacks as well as the sudden death of PIMT−/− mice (Fig.3A,B), suggesting that the morphological analysis in the brain may shed light on the pathological basis for the epileptogenesis of PIMT-deficient mice.
To examine further the neuronal structure of the brain ofPIMT−/− mice, we investigated the morphology of dendrites, somata, and synapses of the neurons by localizing the immunoreactivities for neurofilament (NF), MAP-2, and synaptotagmin. As shown in Figure 6, the immunostaining for NF in cerebral cortices of PIMT−/− mice showed the deformed apical dendrites projecting to the surface of the cerebrum (Fig. 6B) as compared with the straight projections of the control mice (Fig. 6A). The pyramidal neurons with abnormal dendrites were chiefly localized in lateral region of the parietal cortex and began to appear at 5 weeks of age with varying degrees of deformities. In high-power fields, the immunoreactivity for NF showed a twisted conformation in apical dendritic processes of pyramidal neurons in layers II and III of the cerebral cortices fromPIMT−/− mice, whereas the horizontal dendritic fibers in the cerebral cortices showed less-twisted structures (Fig.6D). Immunolocalization of MAP-2 in pyramidal neurons by confocal microscopy confirmed further that the apical dendrites of the pyramidal cells in PIMT−/− mice aberrantly wound and sometimes bifurcated in the proximal portions (Fig.6F), a process not observed in the control mice (Fig.6E). The enigma that still remained to be answered is whether these morphologically abnormal dendrites are functional or dysfunctional and whether the increase in functional dendrites confers the pathological basis for epilepsy. To address these issues, we examined the immunolocalization of synaptotagmin and assessed whether these dendrites retain functional synapses. The result showed that the immunoreactivity for synaptotagmin in PIMT−/− mice was not increased or decreased, with a similar distribution in the brain ofPIMT−/− mice compared with that of the control mice (data not shown), implying that the epileptogenesis of PIMT−/−mice was not a simple case of increased functional dendrites but an aberrant dendritic arborization that may, in large part, be nonfunctional because of the cytoskeletal abnormalities (see below). In this context, it is noteworthy to add that aberrantly arborized mossy fibers with the synaptic reorganization were found in epileptic human temporal lobes (Sutula et al., 1989).
Fig. 6.

Aberrant dendritic arborizations in cortical neurons from PIMT−/− mice. Localization of neurofilament (NF) immunoreactivity in cerebral cortex from PIMT+/+(A, C) and PIMT−/−(B, D) mice is shown. Apical dendrites of pyramidal neurons in layers II and III of the cerebral cortex fromPIMT−/− mice showed the twisted configurations (B, D). Localization of MAP-2 immunoreactivity in cortical pyramidal neurons fromPIMT+/+ (E) andPIMT−/− (F) mice are shown. In PIMT−/− mice, apical dendrites aberrantly wind and bifurcate in the proximal portion (F) Magnification: A,B, 100×; C, D, 500×;E, F, 1600×.
To analyze the dendritic abnormality further, we investigated the ultrastructure of dendrites, axons, and somatic structures of pyramidal cells by electron microscopy. As shown in Figure7, the diameter of axons inPIMT−/− mice was slightly decreased (Fig. 7B) as compared with that in the control mice (Fig. 7A). In an axon hillock of a pyramidal cell, Golgi apparatus (g) and smooth (s) and rough (r) endoplasmic reticulum were aberrantly arranged in the somata, so that the distended hillock was compressed to the nucleus inPIMT−/− mice (Fig. 7D) as compared with the hillock of PIMT+/+ mice (Fig. 7C). In a higher magnification of dendrites, the density of microtubules was locally increased to form high density bundles in the dendritic sections ofPIMT−/− mice (Fig. 7E,F, arrows), whereas the structure of microtubules was evenly distributed in those of PIMT+/+ mice. In PIMT−/− mice, microtubular bundles were unevenly or peripherally localized inside the dendritic fibers (Fig. 7E), which was closely associated with abnormal winding of apical dendrites. The result suggests that the organization of microtubules was altered in PIMT−/− mice and that structural alterations in dendritic and axonal cytoskeletons may give rise to abnormal dendritic arborization of pyramidal neurons inPIMT−/− mice.
Fig. 7.
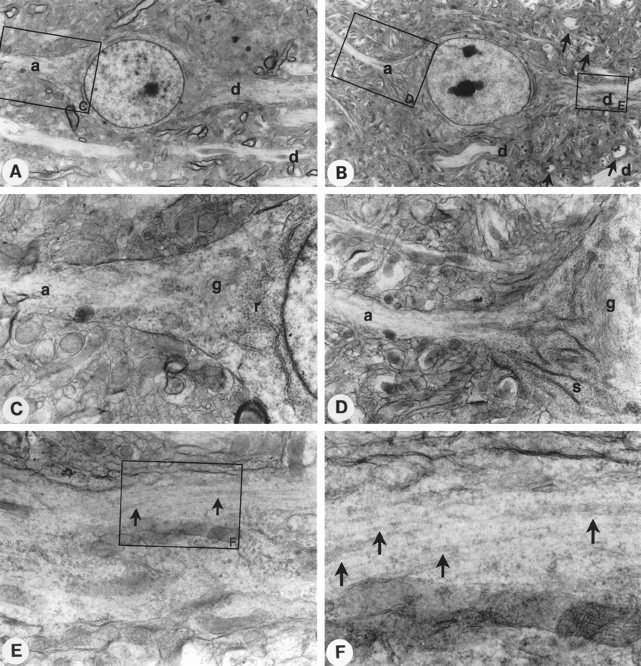
Electron microscopy of cortical neurons fromPIMT+/+ and PIMT−/− mice. A,B, Pyramidal cells in layer III of the cerebral cortex from PIMT+/+ (A) andPIMT−/− (B) mice. Both axonal (a) and dendritic (d) processes were observed. Abnormally arborized dendrites surrounding the somata in PIMT−/− mice are indicated byarrows in B. The large boxes indicate areas shown in subsequent views.C, D, Higher magnification view of an axon hillock of the pyramidal cell from PIMT+/+(C) or PIMT−/−(D) mice. The rough (r) or smooth (s) endoplasmic reticulum and developed Golgi apparatus (g) were observed.E, F, Higher magnification view of a dendrite from a PIMT−/− mouse. High density bundles of microtubules (arrows) were observed in the peripheral dendritic section of a pyramidal neuron from a PIMT−/−mouse. Magnification, A, B, 2100×;C, D, 7500×; E, 14,000×;F, 35,000×.
DISCUSSION
In this paper, we demonstrated that protein methylation is essential for the maintenance of neuronal activity in the CNS and that deficiency of protein methylation leads to fatal progressive epileptic disease. In spite of the fact that PIMT is phylogenetically conserved in various organisms including vertebrates, bacteria, fungi, and plants (Johnson et al., 1991), its biological significance has not been so far well understood. In fact, the repair of damaged aged proteins has been suggested as a biological function of PIMT, this implication solely based on in vitro experiments with various substrates including calmodulin, synapsin, myelin, dentine, and crystalline (Johnson and Aswad, 1985; Potter et al., 1993; Paranandi and Aswad, 1995). The fact that PIMT is highly expressed in various stages of tissues including embryonic and neonatal brains (Shirasawa et al., 1995) suggests role(s) of PIMT in the brain and other tissues in addition to the repair of aged proteins. In this context, the epileptic phenotype of knock-out mice was unexpected but suggestive for the forthcoming studies on PIMT. We discuss here the biochemical alteration of aspartyl residues and the epileptogenic mechanism(s) of PIMT-deficient mice.
l-Isoaspartyl but not d-aspartyl residues accumulate in the brain of PIMT-deficient mice
Previous studies on the specificity of PIMT using synthetic peptides showed that PIMT methylates l-isoaspartyl residues (KASA-l-isoD-LAKY) at a Km value of 0.52 ± 0.08 μm and d-aspartyl residues (KASA-d-D-LAKY) at a Kmvalue of 2700 ± 400 μm with the stoichiometric ratio of d-aspartateKm/l–isoaspartateKm/l being 5190-fold (Lowenson and Clarke, 1992). Based on the computer simulation with these values, Lowenson predicted that an average 100 kDa protein carrying 35l-asparagine and 52 l-aspartate residues, without the methyltransferase activity, should generate 0.52 residues of l-isoaspartate, 0.008 residues ofd-aspartate, and 0.029 residues ofd-isoaspartate per molecule in 10 d. The addition of PIMT activity to this mathematical simulation led to the reduction ofl-isoaspartate to <0.0036 but the increase ofd-aspartate up to 0.020 in 10 d (Lowenson and Clarke, 1992). In the present study, we showed that PIMT represents the major protein methyltransferase in vivo in the brain, testis, and eye, which can transfer the methyl residues into the synthetic peptides carrying l-isoaspartate or ovalbumin (Fig. 3A). Furthermore, we demonstrated that l-isoaspartyl residues were significantly accumulated in the brain from PIMT−/−mice (Fig. 3B) as well as in the testis, heart, muscle, spleen, and eyes of PIMT−/− mice (data not shown) but not in those tissues from PIMT+/− or PIMT+/+ mice. On the other hand, the d-aspartate residue was not increased but rather decreased in the brain from PIMT−/−mice in comparison with those of PIMT+/− mice (Fig.2C). These in vivo data are fully compatible with the previous hypothetical simulation that the complete deficiency of PIMT activity theoretically leads to the accumulation ofl-isoaspartyl residues but a slight decrease ind-aspartyl residues in damaged proteins (Lowenson and Clarke, 1992).
To characterize the substrate proteins accumulated in the brain fromPIMT−/− mice, we labeled brain lysate prepared fromPIMT−/− mice with [14C]methyl-SAM by recombinant PIMT and resolved it on acidic PAGE gel. The labeled molecules in brain lysate of PIMT−/− mice were fractionated in a smear pattern with varying intensities detected; the pattern of these intensities was, in large part, similar to that of control mice, but the intensities in PIMT−/− mice were significantly stronger than those of the control (data not shown), suggesting that the substrates for PIMT may be widely distributed in various proteins, in which the equilibrium amongl-aspartate, l-isoaspartate,d-aspartate, and d-isoaspartate in modified aspartate or asparagine residues was drastically shifted tol-isoaspartate in the substrate molecules ofPIMT−/− mice. Further biochemical investigation on PIMT-deficient mice would clarify the biological significance of the shifted equilibrium in modified aspartate residues of the substrate molecules in vivo.
PIMT deficiency results in a progressive epilepsy with grand mal and myoclonus
Because 10–15% of human epilepsies are suggested to have genetic backgrounds, the identification of mutations in causal genes can be used to define epileptogenic mechanisms. Several human epilepsy genes have been mapped, and some of them were identified recently (Silvestri et al., 1992; Shiang et al., 1993; Steinlein et al., 1995;Pennacchio et al., 1996). However, the causal genes for the majority of epilepsies still remain to be identified (Phillips et al., 1995; Ryan, 1995; Serratosa et al., 1995; Zara et al., 1995; Virtaneva et al., 1996). Moreover, these efforts are complicated not only by clinical and genetic heterogeneity but likely by polygenic inheritance as well. In mice, a number of single-gene mutations that cause epilepsy have been identified by the study of various knock-out mice (Brusa et al., 1995;Li et al., 1995; Toth et al., 1995; Waymire et al., 1995; Matsumoto et al., 1996; Rothstein et al., 1996; Brennan et al., 1997; Homanics et al., 1997; Kim et al., 1997; Luthi et al., 1997), and it is hoped that the identification and molecular analysis of these genes will provide insights into human epilepsy. We show in the present studya mouse model for fatal progressive epilepsy with grand mal and myoclonus and discuss possible novel epileptogenic mechanism(s).
In this study, several lines of evidence strongly suggested that epilepsy was a direct cause of death in PIMT-deficient mice. First, the anticonvulsive drug DPA decreased the frequency of seizures and prolonged the survival of PIMT−/− mice. Second, the video monitor indicated that PIMT−/− mice showed grand mal attacks and myoclonic seizures and died soon after fatal convulsion attacks or from refractory status epilepticus. Third, the postmortem pathology showed otherwise normal tissues in hematoxylin–eosin staining. EEG study indicated that PIMT−/− mice showed generalized tonic–clonic seizure with typical spikes and waves. Taken together, PIMT-deficient mice can be an animal model for a progressive myoclonus epilepsy (PME). PME is a clinical entity with myoclonic seizures, tonic–clonic seizures, and progressive neurological dysfunction such as ataxia or dementia and is classified into two major forms, Unverricht-Lundborg disease and Lafora’s disease. Recently, point mutations in the cystatin B gene have been proven to be responsible for Unverricht-Lundborg disease (Pennacchio et al., 1996). The causal gene for Lafora’s disease, on the other hand, has been mapped to human chromosome 6q23–25 (Serratosa et al., 1995) but has not yet been identified. Interestingly, human PIMT gene was mapped to chromosome 6q22.3–6q24 (MacLaren et al., 1992). Furthermore, the clinical profile of PIMT-deficient mice is consistent with that of Lafora’s disease that develops neurological symptoms during late childhood and leads to a fatal outcome within a decade of the first symptom (Elliott et al., 1992). It is reasonable to speculate that a mutation in the PIMT gene may cause progressive myoclonus epilepsy of the Lafora type in affected pedigrees. Further genetic study on such pedigrees would clarify the causative relationship between PIMT gene mutation and Lafora’s disease.
Cytoskeletal abnormality and accumulation of modified proteins with isoaspartyl residues
To date a number of knock-out or transgenic mice showing epileptic phenotype have shed light on the molecular basis for epileptogenesis (Noebels, 1996). These include (1) ion channel genes such as mkv1.1 orWeaver, (2) genes regulating the synaptic release of neurotransmitters such as synapsin I and II or CAMKII, (3) synaptic receptor genes such as GluRB or 5-HT2C gene, and (4) genes associated with axonogenesis or synaptogenesis such as GAP-43. In our experimental model, the biochemical alteration such as the accumulation of isoaspartyl residues in the brain was clearly associated with the onset of seizure in PIMT−/− mice. It is then speculated that some molecule(s) modified with isoaspartyl residues may confer the epileptogenic basis in PIMT−/− mice. However, it is practically difficult to identify the responsible epileptogenic molecule(s) by means of a biochemical approach because a number of molecules were radiolabeled by recombinant PIMT in the brain lysate prepared from PIMT−/− mice.
Pathological analysis strongly suggested that the enlargement of the brain observed in PIMT-deficient mice after 4 weeks of age may be related to the progression of fatal epilepsy (Fig. 3). However, morphological analyses of light (Fig. 4B) and electron (Fig. 7) microscopic examinations as well as immunohistochemical staining for NF, MAP-2, and synaptotagmin studies (Fig. 6) revealed the swollen pyramidal neurons in layer V of the precentral cortex (Fig. 4C) and the abnormal arborizations of apical dendrites of cortical pyramidal neurons in the parietal lobe of PIMT−/− mice (Fig. 6). This abnormality could, in part, contribute to the enlargement of the brain but fail to explain the 15% increase in the brain size of PIMT−/− mice, suggesting that morphologically invisible alterations such as a generalized increase of extracellular matrix may contribute to the enlargement of the brain in PIMT-deficient mice. Further neurochemical investigation on basic components such as MBP, tubulin, neurofilament, and actin or sphingolipids such as sphingomyelins and cerebrosides may shed light on the biochemical basis for the enlargement of the brain.
EM studies showed the abnormal microtubular organization in the dendrites of pyramidal neurons. The abnormality is especially intriguing because PIMT immunolocalizes to neurofibrillary tangles (NFTs) in the brain of Alzheimer’s disease (T. Shirasawa, unpublished observations). Because NFT is chiefly composed of abnormally phosphorylated tau, it is suggested that tau is one of the candidate substrates for PIMT in degenerating neurons. We then purified tau fromPIMT−/− mice and investigated the isomerization of tau.l-Isoaspartyl residues in tau purified fromPIMT−/− mice were increased 2.7-fold compared with that from PIMT+/+ mice (8.60 vs 3.23 pmol/μg protein), indicating that tau is an in vivo substrate for PIMT. Because microtubule-associated proteins (MAPs) such as tau modulate and regulate the formation of microtubules in neuronal processes, one possible mechanism for the disorganization of microtubules may be the dysfunction of MAPs because of the protein isomerization. In addition,Najbauer et al. (1996) recently reported that tubulin was also isomerized in vivo as well as in vitro, suggesting as another possibility that microtubules fail to organize properly in the neural processes because of the isomerization of tubulin itself. The assembly and disassembly of microtubules inPIMT−/− fibroblasts is now being investigated in our laboratory using a GFP-Tau34 construct (Ludin et al., 1996) to address these issues. As microtubules are involved in the transport of various signal-transducing molecules such as neurotrophines or synaptic molecules, the dysfunction of the transport system may confer the alteration of neuronal excitability in PIMT-deficient mice.
Footnotes
This work was supported by grants from the Sasakawa Health Foundation, the Nagase Science and Technology Foundation, the Brain Science Foundation, the Grant-in-Aid for Scientific Research on Priority Areas (intracellular proteolysis, targeted recombination), the Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture of Japan, CREST, the Japan Science and Technology Corporation, and the Ministry of Health and Welfare of Japan. We thank Dr. R. Mulligan for the pPNT vector and Drs. Akiyama, R. Taniuchi, N. Maruyama, M. Ogawara, H. Nakano, and K. Okumura for their assistance and discussion.
Correspondence should be addressed to Dr. Takuji Shirasawa, Department of Neurophysiology, Tokyo Metropolitan Institute of Gerontology, 35-2 Sakae-cho, Itabashi-ku, Tokyo-173, Japan.
REFERENCES
- 1.Aswad DW. Stoichiometric methylation of porcine adrenocorticotropin by protein carboxyl methyltransferase requires deamidation of asparagine 25. Evidence for methylation at the alpha-carboxyl group of atypical l-isoaspartyl residues. J Biol Chem. 1984;259:10714–10721. [PubMed] [Google Scholar]
- 2.Bradly A. Teratocarcinomas and embryonic stem cells–a practical approach (Robertson EJ, ed). IRL; Oxford: 1987. [Google Scholar]
- 3.Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–390. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- 4.Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270:1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 5.Chirgwin JM, Przylla AE, MacDonald RJ, Runnter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 6.Clark S. Protein carboxyl methyltransferase: two distinct classes of enzymes. Annu Rev Biochem. 1985;54:479–506. doi: 10.1146/annurev.bi.54.070185.002403. [DOI] [PubMed] [Google Scholar]
- 7.Elliott EJ, Talbot JC, Pye IF, Hodges S, Swift PG, Tanner MS. Lafora disease: a progressive myoclonus epilepsy. J Paediatr Child Health. 1992;28:455–458. doi: 10.1111/j.1440-1754.1992.tb02717.x. [DOI] [PubMed] [Google Scholar]
- 8.Fukuda T, Kawano H, Ohyama K, Li H-P, Takeda Y, Ohira A, Kawamura K. Immunohistochemical localization of neurocan and L1 in the formation of thalamocortical pathway of developing rats. J Comp Neurol. 1997;382:141–152. [PubMed] [Google Scholar]
- 9.Geiger T, Clarke S. Deamidation, isomerization, and racemization of asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem. 1987;262:785–794. [PubMed] [Google Scholar]
- 10.Hayashi T, Sasagawa T. A method for identifying the carboxy terminal amino acid of a protein. Anal Biochem. 1993;209:163–168. doi: 10.1006/abio.1993.1097. [DOI] [PubMed] [Google Scholar]
- 11.Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 13.Ingrosso D, Fowler AV, Bleibaum J, Clarke S. Sequence of the d-aspartyl/l-isoaspartyl protein methyltransferase from human erythrocytes. Common sequence motifs for protein, DNA, RNA, and small molecule S-adenosylmethionine-dependent methyltransferases. J Biol Chem. 1989;264:20131–20139. [PubMed] [Google Scholar]
- 14.Johnson BA, Aswad DW. Identification and topography of substrates for protein carboxyl methyltransferase in synaptic membrane and myelin-enriched fractions of bovine and rat brain. J Neurochem. 1985;45:1119–1127. doi: 10.1111/j.1471-4159.1985.tb05531.x. [DOI] [PubMed] [Google Scholar]
- 15.Johnson BA, Langmack EL, Aswad DW. Partial repair of deamidation-damaged calmodulin by protein carboxyl methyltransferase. J Biol Chem. 1987;262:12283–12287. [PubMed] [Google Scholar]
- 16.Johnson BA, Ngo SQ, Aswad DW. Widespread phylogenetic distribution of a protein methyltransferase that modifies l-isoaspartyl residues. Biochem Int. 1991;24:841–847. [PubMed] [Google Scholar]
- 17.Kim D, Jun KS, Lee SB, Kang NG, Min DS, Kim YH, Ryu SH, Suh PG, Shin HS. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–293. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- 18.Kitamura D, Rocs J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 19.Kuida K, Zheng TS, Na S, Kuan C-y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Chin LS, Shipliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowenson JD, Clarke S. Spontaneous degradation and enzymatic repair of aspartyl and asparaginyl residues in aging red cell proteins analyzed by computer simulation. Gerontology. 1991;37:128–151. doi: 10.1159/000213255. [DOI] [PubMed] [Google Scholar]
- 22.Lowenson JD, Clarke S. Recognition of d-aspartyl residues in polypeptides by the erythrocyte l-isoaspartyl/d-aspartyl protein methyltransferase. Implications for the repair hypothesis. J Biol Chem. 1992;267:5985–5995. [PubMed] [Google Scholar]
- 23.Ludin B, Doll T, Meili R, Kaech S, Matus A. Application of novel vectors for GFP-tagging of proteins to study microtubule-associated proteins. Gene. 1996;173:107–111. doi: 10.1016/0378-1119(95)00899-3. [DOI] [PubMed] [Google Scholar]
- 24.Luthi A, Putten H, Botteri FM, Mansuy IM, Meins M, Frey U, Sansig G, Portet C, Schmutz M, Schroder M, Nitsch C, Laurent JP, Monard D. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J Neurosci. 1997;17:4688–4699. doi: 10.1523/JNEUROSCI.17-12-04688.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacLaren DC, O’Connor CM, Xia YR, Mehrabian M, Klisak I, Sparkes RS, Clarke S, Lusis AJ. The l-isoaspartyl/d-aspartyl protein methyltransferase gene (PCMT1) maps to human chromosome 6q22.3–6q24 and the syntenic region of mouse chromosome 10. Genomics. 1992;14:852–856. doi: 10.1016/s0888-7543(05)80104-1. [DOI] [PubMed] [Google Scholar]
- 26.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 28.McFadden PN, Clarke S (1982) Methylation atd-aspartyl residues in erythrocytes: possible step in the repair of aged membrane proteins. Proc Natl Acad Sci USA 2460–2464. [DOI] [PMC free article] [PubMed]
- 29.McFadden PN, Clarke S. Conversion of isoaspartyl peptides to normal peptides: implications for the cellular repair of damaged proteins. Proc Natl Acad Sci USA. 1987;84:2595–2599. doi: 10.1073/pnas.84.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Najbauer J, Orpiszewski J, Aswad DW. Molecular aging of tubulin: accumulation of isoaspartyl sites in vitro and in vivo. Biochemistry. 1996;35:5183–5190. doi: 10.1021/bi953063g. [DOI] [PubMed] [Google Scholar]
- 31.Nakano H, Saito K, Ogashiwa M, Suzuki K. Referential derivation of epidural electroencephalogram in E1 mice. J Vet Med Sci. 1993;55:39–43. doi: 10.1292/jvms.55.39. [DOI] [PubMed] [Google Scholar]
- 32.Nakano H, Saito K, Suzuki K. Chronic implantation technique for monopolar EEG monitoring epileptic seizures in mice. Brain Res Bull. 1994;35:264–268. doi: 10.1016/0361-9230(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 33.Noebels JL. Targeting epilepsy genes. Neuron. 1996;16:241–244. doi: 10.1016/s0896-6273(00)80042-2. [DOI] [PubMed] [Google Scholar]
- 34.O’Connor CM, Aswad DW, Clarke S. Mammalian brain and erythrocyte carboxyl methyltransferases are similar enzymes that recognize both d-aspartyl and l-isoaspartyl residues in structurally altered protein substrates. Proc Natl Acad Sci USA. 1984;81:7757–7761. doi: 10.1073/pnas.81.24.7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paranandi MV, Aswad DW. Spontaneous alterations in the covalent structure of synapsin I during in vitro aging. Biochem Biophys Res Commun. 1995;212:442–448. doi: 10.1006/bbrc.1995.1989. [DOI] [PubMed] [Google Scholar]
- 36.Paranandi MV, Guzzetta AW, Hancock WS, Aswad DW. Deamidation and isoaspartate formation during in vitro aging of recombinant tissue plasminogen activator. J Biol Chem. 1994;269:243–253. [PubMed] [Google Scholar]
- 37.Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, D’Amato E, Ramirez L, Faham M, Koskiniemi M, Warrington JA, Norio R, Chapelle A, Cox DR, Myers RM. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science. 1996;271:1731–1734. doi: 10.1126/science.271.5256.1731. [DOI] [PubMed] [Google Scholar]
- 38.Phillips HA, Scheffer IE, Berkovic SF, Hollway GE, Sutherland GR, Mulley JC. Localization of a gene for autosomal dominant nocturnal frontal lobe epilepsy to chromosome 20q 13.2. Nat Genet. 1995;10:117–118. doi: 10.1038/ng0595-117. [DOI] [PubMed] [Google Scholar]
- 39.Potter SM, Henzel WJ, Aswad DW. In vitro aging of calmodulin generates isoaspartate at multiple Asn–Gly and Asp–Gly sites in calcium-binding domains II, III, and IV. Protein Sci. 1993;2:1648–1663. doi: 10.1002/pro.5560021011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Romanik EA, Ladino CA, Killoy LC, D’Ardenne SC, O’Connor CM. Genomic organization and tissue expression of the murine gene encoding the protein beta-aspartate methyltransferase. Gene. 1992;118:217–222. doi: 10.1016/0378-1119(92)90191-q. [DOI] [PubMed] [Google Scholar]
- 41.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kunel RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 42.Ryan SG. Partial epilepsy: chinks in the armour. Nat Genet. 1995;10:4–6. doi: 10.1038/ng0595-4. [DOI] [PubMed] [Google Scholar]
- 43.Serratosa JM, Delgado-Escueta AV, Posada IS, Shih ID, Berciano J, Zabala JA, Antunez MC, Sparkes RS. The gene for progressive myoclonus epilepsy of the Lafora type maps to chromosome 6q. Hum Mol Genet. 1995;4:1657–1663. doi: 10.1093/hmg/4.9.1657. [DOI] [PubMed] [Google Scholar]
- 44.Shiang R, Ryan SG, Zhu YZ, Hahn AF, O’Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- 45.Shirasawa T, Endoh R, Zeng Y-X, Sakamoto K, Mori H. Protein l-isoaspartyl methyltransferase: developmentally regulated gene expression and protein localization in the central nervous system of aged rat. Neurosci Lett. 1995;187:1–4. doi: 10.1016/0304-3940(95)11389-e. [DOI] [PubMed] [Google Scholar]
- 46.Silvestri G, Moraes CT, Shanske S, Oh SJ, DiMauro S. A new mtDNA mutation in the tRNA(Lys) gene associated with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1992;51:1213–1217. [PMC free article] [PubMed] [Google Scholar]
- 47.Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- 48.Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- 49.Toth M, Grimsby J, Buzsaki G, Donovan GP. Epileptic seizures caused by inactivation of a novel gene, jerky, related to centromere binding protein-B in transgenic mice. Nat Genet. 1995;11:71–75. doi: 10.1038/ng0995-71. [DOI] [PubMed] [Google Scholar]
- 50.Tybulewicz VL, Crawford CD, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 51.Virtaneva K, Miao J, Traskelin AL, Stone N, Warrington JA, Weissenbach J, Myers RM, Cox DR, Sistonen P, Chapelle A, Lehesjoki AE. Progressive myoclonus epilepsy EPM1 locus maps to a 175-kb interval in distal 21q. Am J Hum Genet. 1996;58:1247–1253. [PMC free article] [PubMed] [Google Scholar]
- 52.Waymire KG, Mahuren JD, Jaje JM, Guilarte TR, Coburn SP, MacGregor GR. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. 1995;11:45–51. doi: 10.1038/ng0995-45. [DOI] [PubMed] [Google Scholar]
- 53.Wilkins KE, Gibson DA. The patterns of spinal deformity in Duchenne muscular dystrophy. J Bone Joint Surg. 1976;58:24–32. [PubMed] [Google Scholar]
- 54.Zara F, Bianchi A, Avanzini G, Donato SD, Castellotti B, Patel PI, Pandolfo M. Mapping of genes predisposing to idiopathic generalized epilepsy. Hum Mol Genet. 1995;4:1201–1207. doi: 10.1093/hmg/4.7.1201. [DOI] [PubMed] [Google Scholar]