

Abstract
In the spontaneous ataxic mutant mouse stargazer, there is a selective reduction of brain-derived neurotrophic factor (BDNF) mRNA expression in the cerebellum. BDNF protein levels in the cerebellum are reduced by 70%. Despite normal levels of full-length and truncated TrkB receptor, constitutive and neurotrophin-4/5-induced tyrosine phosphorylation was significantly reduced in several signal transduction molecules, including phospholipase-Cγ1, erk1, and erk2. Morphological examination revealed an increased number of external granule cells at postnatal day 15 and the presence of abnormal neurons resembling immature granule cells in the adult. These abnormalities are associated with a severe impairment in the acquisition of classical eyeblink conditioning, indicating cerebellar malfunction. Our data suggest that normal BDNF expression and TrkB signal transduction in the cerebellum are necessary for learning and plasticity in this model.
Keywords: mutant mice, neurotrophic factor, BDNF, TrkB, signal transduction, eyeblink conditioning, learning, cerebellum
Neurotrophic factors are important regulators of the formation and function of the nervous system during development and are required for maintenance in the adult brain. The family of the neurotrophins includes nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and the neurotrophins (NTs) 3, 4/5, and 6. Although most of the neurotrophins are highly expressed in immature regions of the nervous system during development, the regulation of BDNF expression is most prominent in brain regions in which neurogenesis has already occurred (Maisonpierre et al., 1990). The BDNF level generally increases as these regions mature and remains the most abundant neurotrophin in the adult brain (Leibrock et al., 1989; Maisonpierre et al., 1990). In mouse and rat, the histogenesis and morphogenesis of cerebellum primarily occur within the first postnatal month (Jacobson, 1991; Altman and Bayer, 1997). Correspondingly, BDNF mRNA expression in the cerebellum is not apparent until postnatal day 15 (P15) but then rises quickly and peaks at approximately P20 in rat (Rocamora et al., 1993) and mouse (Qiao et al., 1996), coinciding with granule cell migration and maturation.
The cerebellum plays an important role in movement and posture. Malformation and lesions of the cerebellum disrupt motor coordination and impair balance. In addition to the classical role in the fine motor control, increasing evidence has shown that the cerebellum is also involved in a variety of nonmotor cognitive functions, including sensory discrimination (Gao et al., 1996), attention (Allen et al., 1997), and learning and memory (Thompson, 1986; Andreasen et al., 1995). Classical eyeblink conditioning is a form of sensorimotor learning that depends crucially on the cerebellum (Thompson, 1990; Yeo, 1991). More recent studies in several spontaneous and transgenic mutant mice have shown the importance of both the cerebellar cortex and interpositus nucleus in this learning paradigm (Chen et al., 1995,1996; Shibuki et al., 1996; Kim and Thompson, 1997). Because the neural circuitry involved in this form of learning has been well characterized (Thompson, 1986, 1990), the eyeblink conditioning paradigm allows us to screen for the cellular and molecular substrates, which are required for cerebellar function and plasticity.
Our knowledge of BDNF function in vivo primarily comes from studies of BDNF knock-out transgenic mice (Ernfors et al., 1994;Conover et al., 1995; Liu et al., 1995). However, the high early postnatal mortality and the universal deletion of the gene make it difficult to determine the exact role of BDNF on a specific neuronal type in the brain, especially at late developmental stages. We have reported previously a specific and pronounced deficit in BDNF mRNA expression in the cerebellum of the stargazer(stg) mouse (Qiao et al., 1996), a spontaneous mutant with ataxia and generalized spike-wave seizures. Other brain areas of this mouse express normal levels of BDNF, and mRNA levels for NGF and NT-3 were normal throughout the brain, including the cerebellum. We took advantage of this unique mutant mouse model and its region-specific defect of BDNF expression to study the role of BDNF in cerebellar development and function.
MATERIALS AND METHODS
Animals. Wild-type (C57BL/6J; +/+) and mutant (C3B6Fe+; stg/stg) mice were obtained from the breeding colonies of The Jackson Laboratory (Bar Harbor, ME) and maintained in the University of Southern California Gerontology Center vivarium on a 12 hr light/dark cycle with food and water available ad libitum. Heterozygous males (+/stg) and homozygous females (stg/stg) were mated to produce stgmutant mice. Because we have not found any significant phenotype difference so far between +/+ and +/stg, nonmutant littermates (+/−) including both genotypes were used in developmental and behavioral studies as wild-type controls.
BDNF enzyme immunoassay. For BDNF enzyme immunoassay (EIA), we used modified techniques developed by Nawa et al. (1995). The cerebellar tissues were isolated from adult +/+ and stg/stgmice (n = 3), weighed, and immediately frozen on dry ice. The tissues were homogenized for 60 sec with 100 volumes of homogenization buffer containing 50 mm Tris, 0.3m NaCl, 0.1% Triton X-100, 1% bovine serum albumin (Corn Fraction V; Sigma, St. Louis, MO), aprotinin (200 kallikrein inhibitor units/ml), 0.1 mm phenylmethanesulfonyl fluoride, 0.1 mm benzethonium chloride, and 1 mm benzamidine. Homogenates were centrifuged for 20 min at 15,000 × gat 4°C, and supernatants were stored at −70°C until use. Anti-BDNF antibodies were kindly provided by Josette Carnahan (Amgen, Thousand Oaks, CA). Polystyrene 96-well microtiter plates (Corning, Corning, NY) were coated with 100 ng/well low-affinity antibody in 0.1 mTris buffer, pH 9.0, for 12–18 hr and then blocked with EIA buffer containing 50 mm Tris, 0.3 m NaCl, 0.05% Thimosal, 0.1% Triton X-100, 1% BSA, and 1% gelatin, pH 7.5, at 4°C overnight. One hundred microliters of tissue extracts or BDNF standards (recombinant human BDNF; Peprotech, Rocky Hill, NJ) in EIA buffer were loaded to wells for 12–18 hr. After washing with EIA buffer without BSA, 100 μl of biotinylated high-affinity anti-BDNF antibody (10 ng/ml) in EIA buffer were added and incubated for 12–18 hr at room temperature. Bound biotinylated secondary antibodies were detected by incubation with streptavidin/peroxidase (1:250) for 3 hr. After extensive wash, the enzyme activity retained in each well was measured by incubation with 200 μl of 3,3′,5,5′-tetramethylbenzidine substrates for 30 min. The blue end product was read on a SPECTRAmax 250 microplate spectrophotometer (Molecular Devices, Sunnyvale, CA) at 370 and 655 nm using the SOFTmax computer program. A standard curve in the range of 1–300 pg of recombinant human BDNF was determined for each titer plate.
TrkB receptor signal transduction assay. Standard procedures of TrkB receptor signal transduction assays were followed as described in detail previously (Knusel et al., 1994). Briefly, cerebellums from 2- and 6-month-old wild-type and stg/stg mice were homogenized in 1 ml of lysis buffer (137 mm NaCl, 20 mm Tris, pH 8.0, 1% Triton X-100, 10% glycerol, 1 mm phenylmethylsulfonyl fluoride, and 500 μmorthovanadate). Soluble proteins were separated by centrifugation, and the supernatant was precipitated with wheat germ lectin agarose to partially purify TrkB proteins. The glycoproteins were then subjected to SDS-PAGE and probed by Western blotting with a polyclonal rabbit anti-TrkB antibody directed against extracellular domain sequences (antibody was kindly provided by Dr. David Kaplan, National Institute of Cancer, Frederick Cancer Research and Development Center). Phosphorylation assays were performed on microslices (300 × 300 μm) prepared from cerebellum of 2-month-old +/+ andstg/stg mice according to previously published procedures (Knusel et al., 1994). Briefly, the slices were maintained in gassed Krebs’ buffer and then incubated for 4 min with recombinant human NT-4/5 (kindly provided by Genentech, San Francisco, CA) at 37°C. Immediately after incubation, the tissue was lysed and immunoprecipitated with mouse anti-phospholipase-Cγ1 (PLCγ1; Upstate Biotechnology, Lake Placid, NY). By Western blotting, the precipitate was analyzed for the presence of phosphotyrosine on PLCγ1 with anti-phosphotyrosine mouse monoclonal antibody (4G10; Upstate Biotechnology). The same microslice preparation was used to measure the phosphotyrosine levels of erk1 and erk2, another two signal transduction molecules. After incubation with NT-4/5, the tissue was lysed, and an aliquot of the lysate was separated by SDS-PAGE. Tyrosine-phosphorylated proteins were detected by Western blotting with anti-phosphotyrosine antibody.
Tissue processing for histology. Mice were anesthetized with Avertin (1.25% tribromoethanol–amylalcohol solution) and perfused transcardially with 4% paraformaldehyde in PBS (0.1 msodium phosphate and 0.9% NaCl, pH 7.4). The brains were post-fixed overnight and subsequently equilibrated in 30% sucrose solution. Coronal sections were cut and stained with either 1% cresyl violet and eosin or hematoxylin and eosin.
Cell count and electron microscopy. Mice were perfused transcardially with 0.1 m cacodylate buffer for 2 min and then with fixative of 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m cacodylate buffer with 0.02% CaCl2 for 20 min. The medial cerebellum was sliced into 1 mm translobular (sagittal) sections. The sections were post-fixed in the fixative for 2 d and in 1% osmium tetroxide for 1 hr. Tissues were washed in the buffer, dehydrated, and infiltrated with propylene oxide. Tissues were then embedded in Epon and polymerized overnight at 67°C. Semithin sections (3 μm) were cut for light microscopic evaluation and granule cell count. Sections were deplastic in NaOH ethanol solution before hematoxylin and eosin staining. External granule cells from lobules 5–8 were counted in 10 sections from each animal. Means of granule cells along the pia surface (millimeters) were calculated. Student’s t test was used for statistical significance between the genotypes. Ultrathin sections were then cut from the blocks, counterstained with 2% uranyl acetate and lead citrate, mounted on grids, and examined with a JEOL JEM-1200EX electron microscope.
Behavioral analysis. Eyeblink conditioning procedures were followed as described in detail previously (Chen et al., 1996). Briefly, adult male stg/stg (n = 8) and wild-type littermate (+/−) (n = 8) mice of C3B6Fe+ strain were anesthetized with ketamine (80 mg/kg) and xylazine (20 mg/kg) and implanted with four wires (Teflon-coated stainless steel; A-M Systems, Everett, WA) subcutaneously to the left upper eyelid. The tips of the wires were exposed, and two of the wires were used to record differential electromyography (EMG) from obicularis oculi, and the remaining two wires were used to deliver periorbital shock. A four-pin strip connector to which the wires were soldered was cemented to the animal’s skull with dental acrylic. After 1 d of habituation, animals underwent 6 d of paired behavioral training. The daily paired training consisted of 100 trials grouped in 10 blocks. Each block included one conditioned stimulation (CS) alone trial (80 dB, 1 kHz tone), one unconditioned stimulation (US) alone trial (100 msec shock, 100 Hz biphasic square pulses), and eight CS–US paired trials with a 252 msec interstimulus interval. The US intensity used was the minimal voltage required to elicit an eyeblink and head turn response, and the US intensity was adjusted daily for each animal. The EMG activity was digitized using a window discriminator and stored onto a computer with a sampling rate of 4 msec/bin. The “bad” trials were excluded using the following criteria: (1) during the 252 msec pre-CS period, the average unit counts per bin were >2; (2) the SD was greater than the average unit counts per bin during the pre-CS period; or (3) the average unit count within 28 msec after the CS onset was 1 SD larger than the average of pre-CS activity. The eyeblink conditioned response (CR) was defined as the average unit activity of any consecutive seven bins during the CR period (from 84 msec after the CS onset to the US onset for paired trials or to the termination of the CS for CS alone trials) that was higher than “average plus SD plus 1” of the pre-CS unit activity. The CR onset latency was computed as the first time during the CR period at which the average unit count of any 28 msec was higher than average plus SD plus 1 of the pre-CS unit activity. The unconditioned response (UR) amplitude was defined as the difference between averaged unit activity during the UR period [100 msec period 50 msec after the termination of the unconditioned stimulation (US)] and averaged unit of the pre-CS period.
RESULTS
Lack of BDNF protein in the stg mutant cerebellum
In a previous paper, we have shown a pronounced selective failure of BDNF mRNA expression in cerebellar granule cells of thestg mouse (Qiao et al., 1996). To test whether the lack of BDNF mRNA is reflected at the protein level, BDNF was measured with a two-site EIA for this protein. We found a 70% reduction in BDNF protein in the adult stg cerebellum compared with normal control mice [+/+, 1.20 ± 0.08 ng/gm (mean ± SE);stg/stg, 0.35 ± 0.03 ng/gm; n = 3;p < 0.01]. This result is consistent with the decreased BDNF mRNA level found previously by Northern blotting andin situ hybridization (Qiao et al., 1996).
Deficit on TrkB signal transduction in thestg cerebellum
Our previous study has shown a normal pattern and level of trkB mRNA expression in the cerebellum of stg mouse, despite the striking reduction of its ligand BDNF. To measure the levels of TrkB protein, we performed Western blotting using a polyclonal rabbit anti-TrkB antibody directed against extracellular domain sequences. As shown in Figure 1A, the antibody identified two major bands corresponding in molecular size to full-length TrkB at 145 kDa with the intracellular kinase domain and to truncated TrkB at 95 kDa lacking the kinase domain. In agreement with the mRNA levels, no significant difference was found in either form of TrkB receptor protein between the wild-type and stg mutant cerebellum of the 2- and 6-month-old mice tested. The antibody recognized a third band that was less intense with molecular weight at ∼75 kDa in our blots of wheat germ lectin-precipitated proteins (Fig.1A). Although the molecular identity of this band is unknown at present, the reduced prevalence of this protein instg indicates reduced levels in the mutant cerebellum of a protein with a very similar epitope as TrkB or of a protein that coprecipitates with one or both of the TrkB forms.
Fig. 1.

Western blot analysis of levels of TrkB receptor protein (A) and Trk signaling in cerebellum (B, C) of wild-type andstg/stg mice. A, Glycoproteins were precipitated by incubation with wheat germ lectin and were then probed with a polyclonal rabbit anti-TrkB receptor antibody directed against extracellular domain sequences of TrkB. The antibody identified two major bands at 145 kDa and 95 kDa corresponding to the full-length and truncated forms of TrkB receptor. No significant difference was found between the wild-type and stg mutant cerebellum for either form of TrkB receptor protein at 2 and 6 months of age. The same antibody detected a third band with molecular weight of ∼75 kDa, indicated in the figure with an arrow. This protein was clearly reduced in stg/stg compared with wild type.B, PLCγ1 was immunoprecipitated from untreated and NT-4/5-treated cerebellar slices with anti-PLCγ1 and analyzed by Western blotting with anti-phosphotyrosine. Both constitutive and NT-4/5-induced tyrosine phosphorylation of PLCγ1 was significantly reduced in stg mouse cerebellum when compared with wild-type cerebellum. C, Tyrosine phosphorylated proteins were detected by Western blotting with anti-phosphotyrosine antibody in lysates from untreated and NT-4/5-treated cerebellar slices. In contrast to the equivalent levels of a third band on the same blot, both constitutive and NT-4/5-induced phosphotyrosine signals of two proteins with molecular weights identical to erk1 and erk2 were reduced in stg cerebellum compared with wild-type cerebellum.
To further assess the functional response of TrkB receptors instg, we performed tyrosine phosphorylation assays in freshly prepared microslices from stg cerebellum for several proteins involved in TrkB signal transduction, including PLCγ1, erk1, and erk2 (Knusel et al., 1994). Our results indicate strongly reduced constitutive and NT-4/5-induced tyrosine phosphorylation of PLCγ1 instg cerebellum (Fig. 1B). To confirm that the difference of PLCγ1 signal was not attributable to decreased PLCγ1 level, the PLCγ1 content was measured. Comparable PLCγ1 protein levels were found in stg and wild-type cerebellum (data not shown). Phosphotyrosine signals of untreated and NT-4/5-treated microslices for both erk1 and erk2 were also reduced instg cerebellum compared with controls (Fig. 1C). Together, our results reveal a consistent deficit in constitutive and NT-induced activation of cellular messengers involved in TrkB signal transduction.
Normal cerebellar–vestibular morphology instg homozygotes
We have reported normal foliation and laminar structure instg cerebellar cortex (Qiao et al., 1996). In the present study, we further investigated the cerebellar–vestibular system, which might contribute to the behavioral deficits in motor coordination and in which neurotrophins are known to be involved in development. Coronal brain sections through medial cerebellum revealed a normal layout of cerebellar cortex, cerebellar deep nuclei, and vestibular nuclei instg mouse when compared with the wild type (Fig.2A,B). At high magnification, no qualitative changes were observed in the interpositus nucleus (Fig. 2C), the vestibular nuclei (Fig.2D), as well as the vestibular ganglion (Fig.2E), in the appearance, the density, and the size of the neurons in the adult stg mutants.
Fig. 2.
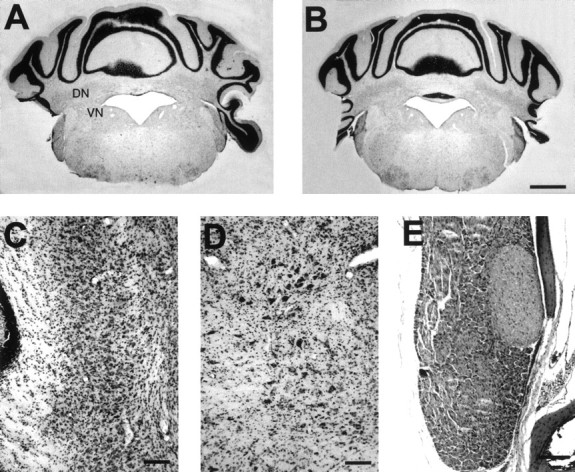
Coronal brain sections through the cerebellar–vestibular system of adult wild-type (A) and stg/stg(B–E) mice. A, B, Comparable gross histological appearance in wild-type (A) and stg(B) mice is evident in cerebellar cortex, cerebellar deep nuclei (DN), and vestibular nuclei (VN). At high magnification, no qualitative changes could be found in the appearance, the density, or the size of the neurons in the interpositus nuclei (C), vestibular nuclei (D), as well as vestibular ganglion (E) instg mutants. C, Top is medial and bottom is lateral. Scale bars:B, 1 mm; C–E, 100 μm.
Increased external granule cells duringstg development
To test the possible influence of BDNF on cerebellar granule cells during development, we examined the cerebellar structure instg/stg and +/− littermates at P15, P20, and P30. At P15, few scattered external granule cells (EGCs) were seen near the surface of the cerebellar cortex in wild-type animals (Fig.3A). In contrast, multiple layers of EGCs were found in the stg cerebellar cortex at the same developmental stage (Fig. 3B). At P20, the EGC layer had virtually disappeared in both stg and wild-type mice (Fig. 3C,D), and no difference was found at P30 (data not shown). Quantitative cell counts of EGCs (n = 3) confirmed significantly more external granule cells in the mutant cerebellum than in the wild type at P15 (226% of control; p < 0.01), whereas no difference at P20 (p > 0.05) was found between the two genotypes (Fig. 3E). This finding is consistent with the observation of delayed disappearance of the EGC layer in the cerebellum of BDNF knock-out mice (Jones et al., 1994), suggesting a regulatory role of BDNF in the cerebellar granule cell migration.
Fig. 3.

Postnatal cerebellar development in wild-type andstg/stg mice. A, B, Horizontal cerebellar sections at P15 reveal few scattered EGCs (arrows) in wild-type mice (A) but multiple layers of EGCs in stg cerebellum (B). C, D, At P20, the EGC layer (arrows) is hardly visible in both the wild-type and stg mouse. E, Quantitative cell counts of EGCs from 10 semithin sagittal sections of medial cerebellum (n = 3 for all the groups) have shown significantly more EGCs (226% of control; p < 0.01) in the mutant cerebellum than the corresponding wild type at P15, whereas there was no difference at P20 (p > 0.05) between the two genotypes. Scale bar, 0.5 mm.
Abnormal granule cell morphology in adultstg cerebellum
BDNF has been proposed to play a direct role in synaptogenesis and adaptive changes in mature brain (Gall, 1992). Excess BDNF has been shown to inhibit ocular dominance column formation in the visual cortex and, therefore, may influence the activity-dependent control of axonal branching during development (Cabelli et al., 1995). To determine whether BDNF is involved in synaptic formation during development, we examined the cerebellar ultrastructure in adult mice from both genotypes by electron microscopy. Qualitative examination of the input and output synapses of granule cells revealed normal synaptic structures in stg cerebellum. Figure4C shows the structure of a typical glomerulus in the granule cell layer of adult stgcerebellum. Large mossy fiber rosettes filled with clear synaptic vesicles and dense core vesicles form asymmetrical synapses with granule cell dendrites. Translobular (sagittal) section through the outer molecular layer in adult stg cerebellum is shown in Figure 4D. Numerous transverse cut parallel fibers are packed in this region. Several parallel fiber varicosities containing a large concentration of tightly packed round synaptic vesicles have asymmetrical contact with presumed Purkinje cell spines featuring a densely staining postsynaptic web. No noticeable abnormality was found in the synapses at either mossy fiber–granule cell or parallel fiber–Purkinje cell sites. Although not excluding the potential contribution of minimal level of BDNF released from neighboring cell types as well as possible subtle quantitative differences in the number of synapses, our findings suggest that a near total lack of BDNF in granule cells has little effect on the synaptic structure of their input and output sites in the cerebellum.
Fig. 4.
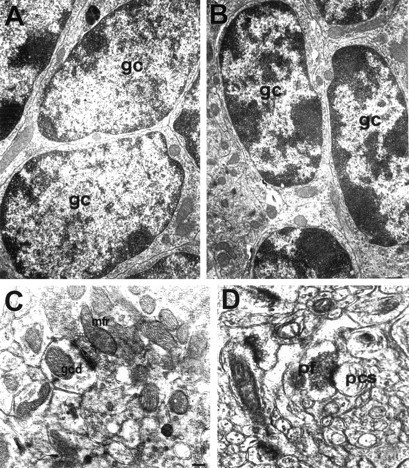
Electron micrographs of cerebellar cortex.A, B, Granule cells from adult +/+ (A) and stg/stg(B) mice. The darkly staining nucleus of granule cells (gc) in the wild-type cerebellum has only few scattered clumps of chromatin around the nucleus membrane and is surrounded by a thin layer of cytoplasm. The cell bodies are packed densely within the granule cell layer. In contrast, some of the granule cells in the mutant cerebellum displayed elongated oval-shaped nuclei studded with clumps of coarse chromatin. Such granule cells resemble immature-looking cells and represent <20% of the granule cell population in the adult stg cerebellum.C, Typical glomerulus in the granule cell layer ofstg cerebellum shows the normal structure of mossy fiber–granule cell synapses, large mossy fiber rosettes (mfr) filled with clear and dense core synaptic vesicles, as well as mitochondria forming asymmetrical synapses with granule cell dendrites (gcd). D, Translobular (sagittal) section through the outer molecular layer instg cerebellum displays numerous transverse cut parallel fibers (pf). Several parallel fiber varicosities containing a large concentration of tightly packed round synaptic vesicles have asymmetrical contact with presumably Purkinje cell spines (pcs) featuring a densely staining postsynaptic web. No noticeable abnormality was found at the parallel fiber–Purkinje cell synapses. Magnification: A,B, 10,000×; C, D, 20,000×.
In contrast to the normal appearance of cerebellar synapses, a noticeable difference was observed in the appearance of granule cell bodies (Fig. 4A,B). In the wild-type cerebellum, the darkly staining nucleus of granule cells has only a few scattered clumps of chromatin around the nucleus membrane and is surrounded by a thin layer of cytoplasm. The cell bodies are packed densely within the granule cell layer (Fig.4A). In contrast, some of the granule cells in the mutant cerebellum displayed elongated oval-shaped nuclei studded with clumps of coarse chromatin (Fig. 4B). The morphology of these granule cells resembles migrating immature granule cells at early postnatal days, as described by Altman (1972). Such abnormal granule cells represented <20% of the granule cell population in the adult stg cerebellum but were never seen in the wild-type cerebellar cortex.
Impaired classical eyeblink conditioning instg mice
To determine whether there is any change in cerebellar function that may be correlated with the biochemical and anatomical abnormalities, we performed classical eyeblink conditioning tests in adult stg mutant. We found that stg mice, when compared with their normal littermates, were severely impaired in acquisition in this paradigm. Figure 5shows the percentage (A) and the amplitude (B) of the eyeblink CR (+/− littermates,n = 8; stg/stg, n = 8). During 6 d of paired training, wild-type mice exhibited a significant level of learning (linear trend analysis, p= 0.00009), whereas stg mutants performed poorly and showed a profound impairment with little sign of learning (p = 0.0558). A two-way ANOVA analysis clearly demonstrated that wild-type mice learn to a significantly higher asymptotic level at a faster learning rate than stg mutants in both the percentage and the amplitude of the eyeblink CRs (CR percentage, group difference, p = 0.0007; learning rate, p = 0.00003; CR amplitude, group difference,p = 0.00024; learning rate, p = 0.00024). Analysis of CR performance in unpaired controls andstg mice fails to reveal any statistic significance (group difference, p = 0.73; learning rate, p= 0.49). The results of CR onset latency analysis are shown in Figure6A. The onset latency of the wild-type littermates exhibited a normal pattern similar to those observed in other mouse strains, such as C57BL6/J and BALBC mice (data not shown). A large number of CR onset latencies were within the 88–144 msec range. However, the stg mice exhibited many fewer CRs with early onset latencies. In addition, essentially no peak in CR onset latency was found in stg mutants, with only a slightly increased number of trials of ∼172 msec. The trial distribution indicates significantly delayed and virtually flat CR onset latencies in stg mutants compared with that in wild types. Sensitivity to the US shocks and URs to the US were also tested. Neither US intensities (Fig. 6B) nor UR amplitudes (Fig. 6C) were significantly different between the two groups.
Fig. 5.

Performance of classical eyeblink conditioning during acquisition training in wild-type (open square),stg (filled square), andtg (filled circle) mice. During 6 d of paired training, wild-type and tg mice exhibited a significant level of learning, whereas stgmutants performed poorly. Both percentage (A) and amplitude (B) of eyeblink CR revealed severe impairment in the acquisition of eyeblink conditioning in thestg mutant.
Fig. 6.
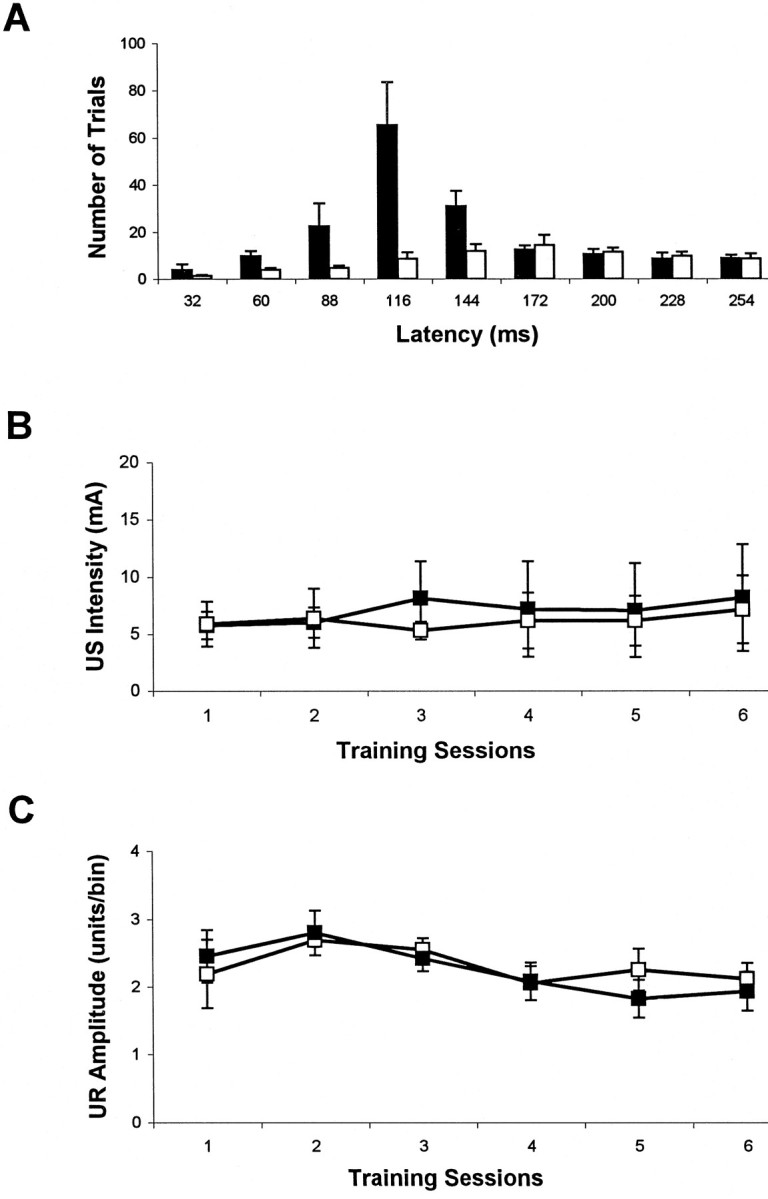
A, Histogram of the CR onset latencies during training. The normal pattern of onset latency in the wild-type (filled bar) littermates is contrasted with the delayed and flat distribution of CR onset latencies instg mutants (open bar). B,C, Sensitivity to the US and UR to the US in the wild-type (open square) and stg(filled square) mice. Neither US intensities (B) nor UR amplitudes (C) were significantly different between the two groups.
To ascertain that the impairment in stg mutants was not attributable to impaired sensitivities to the US or the CS, pain and tone perceptions were tested in stg mice. Tail flick analgesia measurements showed no significant difference between wild-type and stg mice at either 55 or 60°C [55°C, +/−, 20.67 ± 1.45 sec (mean ± SE); stg/stg, 23.08 ± 1.22; p > 0.05; 60°C, +/−, 10.61 ± 0.98; stg/stg, 11.08 ± 0.89; p > 0.05]. Multiple-unit recordings from auditory cortex in stgmouse revealed normal evoked spikes during the presentation of a 350 msec, 80 dB, 1 kHz tone, which is comparable to that of the wild type (data not shown). These results demonstrate normal perceptions in thestg mouse, which in turn strongly suggest that the eyeblink conditioning impairment reflects a functional defect of the cerebellum in stg mutant.
To determine whether the impaired eyeblink conditioning instg mice was linked specifically to the BDNF deficit or to the nonspecific feature of seizure activity, we tested eyeblink conditioning in a second ataxic mutant with the same type of spike wave seizures, the tottering (tg; chromosome 8) mouse. Adult tg/tg mutants display a similar ataxic gait with mild cerebellar hypoplasia (Sidman et al., 1965; Noebels and Sidman, 1979;Qiao and Noebels, 1991) but with normal cerebellar BDNF expression (Qiao et al., 1996). Unlike the stg mutant, thetg mice exhibited normal acquisition of conditioned eyelid responses in comparison to the wild-type mice (CR percentage, group difference, p = 0.13; learning rate, p= 0.019; n = 4.) (Fig. 5A). The result suggests that the impairment in eyeblink conditioning is not related to the secondary effects of either spike wave seizures, which do not involve the cerebellar cortex, or cerebellar hypoplasia. Impaired cerebellar function in stg is more likely linked to the BDNF defect in this mutant.
DISCUSSION
This report extends our previous finding of a specific and pronounced deficit in BDNF expression in stg mouse to the protein level. BDNF protein in this mutant cerebellum was reduced by 70% compared with controls. Protein levels of the BDNF receptor TrkB were normal, but ligand-induced tyrosine phosphorylation of several TrkB-activated signal transduction molecules was reduced. At the morphological level, an increased number of external granule cells was found in the stg cerebellum at P15. This difference to controls had disappeared at P20. Ultrastructural examination of adultstg revealed some immature-looking granule cells in the cerebellum but normal development of the mossy fiber–granule cell and parallel fiber–Purkinje cell synapses. In contrast to the minor effects of the stg mutation on morphological parameters in the cerebellum, our behavioral results show a severe deficit in cerebellar learning in this mouse. Together, our findings support a role of BDNF in functional plasticity in the CNS that is not reflected in gross morphological changes at cellular or synaptic levels.
BDNF effect on TrkB receptor
Neurotrophins have been found to regulate Trk receptor levels. Chronic exposure of neurons to NGF was shown to result in increased levels of the NGF receptor TrkA (Holtzman et al., 1992; Kojima et al., 1994; Venero et al., 1994; Li et al., 1995). Treatment of rat embryonic neurons with BDNF, NT-3, and NT-4/5 leads to reduction of the protein and mRNA levels for the TrkB receptor, as well as a downregulation of Trk tyrosine phosphorylation (Frank et al., 1996; Knusel et al., 1997). However, little is known about the effect of NT-deprivation on Trk receptor levels. We have shown previously that the BDNF expression defect in the granule cells was not associated with any changes in trkB receptor mRNA levels in stg mutant (Qiao et al., 1996). We now showed that the pronounced reduction of the BDNF protein level instg cerebellum does not result in a change in TrkB protein levels. These results suggest that the expression of the BDNF receptor TrkB in vivo does not depend on the tissue level of its ligand, as suggested by the earlier in vitro findings (Frank et al., 1996; Knusel et al., 1997).
Binding of the NTs to Trks stimulates tyrosine phosphorylation of the receptor and of intracellular substrates. The process has been used as an indicator of Trk receptor activation (Kaplan and Stephens, 1994;Knusel et al., 1994). Despite unchanged levels of TrkB protein, our results demonstrate a significant deficit in tyrosine phosphorylation of several signal transduction molecules in stg cerebellum. Our data indicate a regulatory effect of BDNF on the efficiency of TrkB signaling without an effect on TrkB levels. These results agree with a recent report showing that Trk activation is dramatically decreased in granule and Purkinje cells of BDNF knock-out mice when assayed by specific antibodies raised against tyrosine phosphorylated Trk receptors (Schwartz et al., 1997). In a previous study, we observed a decrease in BDNF-, NT-3-, and NT-4/5-induced Trk and Trk substrate tyrosine phosphorylation at P7–P14 (Knusel et al., 1994). This finding indicates the presence of efficient posttranslational regulatory mechanisms to control the responsiveness of NT receptors.
BDNF and cerebellar morphology
Studies of BDNF gene deletion mice have found severe deficiencies in motor coordination and balance, resulting in head bobbing and tilting, circling behavior, and periods of immobility (Ernfors et al., 1994). The behavioral phenotype reflects defects in both cerebellar and vestibular systems. Indeed, neuronal degeneration in vestibular and other sensory ganglia, as well as abnormal cerebellar development, was found in these mutants (Ernfors et al., 1994, 1995; Jones et al., 1994;Conover et al., 1995; Liu et al., 1995; Schwartz et al., 1997). Thestargazer mutants share major elements of the BDNF−/− behavioral abnormalities, including ataxic gait, episodic upward head tilting, and impaired motor coordination (Noebels et al., 1990; Qiao et al., 1996). In the present study, we failed to detect any abnormalities in the vestibular ganglia and vestibular nuclei of stg mice. The normal morphology is correlated with the absence of circling behavior seen in BDNF−/− mice. In contrast, similar presumably delayed granule cell migration and maturation were evident instg cerebellum. Our results are in agreement with the previous finding that BDNF can influence granule cell migration (Gao et al., 1995). These findings also indicate that the stgbehavioral phenotype is primarily, if not solely, the result of a cerebellar defect without direct involvement of the vestibular system.
Jones et al. (1994) reported a slight delay in granule cell migration in BDNF−/− mutants. The same phenomenon was observed during stg cerebellar development, supporting a role for BDNF in cerebellar granule cell migration. In a more recent study, additional cerebellar abnormalities were found in BDNF transgenics, including increased death of granule cells, stunted growth of Purkinje cell dendrites, impaired formation of horizontal layers, and defects in the foliation pattern (Schwartz et al., 1997). However, our present observations in stg cerebellum only revealed mild morphological abnormalities, i.e., <20% abnormal granule cells in the adult. The following two facts may contribute to the differences. First, ∼30% of normal BDNF mRNA and protein levels were still detected in adult stg cerebellum by Northern analysis and BDNF EIA, respectively. Although most of this BDNF might be produced by cell types other than granule cells, it may still influence granule cell development. BDNF mRNA has been localized in the deep cerebellar nuclei in rat (Rocamora et al., 1993). Second, although we did observe a delayed disappearance of the EGC layer during development, the normal life span of stg mice allowed us to perform our experiments in adult mice. In contrast, because of high postnatal mortality, most studies using BDNF−/− mice are limited to the first 2–3 weeks when the cerebellum is not fully developed. Because cerebellar granule cell proliferation normally continues until P20 (Fujita et al., 1966) and migration, neurite outgrowth, and the synaptic formation proceed until >P30 (Altman, 1972), some of the findings with BDNF transgenics might reflect a possible delay in development rather than absolute loss of function.
Potential role of BDNF in cerebellar-dependent learning
NGF administrations have been reported to improve spatial memory in aged rats (Markowska et al., 1994). Anti-NGF antibody application leads to impairment in passive-avoidance learning in neonatal mice (Ricceri et al., 1994). In the present study, we provide evidence that a specific and pronounced deficit of BDNF expression in cerebellar granule cells is associated with a loss of functional plasticity, detected as severely impaired eyeblink conditioning in stgmutant. Unlike in BDNF knock-out mice (Ernfors et al., 1994; Jones et al., 1994), no sensory or vestibular deficiencies were found instargazer. In addition, the normal eyeblink conditioning performance in the related tottering mutant is significant for two reasons. First, it demonstrates that the BDNF defect instg cerebellum is specifically linked to the impairment in learning, because tg mice express normal levels of BDNF mRNA throughout the forebrain and cerebellum (Qiao et al., 1996). Second, it indicates that the neocortical seizures seen in both mutants, which do not spread to the cerebellum (Noebels and Sidman, 1979), do not account for the learning impairment in stg mouse. BDNF defect appears to be critical for the observed deficit in cerebellar learning in stg mutant and suggests that BDNF is essential for normal neuronal plasticity in the learning and memory process of the eyeblink conditioning paradigm.
As previously found in rabbits, rats, and humans (Thompson, 1986;Skelton, 1988; Daum et al., 1993; Lavond et al., 1993), recent studies have shown impaired eyeblink conditioning in cerebellum-lesioned and Purkinje cell degeneration (pcd) mutant mice (Chen et al., 1996). In comparison to pcd mice (i.e., complete functional cerebellar decortication in adult), only limited morphological changes were present in stg cerebellum. However, more severe deficits in eyeblink conditioning are found instg mice than in pcd mutants. Our results suggest that the functional integrity of cerebellum is severely compromised by the BDNF defect in cerebellar granule cells without parallel gross morphological changes to the cerebellar structure.
In addition to its traditional role as a neuronal survival factor, much evidence has shown that BDNF is involved in the modulation of synaptic transmission. Application of BDNF can potentiate the efficacy of developing neuromuscular synapses in culture (Lohof et al., 1993), enhance synaptic strength in the hippocampus (Lessmann et al., 1994;Kang and Schuman, 1995), facilitate long-term potentiation (LTP) induction (Figurov et al., 1996), and inhibit GABAAsynaptic responses in rat hippocampus (Tanaka et al., 1997). Furthermore, BDNF gene deletion impairs both synaptic transmission and LTP in the hippocampus (Korte et al., 1995; Patterson et al., 1996). Recent evidence indicates that BDNF modulates synaptic transmission by postsynaptic activation of Trk tyrosine kinase receptors (Levine et al., 1995; Tanaka et al., 1997) and subsequent phosphorylation of the NMDA receptors (Suen et al., 1997). The presence of inactive TrkB receptor signal transduction in stg cerebellum in conjunction with decreased Trk activation seen in BDNF knock-out transgenics (Schwartz et al., 1997) supports the hypothesis that defects in BDNF–TrkB signal transduction pathway may underlie the behavioral deficiencies in coordination and learning seen in these mutants.
Footnotes
This work was supported by National Institutes of Health Grants NS22933 (F.H.), AG09793 (F.H.), AG10480 (F.H.), NS09985 (X.Q.), and AG05142, National Science Foundation Grant IBN-9215069, Office of Naval Research Grant N00014-95-1152, National Institute of Mental Health Grant 5P01-MH52194, and a grant from the Sankyo Company, Ltd (R.F.T.).
Correspondence should be addressed to Dr. Xiaoxi Qiao, University of Southern California, Hedco Neuroscience Building, 3614 Watt Way, Los Angeles, CA 90089-2520.
REFERENCES
- 1.Allen G, Buxton RB, Wong EC, Courchesne E. Attentional activation of the cerebellum independent of motor involvement. Science. 1997;275:1940–1943. doi: 10.1126/science.275.5308.1940. [DOI] [PubMed] [Google Scholar]
- 2.Altman J. Postnatal development of the cerebellar cortex in the rat. III. Maturation of the components of the granular layer. J Comp Neurol. 1972;145:465–514. doi: 10.1002/cne.901450403. [DOI] [PubMed] [Google Scholar]
- 3.Altman J, Bayer SA. An overview of the postnatal development of the rat cerebellum. In: Altman J, Bayer SA, editors. Development of the cerebellum: in relation to its evolution, structure, and functions. CRC; Boca Raton, FL: 1997. pp. 324–333. [Google Scholar]
- 4.Andreasen NC, O’Leary DS, Arndt S, Cizadlo T, Hurtig R, Rezai K, Watkins GL, Ponto LL, Hichwa RD. Short-term and long-term verbal memory: a positron emission tomography study. Proc Natl Acad Sci USA. 1995;92:5111–5115. doi: 10.1073/pnas.92.11.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Bao S, Lockard JM, Kim JK, Thompson RF. Impaired classical eyeblink conditioning in cerebellar-lesioned and Purkinje cell degeneration (pcd) mutant mice. J Neurosci. 1996;16:2829–2838. doi: 10.1523/JNEUROSCI.16-08-02829.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M, Ip NY, Boland P, Friedman B, Weigand S, Vejsada R, Kato AC, DeChiara TM, Yancopoulos GD. Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature. 1995;375:235–238. doi: 10.1038/375235a0. [DOI] [PubMed] [Google Scholar]
- 9.Daum I, Ackermann H, Schugens MM, Reimold C, Dichgans J, Birbaumer N. The cerebellum and cognitive functions in humans. Behav Neurosci. 1993;107:411–419. doi: 10.1037//0735-7044.107.3.411. [DOI] [PubMed] [Google Scholar]
- 10.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 11.Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- 12.Figurov A, Pozzo Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 13.Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS. BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci. 1996;8:1220–1230. doi: 10.1111/j.1460-9568.1996.tb01290.x. [DOI] [PubMed] [Google Scholar]
- 14.Fujita S, Shimada M, Nakamura T. 3H-Thymidine autoradiographic studies on the cell proliferation and differentiation in the external and internal granular layers of the mouse cerebellum. J Comp Neurol. 1966;128:191–208. doi: 10.1002/cne.901280206. [DOI] [PubMed] [Google Scholar]
- 15.Gall CM. Regulation of brain neurotrophin expression by physiological activity. Trends Pharmacol Sci. 1992;13:401–403. doi: 10.1016/0165-6147(92)90123-n. [DOI] [PubMed] [Google Scholar]
- 16.Gao JH, Parsons LM, Bower JM, Xiong J, Li J, Fox PT. Cerebellum implicated in sensory acquisition and discrimination rather than motor control. Science. 1996;272:545–547. doi: 10.1126/science.272.5261.545. [DOI] [PubMed] [Google Scholar]
- 17.Gao WQ, Zheng JL, Karihaloo M. Neurotrophin-4/5 (NT-4/5) and brain-derived neurotrophic factor (BDNF) act at later stages of cerebellar granule cell differentiation. J Neurosci. 1995;15:2656–2667. doi: 10.1523/JNEUROSCI.15-04-02656.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holtzman DM, Li Y, Parada LF, Kinsman S, Chen CK, Valletta JS, Zhou J, Long JB, Mobley WC. p140trk mRNA marks NGF-responsive forebrain neurons: evidence that trk gene expression is induced by NGF. Neuron. 1992;9:465–478. doi: 10.1016/0896-6273(92)90184-f. [DOI] [PubMed] [Google Scholar]
- 19.Jacobson M. Developmental neurobiology (Jacobson M, ed). Plenum; New York: 1991. [Google Scholar]
- 20.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 23.Kim JJ, Thompson RF. Cerebellar circuits and synaptic mechanisms involved in classical eyeblink conditioning. Trends Neurosci. 1997;20:177–181. doi: 10.1016/s0166-2236(96)10081-3. [DOI] [PubMed] [Google Scholar]
- 24.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knusel B, Gao H, Okazaki T, Yoshida T, Mori N, Hefti F, Kaplan DR. Ligand-induced down-regulation of Trk messenger RNA, protein and tyrosine phosphorylation in rat cortical neurons. Neuroscience. 1997;78:851–862. doi: 10.1016/s0306-4522(96)00616-1. [DOI] [PubMed] [Google Scholar]
- 26.Kojima M, Ikeuchi T, Hatanaka H. Nerve growth factor induces trkA mRNA expression in cultured basal forebrain cholinergic neurons from 17-day fetal rats. Neurosci Lett. 1994;169:47–50. doi: 10.1016/0304-3940(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 27.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavond DG, Kim JJ, Thompson RF. Mammalian brain substrates of aversive classical conditioning. Annu Rev Psychol. 1993;44:317–342. doi: 10.1146/annurev.ps.44.020193.001533. [DOI] [PubMed] [Google Scholar]
- 29.Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 30.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 31.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua Couzens J, Clary DO, Knusel B, Mobley WC. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Ernfors P, Wu H, Jaenisch R. Sensory but not motor neuron deficits in mice lacking NT4 and BDNF. Nature. 1995;375:238–241. doi: 10.1038/375238a0. [DOI] [PubMed] [Google Scholar]
- 34.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 35.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 36.Markowska AL, Koliatsos VE, Breckler SJ, Price DL, Olton DS. Human nerve growth factor improves spatial memory in aged but not in young rats. J Neurosci. 1994;14:4815–4824. doi: 10.1523/JNEUROSCI.14-08-04815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 38.Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science. 1979;204:1334–1336. doi: 10.1126/science.572084. [DOI] [PubMed] [Google Scholar]
- 39.Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT (1990) Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res [Erratum (1992) 11:72] 7:129–135. [DOI] [PubMed]
- 40.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 41.Qiao X, Noebels JL. Genetic and phenotypic heterogeneity of inherited spike-wave epilepsy: two mutant gene loci with independent cerebral excitability defects. Brain Res. 1991;555:43–50. doi: 10.1016/0006-8993(91)90857-r. [DOI] [PubMed] [Google Scholar]
- 42.Qiao X, Hefti F, Knusel B, Noebels JL. Selective failure of brain-derived neurotrophic factor mRNA expression in the cerebellum of stargazer, a mutant mouse with ataxia. J Neurosci. 1996;16:640–648. doi: 10.1523/JNEUROSCI.16-02-00640.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ricceri L, Alleva E, Calamandrei G. Impairment of passive avoidance learning following repeated administrations of antibodies against nerve growth factor in neonatal mice. NeuroReport. 1994;5:1401–1404. [PubMed] [Google Scholar]
- 44.Rocamora N, Garcia Ladona FJ, Palacios JM, Mengod G. Differential expression of brain-derived neurotrophic factor, neurotrophin-3, and low-affinity nerve growth factor receptor during the postnatal development of the rat cerebellar system. Brain Res Mol Brain Res. 1993;17:1–8. doi: 10.1016/0169-328x(93)90065-w. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 46.Shibuki K, Gomi H, Chen L, Bao S, Kim JJ, Wakatsuki H, Fujisaki T, Fujimoto A, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 47.Sidman RL, Green MC, Appel SH. Catalog of the neurological mutants of the mouse. Harvard UP; Cambridge, MA: 1965. [DOI] [PubMed] [Google Scholar]
- 48.Skelton RW. Bilateral cerebellar lesions disrupt conditioned eyelid responses in unrestrained rats. Behav Neurosci. 1988;102:586–590. doi: 10.1037//0735-7044.102.4.586. [DOI] [PubMed] [Google Scholar]
- 49.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson RF. The neurobiology of learning and memory. Science. 1986;233:941–947. doi: 10.1126/science.3738519. [DOI] [PubMed] [Google Scholar]
- 52.Thompson RF. Neural mechanisms of classical conditioning in mammals. Philos Trans R Soc Lond B Biol Sci. 1990;329:161–170. doi: 10.1098/rstb.1990.0161. [DOI] [PubMed] [Google Scholar]
- 53.Venero JL, Beck KD, Hefti F. Intrastriatal infusion of nerve growth factor after quinolinic acid prevents reduction of cellular expression of choline acetyltransferase messenger RNA and trkA messenger RNA, but not glutamate decarboxylase messenger RNA. Neuroscience. 1994;61:257–268. doi: 10.1016/0306-4522(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 54.Yeo CH. Cerebellum and classical conditioning of motor responses. Ann NY Acad Sci. 1991;627:292–304. doi: 10.1111/j.1749-6632.1991.tb25933.x. [DOI] [PubMed] [Google Scholar]