

Abstract
Dopamine (DA) autoreceptors expressed along the somatodendritic extent of midbrain DA neurons modulate impulse activity, whereas those expressed at DA nerve terminals regulate both DA synthesis and release. Considerable evidence has indicated that these DA autoreceptors are of the D2 subtype of DA receptors. However, many pharmacological studies have suggested an autoreceptor role for the DA D3 receptor. This possibility was tested with mice lacking the D3 receptor as a result of gene targeting. The basal firing rates of DA neurons within both the substantia nigra and ventral tegmental area were not different in D3 receptor mutant and wild-type mice. The putative D3receptor-selective agonistR(+)-trans-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-(1)benzopyrano(4,3-b)−1,4-oxazin-9-ol (PD 128907) was equipotent at inhibiting the activity of both populations of midbrain DA neurons in the two groups of mice. In the γ-butyrolactone (GBL) model of DA autoreceptor function, mutant and wild-type mice were identical with respect to striatal DA synthesis and its suppression by PD 128907. In vivo microdialysis studies of DA release in ventral striatum revealed higher basal levels of extracellular DA in mutant mice but similar inhibitory effects of PD 128907 in mutant and wild-type mice. These results suggest that the effects of PD 128907 on dopamine cell function reflect stimulation of D2 as opposed to D3 receptors. Although D3 receptors do not seem to be significantly involved in DA autoreceptor function, they may participate in postsynaptically activated short-loop feedback modulation of DA release.
Keywords: D3 receptors, mutant mice, dopamine autoreceptors, dopamine receptors, dopamine neurons, dopamine release, dopamine synthesis
Discovery of the dopamine (DA) D3 receptor (Sokoloff et al., 1990) generated considerable excitement regarding possible physiological and behavioral functions mediated by this member of the DA D2 receptor subfamily (D2, D3, and D4). Among the speculated roles for the D3 receptor was that of an autoreceptor, the receptors expressed by DA neurons to provide feedback regulatory control. Initial characterization of the cloned D3 receptor identified several D3 receptor-preferring ligands that had been regarded previously as DA autoreceptor-selective (Svensson et al., 1986). In addition, D3 receptor gene transcripts were detected by PCR in the rat ventral tegmental area (VTA) and substantia nigra (SN) and were absent after selective destruction of DA neurons with 6-hydroxydopamine, suggesting localization to DA neurons (Sokoloff et al., 1990). However, more recent studies have either failed to detect D3 receptor mRNA in the rat midbrain (Landwehrmeyer et al., 1993; Meador-Woodruff et al., 1994; Richtand et al., 1995;Healy and Meador-Woodruff, 1996) or have demonstrated low and very restricted expression to lateral regions of the SN and VTA, with only a portion of these cells also expressing tyrosine hydroxylase (Bouthenet et al., 1991; Diaz et al., 1995).
Functionally, three classes of DA autoreceptors can be defined (for review, see Wolf and Roth, 1987). The soma and dendrites of midbrain DA neurons express autoreceptors that modulate rates of impulse activity (Bunney et al., 1973; Aghajanian and Bunney, 1977). Nerve terminals of these neurons express autoreceptors that modulate DA synthesis (Kehr et al., 1972) and DA release (Farnebo and Hamberger, 1971). Each of these three autoreceptors exhibits pharmacological characteristics of the D2 receptor subfamily (for review, see Clark and White, 1987; Wolf and Roth, 1987), and conclusive anatomical evidence indicates that DA neurons express D2 receptor mRNA (Meador-Woodruff et al., 1989; Mengod et al., 1989; Mansour et al., 1995). Despite the limited anatomical evidence of the expression of D3 receptor mRNA in DA neurons (above), many pharmacological studies have suggested that D3autoreceptors modulate DA impulse flow (Devoto et al., 1995; Kreiss et al., 1995; Lejeune and Millan, 1995; Gobert et al., 1996; Tepper et al., 1997), DA synthesis (Meller et al., 1993; Ahlenius and Salmi, 1994; Aretha et al., 1995; Gobert et al., 1995; Pugsley et al., 1995), and DA release (Damsma et al., 1993; Gainetdinov et al., 1994, 1996;Rivet et al., 1994; Gilbert et al., 1995; Pugsley et al., 1995; Gobert et al., 1996; Routledge et al., 1996; Tepper et al., 1997). With notable exceptions (Tang et al., 1994; Nissbrandt et al., 1995; O’Hara et al., 1996; Tepper et al., 1997), most of these studies inferred a role for D3 receptors based on (1) correlations between DA agonist potency and relative binding affinities at cloned D2 and D3 receptors expressed in various cell lines, (2) comparisons between regional differences in DA agonist potency and relative expression of D2 and D3receptors, or (3) antagonism of agonist effects by putative D3 receptor-selective antagonists. These approaches are not conclusive because serious questions exist with respect to the selectivity of D3 receptor ligands (Chio et al., 1994;Large and Stubbs, 1994; Potenza et al., 1994; Burris et al., 1995;Gonzalez and Sibley, 1995; Sautel et al., 1995).
To test possible autoreceptor roles for D3 receptors more directly, we have used homologous recombination in embryonic stem cells to generate mice lacking the intact D3 receptor gene (Xu et al., 1997). We used well characterized in vivo models of DA autoreceptor function to compare the D3 receptor mutant mice with their wild-type littermates with respect to DA cell firing rates, DA synthesis, and DA release and inhibition of these functions by the putative D3 receptor-selective agonistR(+)-trans-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-(1)benzopyrano(4,3-b)−1,4-oxazin-9-ol (PD 128907) (Sokoloff and Schwartz, 1995). We reasoned that if D3 receptors serve autoreceptor functions, then the basal firing rates of DA neurons, as well as levels of DA synthesis and release, should be altered in D3 receptor mutant mice and that autoreceptor-mediated effects of PD 128907 should be absent or reduced in D3 mutant mice. Our results fail to support an autoreceptor role for D3 receptors but suggest that postsynaptic D3 receptors may regulate DA release via short-loop feedback mechanisms.
MATERIALS AND METHODS
Mice. Mice were generated as detailed in Xu et al. (1997). They were shipped to the Chicago Medical School by commercial carrier. Mice were housed in like groups of three to four animals and were allowed 7–8 d to acclimate to the vivarium before use. All experimental procedures were conducted at the Chicago Medical School. All procedures were performed in strict accordance with theNational Research Council Guide for the Care and Use of Laboratory Animals (1996) and were approved by our Institutional Animal Care and Use Committee.
Extracellular single-unit recordings. All methods for extracellular single-cell recordings were similar to those previously reported (White et al., 1995), although modified somewhat for the mouse (Xu et al., 1994). Briefly, mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and mounted in a standard stereotaxic apparatus with a specialized adapter for the mouse. Body temperature was maintained at 36–37.5°C with a thermostatically controlled heating pad. A 28 gauge (3/8 inch) hypodermic needle was placed in a lateral tail vein through which additional anesthetic (as required) and drugs of study were administered. A burr hole was drilled in the skull, and the dura was retracted from the area overlying the VTA and SN, 0.4–1.3 mm anterior to lambda and 0.2–1.0 mm lateral to the midline. Recording electrodes were made by pulling glass tubing [outer diameter (o.d.), 2.0 mm], which was prefilled with fiberglass, and by breaking the tip back to a diameter of 1–2 μm. Electrodes were filled with 2 m NaCl saturated with 1% (w/v) fast green dye and typically exhibitedin vitro impedances of 1–3 MΩ (at 135 Hz). Electrode potentials were passed through a high impedance amplifier/filter and displayed on an oscilloscope. Individual action potentials were discriminated electronically and monitored with an audio amplifier. Integrated rate histograms, generated by the output of the window discriminator, were plotted by a polygraph recorder, whereas digitized counts of cellular activity were obtained for off-line analysis. Electrodes were lowered to a point 0.5 mm above the VTA and SN and then slowly advanced with a hydraulic microdrive through the DA cell regions (3.2–5.0 mm ventral to the cortical surface). DA cells were identified by standard physiological criteria (Bunney et al., 1973; Wang, 1981;Sanghera et al., 1984) and were recorded for 3–6 min to establish a baseline firing rate. To determine the sensitivity of impulse-modulating somatodendritic autoreceptors, we administered PD 128907 to each mouse through the tail vein, using a cumulative dose regimen in which each dose doubled the previous dose, at 60–90 sec intervals. After agonist-induced inhibition, the D2-class receptor antagonist eticlopride was administered (0.1–0.2 mg/kg) to reverse the effect and confirm receptor mediation. At the end of each experiment, the cell location was marked by ejecting fast green dye, and the spot was verified by routine histological assessment (described below).
γ-Butyrolactone experiments. The l-aromatic amino acid decarboxylase inhibitor NSD 1015 was administered 30 min before death (100 mg/kg, i.p.). γ-Butyrolactone (GBL) was administered (750 mg/kg, i.p.) 5 min before NSD 1015 to eliminate impulse flow in DA neurons (Walters and Roth, 1976). PD 128907 was injected 5 min before GBL. Mice were killed by decapitation, and the brains were quickly removed. Dorsal and ventral striatum were dissected on a chilled glass plate with the aid of a mouse brain matrix designed to allow coronal sections to be cut rapidly and reproducibly (Activational Systems, Warren, MI). Two slices (2 mm each) were taken, beginning at the rostral boundary of the olfactory tubercle. The most anterior slice was the source of ventral striatum, whereas both slices were used to obtain dorsal striatum. The ventral striatum was dissected with angular cuts originating at the lateral olfactory tracts and ending at the midline (average weight, 18.5 mg). The remainder of the striatal region from that slice as well as the similar region from the more caudal slice was considered the dorsal striatum (average weight, 35.8 mg). Tissues were kept at −80°C. To measure tissue catechols, we weighed frozen tissues and then sonically disrupted them in a homogenization solution consisting of 100 mmHClO4, 5 mmNa2S2O5, and α-methyl dopa, an internal standard. After centrifugation (25,000 ×g for 10 min), aliquots of the supernatant were processed by alumina extraction as described previously (Galloway et al., 1986). The 3,4-dihydroxyphenylalanine (DOPA) content of samples was determined using an HPLC system consisting of a Bioanalytical Systems (BAS) Phase II ODS 3 μm column (100 × 3.2 mm), a BAS LC4C electrochemical detector, and a Scientific Systems model 222C HPLC pump. The mobile phase consisted of 0.1 mNaH2PO4, 1 mm EDTA, 0.2 mm 1-octane-sulfonic acid, and 3% methanol, adjusted to pH 2.7 with phosphoric acid. DOPA content was quantified based on both internal and external standards.
In vivo microdialysis experiments. Mice used for dialysis experiments weighed 28–35 gm. Concentric microdialysis probes were constructed as described previously, with fused-silica inlet and outlet lines (Wolf et al., 1994). Dialysis membrane (molecular weight cutoff, 6000; o.d., 250 μm) was obtained from Spectrum (Los Angeles, CA). Data were not corrected for in vitro probe recovery because probes may suffer differential alterations during insertion. Consistent with this assumption, data sets corrected for recovery often exhibit greater variability than do those that are uncorrected (Xue et al., 1996). Probes were stereotaxically implanted under sodium Brevital (8 mg/kg, i.p.). Stereotaxic coordinates were, relative to bregma, anterior, 1.5 mm; lateral, 1.5 mm; and ventral, 2.7–4.7 mm. Ventral coordinates indicate exposed regions of dialysis membrane (2 mm). After surgery, mice were placed in Plexiglas cages (23 × 46 mm) and allowed to recover overnight. Food and water were available ad libitum. Dialysis cages were equipped with balance arms (Instech, Plymouth Meeting, PA), and homemade liquid swivels and tethers were constructed from plastic syringes and tubing. These were used because commercially available swivels were too heavy for use with mice. Probes were perfused overnight at 0.3 μl/min with artificial CSF (aCSF) consisting of (in mm): 2.7 KCl, 140 NaCl, 1.2 CaCl2, 1 MgCl2, 0.3 NaH2PO4, and 1.7 Na2HPO4, pH 7.4. The next morning, the perfusion rate was increased to 2 μl/min for 2–3 hr before the experiment was begun. Experiments consisted of 1 hr of perfusion with control aCSF to determine basal DA efflux, 1 hr of perfusion with aCSF containing PD 128907, and a 1 hr recovery period during which control aCSF was perfused. Thus, administration of PD 128907 occurred ∼20 hr after probe implantation. Fractions were collected every 20 min. After each experiment, mice were anesthetized and perfused intracardially with normal saline followed by 10% formalin. Probe placement was examined in sections stained with cresyl violet. Only data from mice with verified probe placements were included in the analysis. Dialysates were analyzed for DA content using the HPLC system described for GBL experiments. Chromatographic conditions were optimized for early elution of DA to obtain maximum sensitivity. The mobile phase consisted of 0.1 m NaH2PO4, 0.5 mm EDTA, 2 mm 1-octane-sulfonic acid, and 16% methanol, adjusted to pH 4.9. Peaks were recorded using a dual-channel chart recorder and were quantified by comparison with the peak heights of external standards run with every experiment.
Drugs. PD 128907, 7-hydroxy-2-(di-n-propylamino)tetralin (7-OH-DPAT), quinpirole, and eticlopride were obtained from Research Biochemicals (Natick, MA). GBL was obtained from VWR Scientific (Chicago, IL). NSD 1015 and alumina for column chromatography were obtained from Sigma (St. Louis, MO).
Statistics. Two-way repeated measures ANOVA (groups and dose) was used to analyze the electrophysiological and DOPA synthesis results when PD 128907 was administered. In both cases, dose was the repeated measure. Planned comparisons were made with Dunnett’s test. Results from basal DOPA synthesis measures were analyzed with a two-way ANOVA (groups and treatment) with the treatments being NSD 1015 and NSD 1015 plus GBL. Basal levels of extracellular DA obtained with microdialysis were compared with a one-way ANOVA. Because of the significant difference between the two groups of mice on this measure, we used two-way (groups × fraction) repeated measures (fraction) analysis of covariance (ANCOVA) with basal DA as the covariate to compare the effects of PD 128907 on DA release.
RESULTS
Lack of evidence of impulse-modulating DA D3 autoreceptors
PD 128907 produced a potent, dose-dependent suppression of firing of DA neurons in both the VTA and SN and did so equally well in mutant and wild-type mice (Fig. 1). Similar experiments conducted with other putative D3receptor-selective agonists, 7-OH-DPAT and quinpirole, produced identical results (data not shown). We also observed no difference in the basal firing rates of DA neurons recorded in the two groups of mice. Such a difference might be expected if D3 receptors exert a tonic inhibitory influence on neuronal activity. These findings indicate that inhibition of DA cell activity by putative D3receptor-selective agonists is not mediated by stimulation of D3 receptors and thus must be mediated by D2receptors. Because in situ hybridization histochemistry indicates restricted expression of D3 mRNA in lateral portions of the rat SN and VTA (Diaz et al., 1995), we determined the sites of our recorded DA neurons using routine histological procedures and confirmed that DA neurons from these regions of the midbrain were included in our samples (Fig. 2).
Fig. 1.
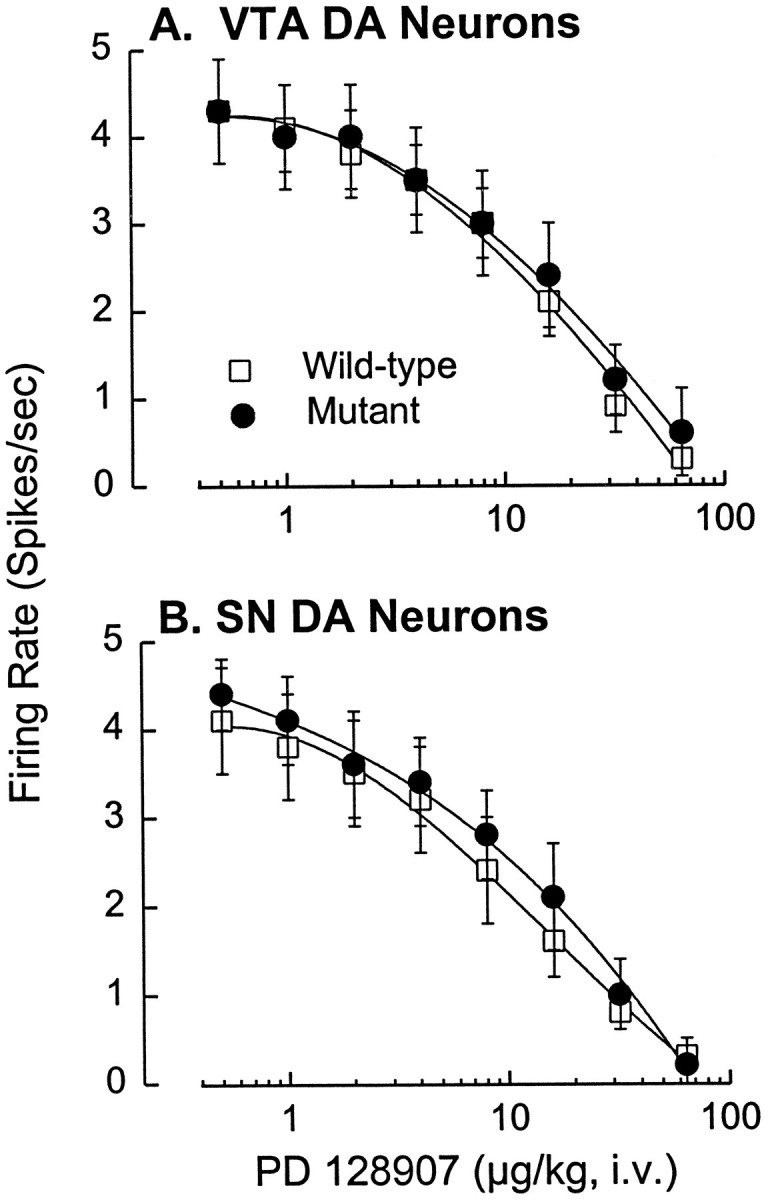
Similar inhibition of midbrain DA neurons by PD 128907 in D3 receptor mutant and wild-type mice.A, Cumulative dose–response curves showing the significant dose-dependent suppression of VTA DA neuronal activity by PD 128907 (F6,120 = 62.5;p < 0.001) and the lack of difference between the wild-type and mutant mice with respect to this effect. There was also no significant difference between the groups with respect to basal firing rates, 4.3 ± 0.6 spikes/sec (mean ± SEM) in wild-type mice (n = 12) and 4.3 ± 0.5 spikes/sec in the mutant mice (n = 10). Eticlopride reversed the inhibition to 85–110% of the basal firing rate in every cell tested. B, Similar dose–response curves indicating dose-dependent inhibition of SN DA neurons by PD 128907 (F6,138 = 51.5; p < 0.001) and the lack of difference between wild-type and mutant mice with respect to this effect. There was also no significant difference in the basal firing rates for SN DA neurons in wild-type (4.1 ± 0.6 spikes/sec; n = 12) and mutant (4.4 ± 0.4 spikes/sec; n = 13) mice. Eticlopride reversed the agonist-induced inhibition to 82–112% of the basal firing rate in every cell tested. Each point represents the mean ± SEM.
Fig. 2.
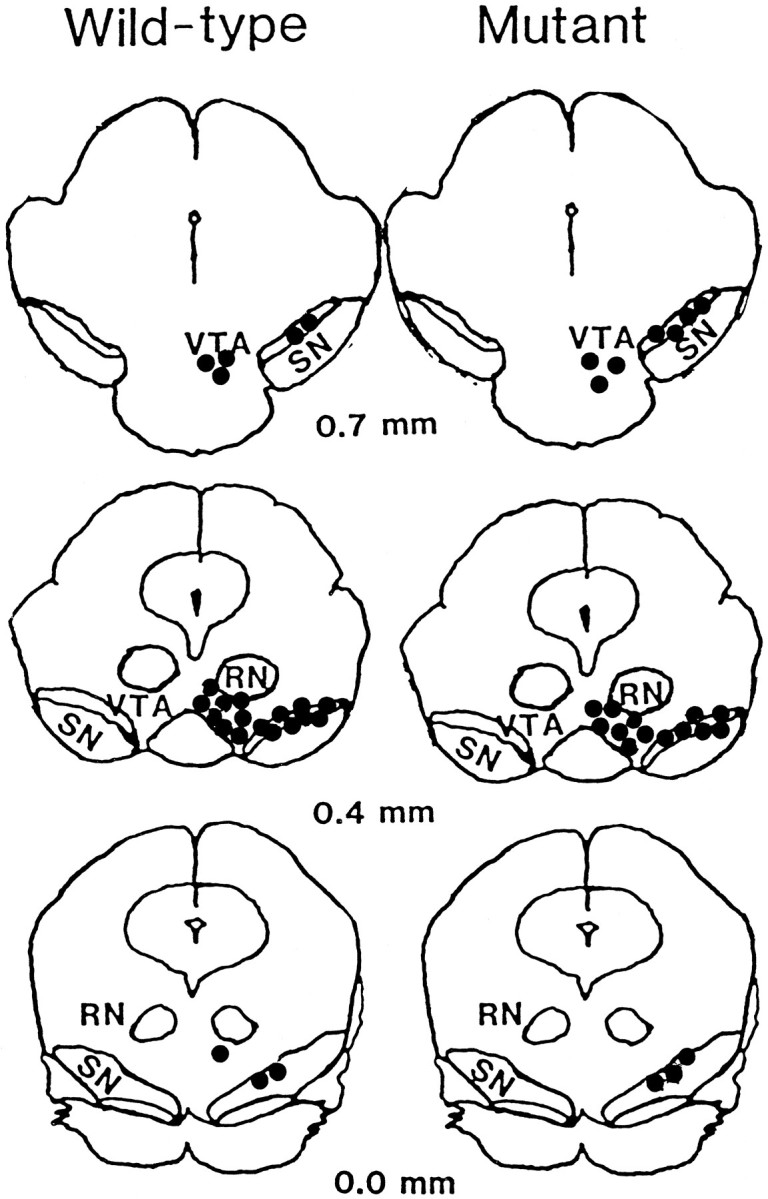
Recording sites for DA neurons within the mouse midbrain. Approximate recording sites (filled circles) for DA neurons within the ventral tegmental area (VTA) and substantia nigra (SN) of wild-type (left) and D3 receptor mutant (right) mice. Coordinates are expressed as anterior to lambda. RN, Red nucleus.
Regulation of DA synthesis is unaltered in D3 receptor mutant mice
We used the GBL model to determine whether the D3receptor null mutation altered control of DA synthesis by nerve terminal autoreceptors. In this model, GBL is used to inhibit impulse flow in DA neurons. Under these conditions, DA agonists can inhibit DA synthesis only via nerve terminal autoreceptors and not via long-loop feedback pathways or stimulation of somatodendritic impulse-modulating DA autoreceptors (Walters and Roth, 1976). Accumulation of the DA precursor DOPA was measured after administration of thel-aromatic amino acid decarboxylase inhibitor NSD 1015 to prevent conversion of DOPA to DA. DOPA accumulation was used as an index of the rate of tyrosine hydroxylation, the rate-limiting step in DA biosynthesis. Studies were performed in both dorsal and ventral striatum (including nucleus accumbens, olfactory tubercles, and islands of Calleja) because of reported differences in D3 receptor expression in these terminal fields (for review, see Sokoloff and Schwartz, 1995).
Administration of GBL (with NSD 1015) increases DOPA formation in DA terminal fields because of a reduction in impulse-dependent DA release from nerve terminals and a resultant reduction in DA autoreceptor stimulation. The magnitude of the GBL-induced increase in DOPA formation therefore provides a measure of the magnitude of ongoing (tonic) suppression of DA synthesis by synthesis-modulating DA autoreceptors. We found that the magnitude of the GBL-induced increase in DOPA formation did not differ between D3 receptor mutant and wild-type mice in either the dorsal or ventral striatum (Fig.3). This argues strongly against a contribution of D3 receptors to tonic autoreceptor-mediated modulation of DA synthesis. Furthermore, PD 128907 was equally potent at reversing GBL-induced DOPA formation in the mutant and wild-type mice (Fig. 4), indicating that PD 128907 does not inhibit DA synthesis by stimulating D3autoreceptors but must do so via D2 autoreceptors. In fact, at the highest dose tested, PD 128907 appeared somewhat more potent in the mutant compared with the wild-type mice (Fig. 4).
Fig. 3.

Similar GBL-induced increase in striatal DOPA formation in D3 receptor mutant and wild-type mice. Basal levels of DOPA, measured after inhibition of l-aromatic amino acid decarboxylase with NSD 1015, did not significantly differ in wild-type and D3 receptor mutant mice in either the ventral or dorsal striatum. GBL significantly increased DOPA formation in both ventral (F1,20 = 58.58;p < 0.001) and dorsal (F1,19 = 261.79; p < 0.001) striatum, as a result of the cessation of impulse-dependent DA release and the relief of tonic autoreceptor-mediated inhibition of tyrosine hydroxylase activity. Again, there were no significant differences in the GBL-induced increase in the two groups of mice. Error bars indicate SEM. Sample size is six for all groups except for the NSD alone group (dorsal striatum in mutants) wheren = 5.
Fig. 4.
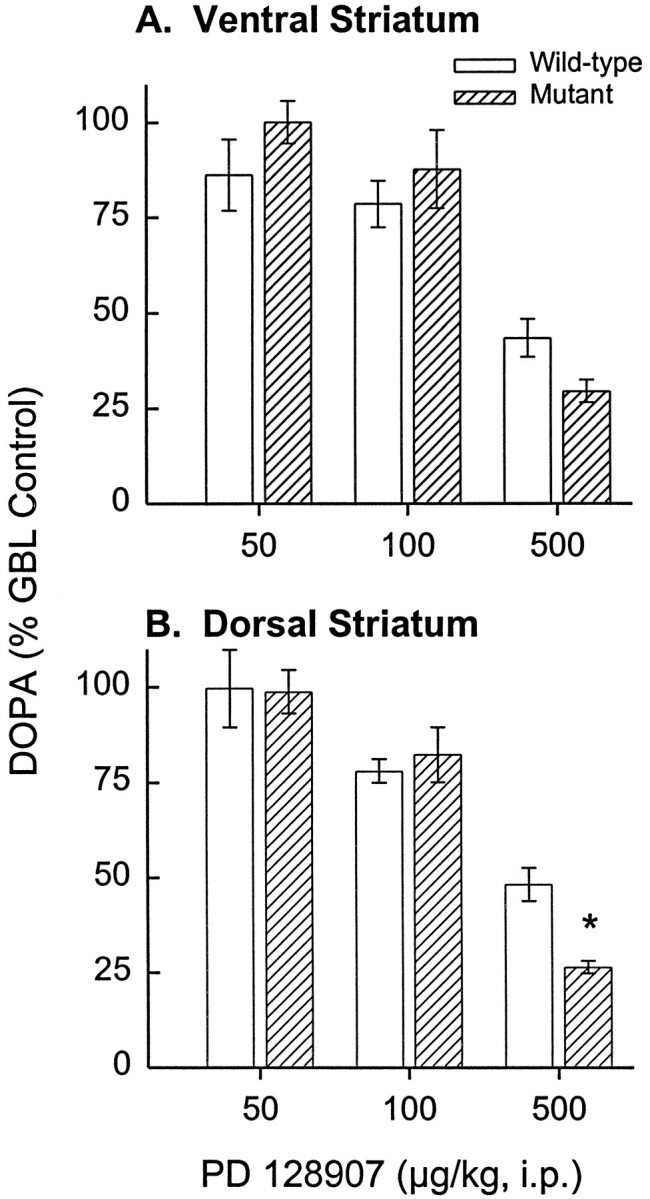
Similar inhibition of GBL-induced striatal DOPA formation by PD 128907 in D3 receptor mutant and wild-type mice. A, Reversal of GBL-induced DOPA synthesis by PD 128907 (F3,57 = 29.97; p< 0.001) in the ventral striatum was not significantly different in D3 receptor mutant and wild-type mice. Data are presented as the percent of DOPA levels measured in mice administered GBL plus NSD without PD 128907 (wild type, 2.51 μg/gm/30 min; mutant, 2.53 μg/gm/30 min). Sample sizes for the 0, 0.05, 0.1, and 0.5 mg/kg doses of PD 128907 are 16, 4, 6, and 6 for the mutant and 19, 3, 5, and 6 for the wild-type mice, respectively. B, Reversal of GBL-induced DOPA synthesis by PD 128907 (F3,57 = 25.23; p < 0.001) in the dorsal striatum was also not significantly different in the two groups of mice; basal levels were 3.26 ng/gm/30 min for the wild-type and 3.40 ng/gm/30 min for the mutant mice. Error bars indicate SEM. Sample sizes for the 0, 0.05, 0.1, and 0.5 mg/kg doses of PD 128907 are 21, 4, 6, and 6 for the mutant and 14, 3, 5, and 5 for the wild-type mice, respectively. Note that the effect of the highest dose of PD 128907 on DA synthesis within the dorsal striatum was actually significantly greater in the mutant than in the wild-type mice (*p < 0.05; Dunnett’s test).
Regulation of DA release, but not PD 128907-induced suppression of DA release, is altered in D3 receptor mutant mice
Extracellular DA levels were measured using in vivomicrodialysis in the ventral striatum of freely moving mice. A representative probe placement is shown in Figure5. To determine basal levels of DA efflux, we collected baseline samples of DA for 1 hr before administration of PD 128907. Basal DA efflux was consistently and significantly higher in mutant mice (Fig.6A). After collection of baseline samples, PD 128907 was applied by reverse dialysis for 1 hr, followed by a 1 hr recovery period. Initial experiments in wild-type mice indicated a small (15%), although statistically significant, decrease in DA efflux during perfusion with 100 nm PD 128907 (data not shown); however, to maximize our ability to detect potential differences between the wild-type and mutant mice, we used a higher concentration of PD 128907 (1 μM). At this concentration, PD 128907 significantly decreased DA efflux in both mutant and wild-type mice, as determined by comparisons of fractions during drug infusion to the weighted mean of three baseline fractions (Fig. 6A). When these data were expressed as the percent of basal DA release (Fig. 6B), an apparent group difference was observed. To determine whether this was attributable to the difference in basal DA efflux or to a blunting of the inhibitory effects of PD 128907 in mutant mice, we performed an analysis of covariance with basal DA as the covariate. The results confirmed the significant difference in basal DA levels but indicated that when this covariate was controlled, there was no significant difference between mutant and wild-type mice with respect to the suppression of DA release by PD 128907, i.e., the absolute decrease in DA release produced by the agonist was not different in the two groups of mice. Nevertheless, it is apparent that there was a lag in the onset of the drug-induced suppression in the mutant mice; thus, marked decreases in DA efflux were observed during the first 20 min of PD 128907 application in the wild-type mice, whereas no effect was observed during this period in the mutants (Fig. 6).
Fig. 5.
Localization of microdialysis probe in the ventral striatum. In this coronal section of the mouse forebrain, thetract left by the microdialysis probe can be seen traversing the ventral striatum. Only the bottom 2 mm of the probe (located between the arrowheads) consisted of open dialysis membrane. NAc, Nucleus accumbens;CPu, caudate-putamen; LV, lateral ventricle; ICj, island of Calleja; ac, anterior commisure; cc, corpus callosum.
Fig. 6.
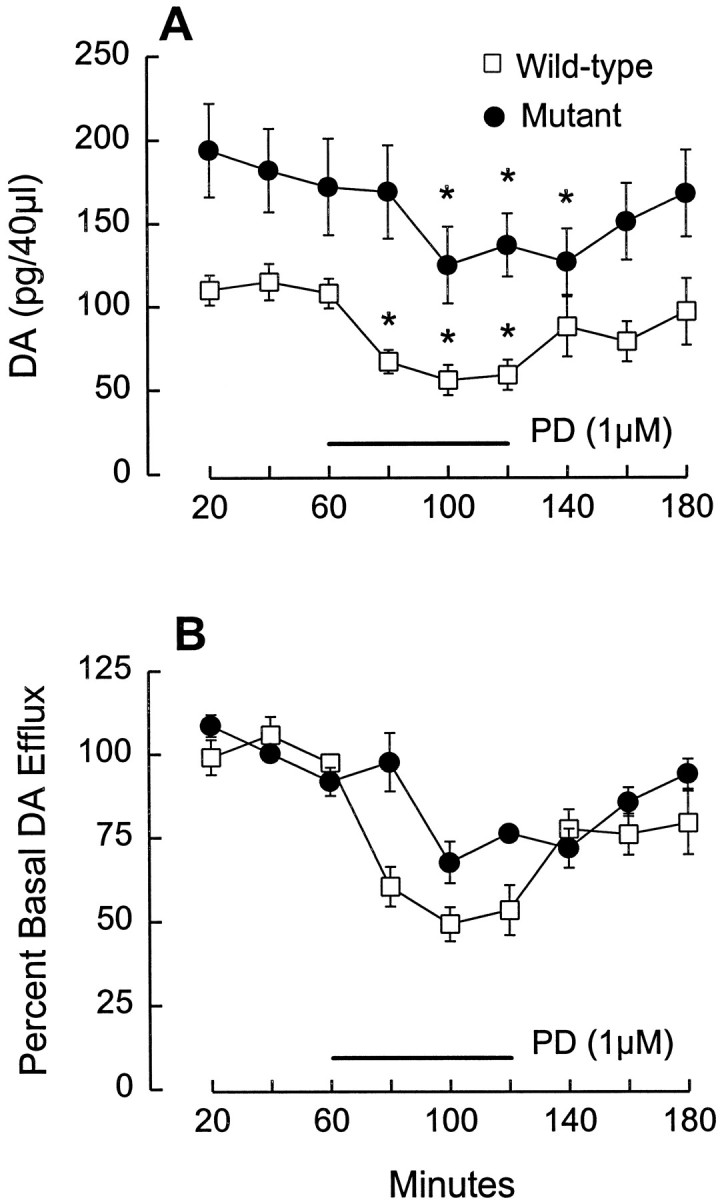
Regulation of basal DA efflux, but not PD 128907-induced suppression of DA release, is altered in D3receptor mutant mice. A, After 2–3 hr of perfusion, three baseline fractions (20 min each) were collected (0–60 min). Basal DA efflux was significantly higher in mutant mice (F1,9 = 26.9; p = 0.0008). Values for basal levels in wild-type mice were ∼13 nm, which is comparable with values obtained in our previously published studies of rat NAc (e.g., see Wolf et al., 1994). PD 128907 was applied for 60 min (horizontal bar), beginning after collection of the third baseline fraction. PD 128907 significantly reduced DA efflux in both wild-type and mutant mice (F1,10 = 7.798; p = 0.019).Asterisks indicate points that differ significantly (p < 0.01; Dunnett’s test) when compared with the weighted mean of the three predrug baseline values. B, When the results shown in A are expressed as percent of basal DA efflux, the inhibitory effect of PD 128907 appears blunted in the mutant mice. However, ANCOVA indicated that this apparent effect was solely attributable to the initial differences in basal DA efflux, i.e., there was no difference between the two groups with respect to the release-suppressing effects of PD 128097 when basal DA levels were controlled as a covariate. Results are expressed as mean ± SEM (n = 6/group).
DISCUSSION
Autoreceptor regulation of impulse flow and DA synthesis is mediated primarily by DA D2 receptors
Our results indicate that D3 receptors may be involved in the regulation of DA release but not via autoreceptor mechanisms. Moreover, D3 receptors do not seem to contribute significantly to functional pools of either somatodendritic impulse-modulating or nerve terminal synthesis- or release-modulating DA autoreceptors. The putative D3 receptor-selective agonist PD 128907 was equally potent at inhibiting DA cell firing and decreasing DA synthesis in D3 receptor mutant mice and their wild-type littermates. Therefore, the ability of PD 128907 and other putative D3 receptor-selective ligands to regulate DA neuronal activity and DA synthesis, demonstrated by many laboratories (see introductory remarks), could not have been by activation of D3 receptors but must have been mediated by the well-accepted D2 autoreceptor population. With respect to impulse-regulating DA autoreceptors, our findings are consistent with a recent report indicating a complete loss of autoreceptor-mediated hyperpolarization and suppression of activity of midbrain DA neurons in mice lacking the DA D2 receptor gene (Mercuri et al., 1997). Taken together, these findings demonstrate the power of the gene knock-out approach in the identification of functions mediated by individual members of a receptor family.
Our findings stand in contrast to recent reports in which antisense oligodeoxynucleotides have been used to reduce D3 receptor expression (Nissbrandt et al., 1995; Tepper et al., 1997). Nissbrandt and colleagues (1995) found that intracerebroventricular administration of a D3 receptor antisense oligodeoxynucleotide enhanced basal levels of DA synthesis in the nucleus accumbens but failed to affect the suppression of synthesis by the nonselective DA receptor agonist apomorphine. More recently, Tepper and coworkers (1997)reported a nearly equivalent (50%) reduction in the inhibitory effects of apomorphine on the activity of rat SN DA neurons after intra-SN infusion of antisense oligodeoxynucleotides complementary to the initial coding region of either the DA D2 or D3receptor. Despite considerably greater expression of D2than of D3 receptors in the SN, Tepper et al. (1997) argued that D3 receptor coupling to transduction events may be more efficient such that D2 and D3 receptors play equivalent functional roles as DA autoreceptors.
The results of these antisense oligodeoxynucleotide studies raise the possibility that in our D3 receptor mutant mice, D2 receptors had compensated for the loss of D3receptors and thereby masked any functional loss in our assays. If this is true, then it is interesting that the reciprocal does not seem to be true, i.e., that D3 receptors are not able to compensate for the loss of D2 receptors (Mercuri et al., 1997). This would suggest that D2 receptors are capable of autoregulation independently, whereas D3 receptor autoregulatory activity would be dependent on concurrent D2autoreceptor stimulation. Although there are no definitive findings that eliminate the possibility of compensation of D3receptor function by D2 autoreceptors, we think that this explanation is unlikely because of the following: (1) such compensation would have to be precise given the almost identical effects observed in the mutant and wild-type mice during electrophysiological and neurochemical experiments, (2) such compensation would have to be specific to certain receptor functions given that differences between mutant and wild-type mice were observed in basal DA efflux and in certain behavioral measures (Accili et al., 1996; Xu et al., 1997), (3) there are no increases in D2 receptor binding densities caused by the D3 receptor mutation (Accili et al., 1996; Xu et al., 1997), (4) knock-out of the DA D2 receptor leads to a complete loss of autoreceptor function in the SN (Mercuri et al., 1997), and (5) there have been no documented cases of compensatory upregulation of related receptor subtypes after deletion of G-protein coupled receptors (for review, see Tecott et al., 1996).
It is also possible that the difference between findings from antisense knock-down and genetic knock-out studies reflects differences between mice and rats with respect to D3 receptor contributions to autoregulation. However, there does not seem to be a difference in the levels of D3 receptors expressed in the midbrain of these two species (Mercuri et al., 1997). Moreover, recent studies have demonstrated that putative D3 autoreceptor effects in rats are in fact caused by D2 receptors, including hyperpolarization and suppression of DA neuron activity (Bowery et al., 1996). Given the low abundance and highly restricted expression of D3 mRNA by DA neurons (Diaz et al., 1995), it is certainly possible that a small number of DA neurons express functional D3 autoreceptors. However, any autoreceptor role occurring in such a small neuronal population is unlikely to exert significant impact on the in vivo activity of the mesotelencephalic DA systems as a whole, unless such a role is accomplished by low levels of D3 receptors the mRNA of which is below the detection limit of current in situ hybridization (Bouthenet et al., 1991;Diaz et al., 1995), reverse transcriptase-PCR (Valerio et al., 1994), and ribonuclease protection (Richtand et al., 1995) assays. Although we cannot exclude this possibility and the findings of Tepper et al. (1997) certainly support it, we believe the bulk of the evidence argues that D2 receptors are primarily responsible for autoregulation of DA neuronal function.
D3 receptors may contribute to local regulation of DA release
Our microdialysis studies indicated a significant increase in basal DA efflux in the ventral striatum of D3 receptor mutant mice compared with their wild-type littermates. Although “no-net flux” dialysis studies are often needed to quantify differences in basal transmitter efflux in vivo (for review, see Justice, 1993), the difference between D3 receptor mutant and wild-type mice was quite robust, with all but one mutant mouse showing higher basal DA efflux than any of the wild-type mice. Increased basal DA efflux might be expected if nerve terminal D3 autoreceptors normally exert a tonic inhibitory influence on DA release. However, the results obtained with PD 128907 are incompatible with this simple interpretation because this putative D3 receptor-selective agonist inhibited DA release by the same absolute amount in D3 receptor mutant and wild-type mice. Thus, no reduction of DA autoreceptor modulation occurred as a result of the D3 receptor mutation. Although the percent decrease caused by PD 128907 was smaller in the mutant mice, this effect was attributable to their higher basal DA levels, a factor known to reduce the ability of DA agonists to suppress DA release (Cubeddu and Hoffman, 1982; Dwoskin and Zahniser, 1986; for review, see Wolf and Roth, 1987).
An alternative explanation for our findings is that DA release is normally modulated both by D2 release-modulating autoreceptors and by negative feedback pathways engaged by postsynaptic D3 (and other D2-class) receptors. Loss of D3 receptor-mediated inhibitory feedback would explain increased basal DA efflux, whereas activation of D2release-modulating autoreceptors by PD 128907 would explain the normal inhibitory effects on DA release. This model is consistent with the D3 mRNA findings indicating that D3 receptors are distributed primarily within target neurons of the ascending DA systems. In ventral striatal terminal fields where the D3receptor is most highly expressed (nucleus accumbens shell, olfactory tubercle, and islands of Calleja), there is good agreement between levels of D3 receptor mRNA and D3 receptor binding sites (Diaz et al., 1995), suggesting that most D3receptors exist on dendrites, soma, or local terminals of intrinsic neurons. If postsynaptic D3 receptors are coupled to feedback pathways that normally exert an inhibitory influence on DA release, the loss of such feedback might lead to increased basal DA efflux but leave D2 autoreceptor-mediated effects intact.
Negative feedback pathways engaged by postsynaptic D3receptors might involve retrograde messengers, short-loop paths (either mono- or multisynaptic) terminating on DA terminals, or striatomesencephalic (long-loop) projections regulating impulse flow at the level of DA cell bodies and thereby influencing impulse-dependent DA release from nerve terminals. We do not favor the latter mechanism because (1) we observed no differences in basal firing rates of DA neurons in mutant and wild-type mice, and (2) previous work has shown that local application of DA agonists by reverse dialysis inhibits DA release via local mechanisms and not via long-loop feedback effects on DA impulse flow (Timmerman et al., 1990). As for the first two possibilities, it has long been known that intrinsic striatal neurotransmitters (e.g., acetylcholine, GABA, enkephalin, and substance P) as well as corticostriatal afferents (e.g., glutamate) may modulate DA release (for review, see Chesselet, 1984), and similar effects of the retrograde messenger nitric oxide have recently been demonstrated (Lonart et al., 1993). DA agonists, when applied locally by reverse dialysis, could conceivably engage such local mechanisms, in addition to exerting effects via nerve terminal DA autoreceptors. Indeed, the slower onset of the PD 128907-induced suppression of DA release in the mutants suggests that at least two processes, one of which is faster and involves D3 receptors, are responsible for this effect in the wild-type mice.
Conclusions
Our findings are incongruent with recent claims that D3 receptors function as DA autoreceptors. D3receptors may contribute to regulation of DA release, but the mechanism may involve postsynaptically activated feedback as opposed to nerve terminal autoreceptors. If so, then D3 receptors would not only be subject to anterograde regulation by unknown factors released by DA neurons (Lévesque et al., 1995) but might also engage feedback mechanisms to control DA release. In conclusion, the results obtained with D3 mutant mice support a large body of earlier work suggesting that D2 receptors constitute the vast majority of DA autoreceptors and that the ability of D3-preferring ligands to alter these functions in vivo, as demonstrated by numerous studies in normal rats (see introductory remarks), reflects their lack of selectivity for D3 receptors and must be attributed to stimulation of D2 receptors.
Footnotes
This work was supported by United States Public Health Service Grants DA07735 (M.E.W.) and DA04093 (F.J.W.) and by the Shionogi Institute for Medical Sciences (S.T.). F.J.W. is a recipient of a Research Scientist Development Award DA00207 from the National Institute on Drug Abuse. T.E.K. (DA 05815) and D.C.C. (DA 05794) are supported by National Research Service awards from the National Institute on Drug Abuse. We thank Lorinda Baker and Chang-Jiang Xue for excellent technical assistance.
Correspondence should be addressed to Dr. Francis J. White, Finch University of Health Sciences, Chicago Medical School, Department of Neuroscience, 3333 Green Bay Road, North Chicago, IL 60064-3095.
Dr. Xu’s present address: Department of Cell Biology, Neurobiology, and Anatomy, University of Cincinnati College of Medicine, 231 Bethesda Avenue, Cincinnati, OH 45267-0521.
REFERENCES
- 1.Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, Gauda EB, Lee EJ, Cool MH, Sibley DR, Gerfen CR, Westphal H, Fuchs S. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci USA. 1996;93:1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aghajanian GK, Bunney BS. Dopamine “autoreceptors”: pharmacological characterization by microiontophoretic single unit recording studies. Naunyn Schmiedebergs Arch Pharmacol. 1977;297:1–7. doi: 10.1007/BF00508803. [DOI] [PubMed] [Google Scholar]
- 3.Ahlenius S, Salmi P. Behavioral and biochemical effects of the dopamine D3 receptor-selective ligand, 7-OH-DPAT, in the normal and the reserpine-treated rat. Eur J Pharmacol. 1994;260:177–181. doi: 10.1016/0014-2999(94)90335-2. [DOI] [PubMed] [Google Scholar]
- 4.Aretha CW, Sinha A, Galloway MP. Dopamine D3-preferring ligands act at synthesis modulating autoreceptors. J Pharmacol Exp Ther. 1995;274:609–613. [PubMed] [Google Scholar]
- 5.Bouthenet M-L, Souil E, Martres M-P, Sokoloff P, Giros B, Schwartz J-C. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res. 1991;564:203–219. doi: 10.1016/0006-8993(91)91456-b. [DOI] [PubMed] [Google Scholar]
- 6.Bowery BJ, Razzaque Z, Emms F, Patel S, Freedman S, Bristow L, Kulagowski J, Seabrook GR. Antagonism of the effects of (+)-PD 128907 on midbrain dopamine neurones in rat brain slices by a selective D2 receptor antagonist L-741,626. Br J Pharmacol. 1996;119:1491–1497. doi: 10.1111/j.1476-5381.1996.tb16063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunney BS, Walters JR, Roth RH, Aghajanian GK. Dopaminergic neurons: effect of antipsychotic drugs and amphetamine on single cell activity. J Pharmacol Exp Ther. 1973;185:560–571. [PubMed] [Google Scholar]
- 8.Burris KD, Pacheco MA, Filtz TM, Kung M-P, Kung HF, Molinoff PB. Lack of discrimination by agonists for D2 and D3 dopamine receptors. Neuropsychopharmacology. 1995;12:335–345. doi: 10.1016/0893-133X(94)00099-L. [DOI] [PubMed] [Google Scholar]
- 9.Chesselet M-F. Presynaptic regulation of neurotransmitter release in the brain: facts and hypothesis. Neuroscience. 1984;12:347–375. doi: 10.1016/0306-4522(84)90058-7. [DOI] [PubMed] [Google Scholar]
- 10.Chio CL, Lajiness ME, Huff RM. Activation of heterologously expressed D3 dopamine receptors: comparison with D2 dopamine receptors. Mol Pharmacol. 1994;45:51–60. [PubMed] [Google Scholar]
- 11.Clark D, White FJ. Review: D1 dopamine receptor–the search for a function: a critical evaluation of the D1/D2 dopamine receptor classification and its functional implications. Synapse. 1987;1:347–388. doi: 10.1002/syn.890010408. [DOI] [PubMed] [Google Scholar]
- 12.Cubeddu LX, Hoffman IS. Operational characteristics of the inhibitory feedback mechanism for regulation of dopamine release via presynaptic receptors. J Pharmacol Exp Ther. 1982;223:497–501. [PubMed] [Google Scholar]
- 13.Damsma G, Bottema T, Westerink BHC, Tepper PG, Dijkstra D, Pugsley TA, Mackenzie RG, Heffner TG, Wikström H. Pharmacological aspects of R-(+)-7-OH-DPAT, a putative dopamine D3 receptor ligand. Eur J Pharmacol. 1993;249:R9–R10. doi: 10.1016/0014-2999(93)90533-n. [DOI] [PubMed] [Google Scholar]
- 14.Devoto P, Collu M, Muntoni AL, Pistis M, Serra G, Gessa GL, Diana M. Biochemical and electrophysiological effects of 7-OH-DPAT on the mesolimbic dopaminergic system. Synapse. 1995;20:153–155. doi: 10.1002/syn.890200209. [DOI] [PubMed] [Google Scholar]
- 15.Diaz J, Lévesque D, Lammers CH, Griffon N, Martres M-P, Schwartz J-C, Sokoloff P. Phenotypical characterization of neurons expressing the dopamine D3 receptor in the rat brain. Neuroscience. 1995;65:731–745. doi: 10.1016/0306-4522(94)00527-c. [DOI] [PubMed] [Google Scholar]
- 16.Dwoskin LP, Zahniser NR. Robust modulation of [3H]dopamine release from rat striatal slices by D-2 dopamine receptors. J Pharmacol Exp Ther. 1986;239:442–453. [PubMed] [Google Scholar]
- 17.Farnebo L-O, Hamberger B. Drug-induced changes in the release of 3H-monoamines from field stimulated rat brain slices. Acta Physiol Scand Suppl. 1971;371:35–44. doi: 10.1111/j.1748-1716.1971.tb05213.x. [DOI] [PubMed] [Google Scholar]
- 18.Gainetdinov RR, Grekhova TV, Sotnikova TD, Rayevsky KS. Dopamine D2 and D3 receptor preferring antagonists differentially affect striatal release and metabolism in conscious rats. Eur J Pharmacol. 1994;261:327–331. doi: 10.1016/0014-2999(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 19.Gainetdinov RR, Sotnikova TD, Grekhova TV, Rayevsky KS. In vivo evidence for preferential role of dopamine D3 receptor in the presynaptic regulation of dopamine release but not synthesis. Eur J Pharmacol. 1996;308:261–269. doi: 10.1016/0014-2999(96)00300-7. [DOI] [PubMed] [Google Scholar]
- 20.Galloway MP, Wolf ME, Roth RH. Regulation of dopamine synthesis in the medial prefrontal cortex is mediated by release modulating autoreceptors: studies in vivo. J Pharmacol Exp Ther. 1986;236:689–698. [PubMed] [Google Scholar]
- 21.Gilbert DB, Millar J, Cooper SJ. The putative dopamine D3 agonist, 7-OH-DPAT, reduces dopamine release in the nucleus accumbens and electrical self-stimulation to the ventral tegmentum. Brain Res. 1995;681:1–7. doi: 10.1016/0006-8993(95)00247-n. [DOI] [PubMed] [Google Scholar]
- 22.Gobert A, Rivet JM, Audinot V, Cistarelli L, Spedding M, Vian J, Peglion JL, Millan MJ. Functional correlates of dopamine D3 receptor activation in the rat in vivo and their modulation by the selective antagonist, (+)-S 14297. 2. Both D2 and “Silent” D3 autoreceptors control synthesis and release in mesolimbic, mesocortical and nigrostriatal pathways. J Pharmacol Exp Ther. 1995;275:899–913. [PubMed] [Google Scholar]
- 23.Gobert A, Lejeune F, Rivet JM, Cistarelli L, Millan MJ. Dopamine D3 (auto) receptors inhibit dopamine release in the frontal cortex of freely moving rats in vivo. J Neurochem. 1996;66:2209–2212. doi: 10.1046/j.1471-4159.1996.66052209.x. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez AM, Sibley DR. [3H]7-OH-DPAT is capable of labeling dopamine D2 as well as D3 receptors. Eur J Pharmacol. 1995;272:R1–R3. doi: 10.1016/0014-2999(94)00738-s. [DOI] [PubMed] [Google Scholar]
- 25.Healy DJ, Meador-Woodruff JH. Differential regulation, by MK-801, of dopamine receptor gene expression in rat nigrostriatal and mesocorticolimbic systems. Brain Res. 1996;708:38–44. doi: 10.1016/0006-8993(95)01241-9. [DOI] [PubMed] [Google Scholar]
- 26.Justice JB., Jr Quantitative microdialysis of neurotransmitters. J Neurosci Methods. 1993;48:263–276. doi: 10.1016/0165-0270(93)90097-b. [DOI] [PubMed] [Google Scholar]
- 27.Kehr W, Carlsson A, Lindqvist M, Magnusson T, Atack C. Evidence for a receptor-mediated feedback control of striatal tyrosine hydroxylase activity. J Pharm Pharmacol. 1972;24:744–746. doi: 10.1111/j.2042-7158.1972.tb09104.x. [DOI] [PubMed] [Google Scholar]
- 28.Kreiss DS, Bergstrom DA, Gonzalez AM, Huang K-X, Sibley DR, Walters JR. Dopamine receptor agonist potencies for inhibition of cell firing correlate with dopamine D3 receptor binding affinities. Eur J Pharmacol. 1995;277:209–214. doi: 10.1016/0014-2999(95)00069-w. [DOI] [PubMed] [Google Scholar]
- 29.Landwehrmeyer B, Mengod G, Palacios JM. Differential visualization of dopamine D2 and D3 receptor sites in rat brain. A comparative study using in situ hybridization histochemistry and ligand binding autoradiography. Eur J Neurosci. 1993;5:145–153. doi: 10.1111/j.1460-9568.1993.tb00480.x. [DOI] [PubMed] [Google Scholar]
- 30.Large CH, Stubbs CM. The dopamine D3 receptor: Chinese hamsters or Chinese whispers. Trends Pharmacol. 1994;15:46–47. doi: 10.1016/0165-6147(94)90108-2. [DOI] [PubMed] [Google Scholar]
- 31.Lejeune F, Millan MJ. Activation of dopamine D3 autoreceptors inhibits firing of ventral tegmental dopaminergic neurones in vivo. Eur J Pharmacol. 1995;275:R7–R9. doi: 10.1016/0014-2999(95)00106-u. [DOI] [PubMed] [Google Scholar]
- 32.Lévesque D, Martres M-P, Diaz J, Griffon N, Lammers CH, Sokoloff P, Schwartz J-C. A paradoxical regulation of the dopamine D3 receptor expression suggests the involvement of an anterograde factor from dopamine neurons. Proc Natl Acad Sci USA. 1995;92:1719–1723. doi: 10.1073/pnas.92.5.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lonart G, Cassels KL, Johnson KM. Nitric oxide induces calcium-dependent [3H]dopamine release from striatal slices. J Neurosci Res. 1993;35:192–198. doi: 10.1002/jnr.490350210. [DOI] [PubMed] [Google Scholar]
- 34.Mansour A, Meador-Woodruff JH, Bunzow JR, Civelli O, Akil H, Watson SJ. Localization of dopamine D2 receptor mRNA and D1 and D2 receptor binding in the rat brain and pituitary: an in situ hybridization-receptor autoradiographic analysis. J Neurosci. 1990;10:2587–2600. doi: 10.1523/JNEUROSCI.10-08-02587.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meador-Woodruff JH, Mansour A, Bunzow JR, Van Tol HHM, Watson SJ, Civelli O. Distribution of D2 dopamine receptor mRNA in rat brain. Proc Natl Acad Sci USA. 1989;86:7625–7628. doi: 10.1073/pnas.86.19.7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meador-Woodruff JH, Damask SP, Watson SJ., Jr Differential expression of autoreceptors in the ascending dopamine systems of the human brain. Proc Natl Acad Sci USA. 1994;91:8297–8301. doi: 10.1073/pnas.91.17.8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meller E, Bohmaker K, Goldstein M, Basham DA. Evidence that striatal synthesis-inhibiting autoreceptors are dopamine D3 receptors. Eur J Pharmacol. 1993;249:R5–R6. doi: 10.1016/0014-2999(93)90674-7. [DOI] [PubMed] [Google Scholar]
- 38.Mengod G, Martinez-Mir MI, Vilaro MT, Palacios JM. Localization of the mRNA for the dopamine D2 receptor in the rat brain by in situ hybridization histochemistry. Proc Natl Acad Sci USA. 1989;86:8560–8564. doi: 10.1073/pnas.86.21.8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience. 1997;79:323–327. doi: 10.1016/s0306-4522(97)00135-8. [DOI] [PubMed] [Google Scholar]
- 40.Nissbrandt H, Ekman A, Eriksson E, Heilig M. Dopamine D3 receptor antisense influences dopamine synthesis in rat brain. NeuroReport. 1995;6:573–576. doi: 10.1097/00001756-199502000-00041. [DOI] [PubMed] [Google Scholar]
- 41.O’Hara CM, Uhland-Smith A, O’Malley KL, Todd RD. Inhibition of dopamine synthesis by dopamine D2 and D3 but not D4 receptors. J Pharmacol Exp Ther. 1996;277:186–192. [PubMed] [Google Scholar]
- 42.Potenza MN, Graminski GF, Schmauss C, Lerner MR. Functional expression and characterization of human D2 and D3 dopamine receptors. J Neurosci. 1994;14:1463–1476. doi: 10.1523/JNEUROSCI.14-03-01463.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pugsley TA, Davis MD, Akunne HC, Mackenzie RG, Shih YH, Damsma G, Wikstrom H, Whetzel SZ, Georgic LM, Cooke LW, Demattos SB, Corbin AE, Glase SA, Wise LD, Dijkstra D, Heffner TG. Neurochemical and functional characterization of the preferentially selective dopamine D3 agonist PD 128907. J Pharmacol Exp Ther. 1995;275:1355–1366. [PubMed] [Google Scholar]
- 44.Richtand NM, Kelsoe JR, Segal DS, Kuczenski R. Regional quantification of D1, D2, and D3 dopamine receptor mRNA in rat brain using a ribonuclease protection assay. Mol Brain Res. 1995;33:97–103. doi: 10.1016/0169-328x(95)00112-6. [DOI] [PubMed] [Google Scholar]
- 45.Rivet J-M, Audinot V, Gobert A, Peglion J-L, Millan MJ. Modulation of mesolimbic dopamine release by the selective dopamine D3 receptor antagonist, (+)-S 14297. Eur J Pharmacol. 1994;265:175–177. doi: 10.1016/0014-2999(94)90429-4. [DOI] [PubMed] [Google Scholar]
- 46.Routledge C, Thorn L, Ashmeade T, Taylor S. Elucidation of D3 receptor function in vivo: do D3 receptors mediate inhibition of dopamine neuronal activity? Biochem Soc Trans. 1996;24:199–201. doi: 10.1042/bst0240199. [DOI] [PubMed] [Google Scholar]
- 47.Sanghera MK, Trulson ME, German DC. Electrophysiological properties of mouse dopamine neurons: in vivo and in vitro studies. Neuroscience. 1984;12:793–801. doi: 10.1016/0306-4522(84)90171-4. [DOI] [PubMed] [Google Scholar]
- 48.Sautel F, Griffon N, Lévesque D, Pilon C, Schwartz J-C, Sokoloff P. A functional test identifies dopamine agonists selective for D3 versus D2 receptors. NeuroReport. 1995;6:329–332. doi: 10.1097/00001756-199501000-00026. [DOI] [PubMed] [Google Scholar]
- 49.Sokoloff P, Schwartz J-C. Novel dopamine receptors half a decade later. Trends Pharmacol. 1995;16:270–275. doi: 10.1016/s0165-6147(00)89044-6. [DOI] [PubMed] [Google Scholar]
- 50.Sokoloff P, Giros B, Martres M-P, Bouthenet M-L, Schwartz J-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 51.Svensson K, Johansson AM, Magnusson T, Carlsson A. (+)-AJ 76 and (+)-UH 232: central stimulants acting as preferential dopamine autoreceptor antagonists. Naunyn Schmiedebergs Arch Pharmacol. 1986;334:234–245. doi: 10.1007/BF00508777. [DOI] [PubMed] [Google Scholar]
- 52.Tang L, Todd RD, O’Malley KL. Dopamine D2 and D3 receptors inhibit dopamine release. J Pharmacol Exp Ther. 1994;270:475–479. [PubMed] [Google Scholar]
- 53.Tecott LH, Brennan TJ, Guh L. Gene targeting approaches to analyze neurotransmitter receptor function. Semin Neurosci. 1996;8:145–152. [Google Scholar]
- 54.Tepper JM, Sun BC, Martin LP, Creese I. Functional roles of dopamine D2 and D3 autoreceptors on nigrostriatal neurons analyzed by antisense knockdown in vivo. J Neurosci. 1997;17:2519–2530. doi: 10.1523/JNEUROSCI.17-07-02519.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Timmerman W, De Vries JB, Westerink BHC. Effects of D-2 agonists on the release of dopamine: localization of the mechanism of action. Naunyn Schmiedebergs Arch Pharmacol. 1990;342:650–654. doi: 10.1007/BF00175707. [DOI] [PubMed] [Google Scholar]
- 56.Valerio A, Belloni M, Gorno ML, Tinti C, Memo M, Spano P. Dopamine D2, D3, and D4 receptor mRNA levels in rat brain and pituitary during aging. Neurobiol Aging. 1994;15:713–719. doi: 10.1016/0197-4580(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 57.Walters JR, Roth RH. Dopaminergic neurons: an in vivo system for measuring drug interactions with presynaptic receptors. Naunyn Schmiedebergs Arch Pharmacol. 1976;296:5–14. doi: 10.1007/BF00498834. [DOI] [PubMed] [Google Scholar]
- 58.Wang RY. Dopaminergic neurons in the rat ventral tegmental area. I. Identification and characterization. Brain Res Rev. 1981;3:123–140. [Google Scholar]
- 59.White FJ, Hu X-T, Zhang X-F, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther. 1995;273:445–454. [PubMed] [Google Scholar]
- 60.Wolf ME, Roth RH. Dopamine autoreceptors. In: Creese I, Fraser CM, editors. Dopamine receptors. Liss; New York: 1987. pp. 45–96. [Google Scholar]
- 61.Wolf ME, White FJ, Hu X-T. MK-801 prevents alterations in the mesoaccumbens dopamine system associated with behavioral sensitization to amphetamine. J Neurosci. 1994;14:1735–1745. doi: 10.1523/JNEUROSCI.14-03-01735.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu M, Hu X-T, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S. Elimination of cocaine-induced hyperactivity and dopamine-mediated neurophysiological effects in dopamine D1 receptor deficient mice. Cell. 1994;79:945–955. doi: 10.1016/0092-8674(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 63.Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu X-T, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:838–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]
- 64.Xue CJ, Ng JP, Li Y, Wolf ME. Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem. 1996;67:352–363. doi: 10.1046/j.1471-4159.1996.67010352.x. [DOI] [PubMed] [Google Scholar]