

Abstract
Steroid hormones exert dramatic effects on neuronal expression of genes that encode neuropeptides. Expression of the neurotensin/neuromedin (NT/N) gene in preoptic area neurons is dramatically enhanced by estrogen in vivo, even though its promoter lacks palindromic estrogen response elements. We report here that estrogen promotes transcription of this gene by interactions with the cAMP cascade in a neuronal cell line, SK-N-SH, and in a mouse model. In neuroblastoma cells, estrogen increases cAMP and the phosphorylation of the cAMP response element-binding protein in a time frame that precedes induction of NT/N gene transcription. Interference with the cAMP/protein kinase A signal transduction cascade blocks the ability of estrogen to elicit increases in transcription of this gene. Furthermore, in studies performed in vivo using mice deficient in protein kinase A, estrogen fails to induce increases in NT/N mRNA but retains its ability to promote estrogen response element-dependent progesterone receptor gene transcription. These data represent the first report of a nonclassical effect of estrogen on the expression of an endogenous estrogen-regulated neuropeptide gene through cAMP-mediated mechanisms both in a neuroblastoma cell line and in hypothalamic neurons. More importantly, this “cross-talk” may represent a more generalized mechanism by which steroid hormones act through other signal transduction cascades to regulate the expression of other genes in the brain.
Keywords: estrogen, neurotensin, cAMP, nonclassical, gene-transcription, mouse brain, signal transduction, SK-N-SH cells
Estrogen exerts many of its effects by the well characterized mechanism of transactivation, involving nuclear receptor dimerization and binding to consensus estrogen response elements (EREs) (for review, see Malayer and Gorski, 1993). However, there is accumulating evidence to suggest that estrogen might promote gene transcription by signaling through pathways other than those traditionally associated with steroid hormone-induced gene transcription. This is particularly relevant to understanding how estrogen might influence the expression of genes whose promoters do not contain recognizable EREs. We show here that in some cases, steroid hormone modulation of peptidergic neurotransmission involves an ability to influence pathways leading cAMP-dependent gene transcription, bothin vitro and in vivo.
A link between estrogen exposure and accumulation of intracellular cAMP in uterine tissue has been suggested since the early 1960s; however, the mechanism by which the hormone elicits this effect is still unknown. Recent reports indicate that estrogen treatment of various cultured peripheral cell types induces the accumulation of cAMP. The accumulation of cAMP has been noted in MCF-7 cells, a human breast cancer cell line, in which 1 nm 17β-estradiol induced maximal cAMP production within 1 hr of treatment (Aronica et al., 1994). Similar effects have been observed in human prostate cells (Nakhla et al., 1994). Most recently, the involvement of cAMP and protein kinase A (PKA) have been noted in estradiol-induced dendritic spine outgrowth in cultured hippocampal neurons (Murphy and Segal, 1997). Transcriptional effects of estrogen involving cAMP-dependent signaling, however, have not been linked previously to the expression of a neurotransmitter gene in brain neurons.
Neurotensin/neuromedin (NT/N) is one of several neuropeptide genes dramatically regulated by estrogen exposure in vivo(Alexander et al., 1989a,b, 1991; Brot et al., 1993; Alexander and Leeman, 1994; Szot and Dorsa, 1994). Neurotensin is a 13-amino acid peptide, thought to act as a neurotransmitter or neuromodulator in the CNS (Leeman et al., 1982). It is involved in stimulation of prolactin release and may participate in the preovulatory leutenizing hormone surge (Alexander et al., 1989a, 1991; Alexander and Leeman, 1994). NT/N is expressed in an estrogen-dependent manner in the medial preoptic nucleus (MPON) of the rodent hypothalamus (Axelson et al., 1992) and also in the bed nucleus of the stria terminalis (our unpublished observations), two nuclei that are rich in nuclear estrogen receptor protein. In the female, NT/N mRNA levels in the MPON reflect the changes in circulating estrogen during the estrous cycle (Alexander et al., 1989a, 1991). We have shown recently that a single dose of estrogen administered to rats rapidly induces the persistent phosphorylation of the cAMP response element-binding protein (CREB) in these brain regions (Zhou et al., 1996). The promoter of the neurotensin gene contains CREB recognition sites, in addition to other potentially important transcription factor binding sites such as those for AP-1 complexes and glucocorticoid receptors (Kislauskis et al., 1988). In light of the relationship between estrogen and cAMP generation in peripheral cells, and the fact that estrogen can induce the phosphorylation of the CREB protein in brain regions in which the neurotensin gene is regulated by estrogen, we have examined the possibility that estrogen might modulate the transcription of this gene through activation of the cAMP/PKA pathway.
MATERIALS AND METHODS
Animals. Female C57/BL6 mice at 6 weeks of age were obtained from Simonsen Laboratories (Gilroy, CA), maintained on a 12 hr light/dark cycle, and housed at an American Association for the Accreditation of Laboratory Animal Care-accredited research facility. RIIβ knock-out (Adams et al., 1997), Cβ1knock-out (Qi et al., 1996), and wild-type mice were used between 6 and 8 weeks of age and were treated similarly to the C57/BL6 mice. Animals were bilaterally ovariectomized (OVX) and allowed to recover for 1 week before hormone replacement. Mice weighed ∼20–25 gm and were intraperitoneally injected with either 1 μg of estradiol benzoate (EB) or 100 μg of tamoxifen (T) in 100 μl of sesame oil vehicle. Animals treated with both EB and T were given T 30 min before estrogen exposure, and animals receiving EB or T or vehicle alone received vehicle at the 30 min time point to ensure that all animals were injected with equal amounts of solution at equal time periods and to control for effects caused by the injection. Intact mice received 100 μl of sesame oil vehicle at both times. One group of RIIβ and Cβ knock-out animals received 10 μg of EB daily for 3 d before killing to evaluate a very large, chronic dose of estrogen treatment. Animals were killed at the indicated times after injection by cervical dislocation. Brains were removed and immediately frozen on dry ice. They were then sectioned by cryostat in 20-μm-thick sections and thaw-mounted onto RNase-free positively charged slides (Fisher Scientific, Santa Clara, CA) and frozen at −80°C before being assayed.
In situ hybridization. In situ hybridization was performed on slide-mounted brain sections following a processing procedure described previously by Miller et al. (1988). Tissue slices were hybridized with a 35S-UTP-labeled riboprobe complementary to the coding region of the mouse NT/N gene (generously provided by Dr. Gene Erwin, University of Colorado, Denver, CO) or with a riboprobe complementary to the ligand-binding domain of the rat progesterone receptor (PR; kindly provided by Dr. OK-Kyong Parke-Sarge, Northwestern University). NT/N antisense riboprobe was synthesized as described previously (Adams et al., 1997). Rat PR riboprobe was synthesized as described previously (Park and Mayo, 1991). Optical density was determined using the MicroComputer Imaging Device (MCID; Imaging Research Inc., St. Catherine’s, Ontario, Canada). Sense riboprobe revealed no specific labeling of any brain region (data not shown). For optical density measurements, tissue background was subtracted from each reading, and both left and right MPON readings from two consecutive sections from each animal were measured (bregma, −0.1 mm) (Franklin and Paxinos, 1997).
Reporter gene constructs. The NT/N promoter–reporter construct used in our experiments is identical to that used previously (Harrison et al., 1995). The construct contains the first 216 nucleotides of the rat NT/N gene promoter, previously shown to be the minimal fragment necessary to induce NT/N gene expression by various agents, including cAMP (Kislauskis and Dobner, 1990). pCH110 was purchased from Pharmacia (Uppsala, Sweden) and used as a control against which to normalize for transfection efficiency. Dominant negative CREB (KCREB) was provided by Dr. Richard Goodman (Vollum Institute, Portland, OR). PKI was provided by Dr. Richard Maurer (Vollum Institute).
Cell culture. SK-N-SH cells were obtained from the American Type Culture Collection (Rockville, MD). Cells were grown as reported previously (Watters et al., 1997). Passages 3–9 were used for these experiments because the use of early passages was noted to be essential for estrogen effects. Data shown are representative experiments. Each experiment was repeated at least five times. Procedures used for transfection are as reported previously (Watters et al., 1997), exceptN-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate was the method used for transfection (Boehringer Mannheim, Indianapolis, IN). SK-N-SH cells were transfected in six-well plates at ∼80–90% confluency. Luciferase assays were done according to the manufacturer’s protocol for cell lysis and luciferase measurement using a kit from Promega (Madison, WI). Luciferase measurements were done in a luminometer. β-Galactosidase assays were performed in duplicate using the cell lysates used for luciferase measurement in 96-well titer plates. The substrate o-nitrophenyl β-D-galactopyranoside (Sigma, St. Louis, MO) was in accordance with the kit from Promega. Average normalized luciferase activity units for a representative experiment would be ∼17,000 for vehicle treatment of the NT–luciferase reporter construct, and estrogen induction would be ∼35,000 luciferase units. One microgram each of PKI, REVAB, and KCREB constructs was added to existing NT/pCH110 concentrations, and 1 μg of pGEM7Z was added to wells not receiving these DNA constructs to ensure equal amounts of DNA. Cells were treated with the various doses of water-soluble 17β-estradiol (Sigma), 10 μm forskolin (Sigma), 1 μm tamoxifen (Research Biochemicals, Natick, MA), or 1 μm ICI 182,780 (Zeneca Ltd., London, England) for 8 hr. Protein kinase inhibitors H89, bisindolylmaleimide II (BIM; Calbiochem, San Diego, CA), and KN-62 (Research Biochemicals) were used at a final concentration of 5 μm, and cells were pretreated for 1 hr before stimulation. The doses of inhibitor used were the same as those determined to block the effects of forskolin and phorbol ester on NT/N gene transcription.
cAMP measurement. Cells for cAMP determination were grown as for transfection and labeled for 3 hr in phenol red-free MEM (with supplements) and 10% charcoal-stripped calf serum with [3H]adenine (NEN, Natick, MA). After labeling, cells were washed twice with PBS and replaced with fresh medium for 2 hr and then treated in triplicate for various times with 3 nm 17β-estradiol and 200 μmisobutylmethylxanthine (IBMX; Sigma), a phosphodiesterase inhibitor. After estrogen treatment, medium was removed, 5% TCA containing 1 μm cold cAMP was added, and cells were allowed to precipitate overnight at 4°C. cAMP assays were performed by the method described previously (Wong et al., 1991). Actinomycin D experiments were performed as above, except cells were pretreated for 30 min with 1 μm actinomycin D before stimulation of cAMP with estrogen.
Protein kinase A activity assay. PKA kinase activity was assayed on cell homogenates as described (Clegg et al., 1987) using Kemptide (Peninsula Laboratories, Belmont, CA) as a substrate for the enzyme. Assays were done in the presence or absence of 5 μm cAMP. Residual kinase activity measured in the presence of 4 μg/ml PKI peptide (Sigma) was subtracted. Hypothalami from wild-type controls, Cβ knock-out, and RIIβ knock-out mice were dissected (n ≥ 3), immediately frozen on dry ice, and stored at −80°C until the day of assay.
Western blotting. SK-N-SH cells grown as above were treated with 200 μm IBMX and 3 nm water-soluble 17β-estradiol for the times indicated or with water vehicle in the presence of IBMX. Mice were treated intraperitoneally with 100 μg of tamoxifen for 15 or 75 min. Nuclear extracts from MPON and SK-N-SH cells were obtained as reported previously (Watters et al., 1996), except 5 μm Microcystin L-R (Sigma) was added to buffers A and B just before use. Western blots were performed as described bySambrook et al. (1989). A total of 10 or 15 μg of nuclear extract protein were loaded per well, and gels were transferred to nitrocellulose membranes (Amersham, Arlington Heights, IL). Anti-PCREB antisera 8466 kindly provided by Dr. David Ginty (Johns Hopkins, Baltimore, MD) and the anti-CREB antibodies (Upstate Biotechnology, Lake Placid, NY) were used at a final dilution of 1:5000. Methods used have been reported elsewhere (Ginty et al., 1993). Bands were visualized using the ECL reagent (Amersham). PCREB immunoreactivity was normalized to CREB immunoreactivity to control for unequal protein loading. Bands for tamoxifen induction of PCREB in vivo were quantitated using the MCID system, as above. Quantitation of estrogen induction of PCREB in SK-N-SH cells was performed using the NIH Image software program (National Institutes of Health, Bethesda, MD).
Statistics. Statistical analyses were performed using the ANOVA pre hoc test and the Scheffe F test or Fisher PLSD tests for post hoc significance. Significance levels were set at 95% confidence limits. Data are represented as means ± SEM.
RESULTS
Rapid effects of estrogen on NT/N expression in the MPON of mouse brain are unresponsive to estrogen receptor antagonism
Tamoxifen is one of the few antagonists that have been previously used to antagonize the effects of estrogen in the brain after peripheral administration (Wade et al., 1993a,b). NT/N mRNA levels were significantly reduced (p < 0.05) in ovariectomized vehicle-replaced mice when compared with all other treatments, including intact controls (Fig.1A,B). EB rapidly increased (within 2 hr) NT/N mRNA levels above those of ovariectomized animals, which remained elevated when measured 8 hr after injection. Tamoxifen alone acted agonistically to increase NT/N mRNA levels at both the 3 and 8 hr time points. Furthermore, when administered before EB, it failed to block the effects of estrogen on NT/N gene expression. These data imply that the in vivo effects of estrogen on the NT/N gene are not mediated by the classical mechanism of estrogen action. Tamoxifen interferes with estrogen binding to the ligand-binding domain of the estrogen receptor protein. Although estrogenic activity of tamoxifen has been noted in other tissues (for review, see Kuo and Runowicz, 1995), these effects have been attributed to effects on estrogen receptor function that involve actions other than those mediated by estrogen receptor–ERE (Webb et al., 1995). The inability of tamoxifen to block the effects of estrogen on this gene, and its apparent potent agonistic activity, suggest that cross-talk with other signaling pathways could be involved. An alternate explanation may involve the recent finding that estrogen receptor antagonists such as tamoxifen interacting with the estrogen receptor β (ERβ) cause transactivation at AP-1 sites (Paech et al., 1997). This is most likely not the mechanism used here, because cotransfection of ERβ along with NT in our cell model system failed to augment the estrogen responsiveness of the NT reporter construct (data not shown).
Fig. 1.

A, Effect of estrogen and estrogen antagonists on NT/N mRNA measured by in situhybridization in the MPON of the mouse brain. EB (1 μg) was administered intraperitoneally to bilaterally ovariectomized (OVX) females. Mice were pretreated with T (100 μg) for 30 min before EB exposure. Intact animals received injections of sesame oil vehicle and were killed 8 hr after treatment. EB- and T-treated animals were killed at the times indicated after agonist or antagonist exposure. A, Intact; B, OVX and vehicle-replaced; C, OVX + EB treatment for 2 hr;D, OVX + EB treatment for 8 hr; E, OVX + T treatment for 3 hr; F, OVX + T treatment for 8 hr;G, OVX + EB + T treatment for 8 hr. B, Graphical depiction of NT/N mRNA autoradiograms. They-axis denotes optical density of MPON after subtraction of background. n ≥ 3 animals per treatment group. *p < 0.05 from all other treatments.C, Western blot indicating PCREB immunoreactivity in the MPON of mouse brain after tamoxifen exposure for 15 and 75 min. Eachlane represents 15 μg of protein from a pool of MPON nuclear extract from three or more mice per group. Top panel, PCREB immunoreactivity level; bottom panel, CREB immunoreactivity level. D, Graphical representation of the optical density of the ratio of PCREB to CREB-IR after tamoxifen treatment of mice.
The estrogen receptor antagonist tamoxifen induces phosphorylation of the CREB in mouse brain
Because tamoxifen treatment was unable to block the effects of estrogen on NT/N gene transcription and was in fact an agonist, we investigated the ability of this estrogen receptor antagonist to induce the phosphorylation of CREB in the MPON of mice. Tamoxifen increased the phosphorylation of CREB within 15 min, and the increase persisted at the 75 min time point (Fig. 1C,D). Levels of CREB protein itself were not altered over this time course. The antisera recognizes the phosphorylation of Ser-133 that is essential for transcriptional activation by the CREB protein.
In vitro modeling of estrogen-induced transcription of NT/N- and CRE-containing constructs in the SK-N-SH neural cell line
Figure 2A depicts a dose–response curve for the effects of estrogen on an NT/N promoter–luciferase construct transiently transfected into SK-N-SH cells. The lowest dose to elicit a maximal effect on NT/N reporter gene expression was 3 nm 17β-estradiol. Also shown is the effect of forskolin, a direct activator of adenylate cyclase, on the expression of the NT/N reporter construct. It elicited a threefold to fourfold induction of the NT/N reporter, whereas estrogen elicited a twofold to threefold stimulation. Figure 2Billustrates the effect of E2 on a luciferase reporter construct containing the promoter of the α subunit of the glycoprotein hormones, α168, a gene well documented to be regulated by cAMP-dependent mechanisms (Delegeane et al., 1987). The α168 reporter construct contains 168 nucleotides of the promoter region of the α subunit gene, which includes the cAMP-responsive elements. As was noted with the NT/N promoter, estradiol induces the expression of this gene at a similar concentration.
Fig. 2.
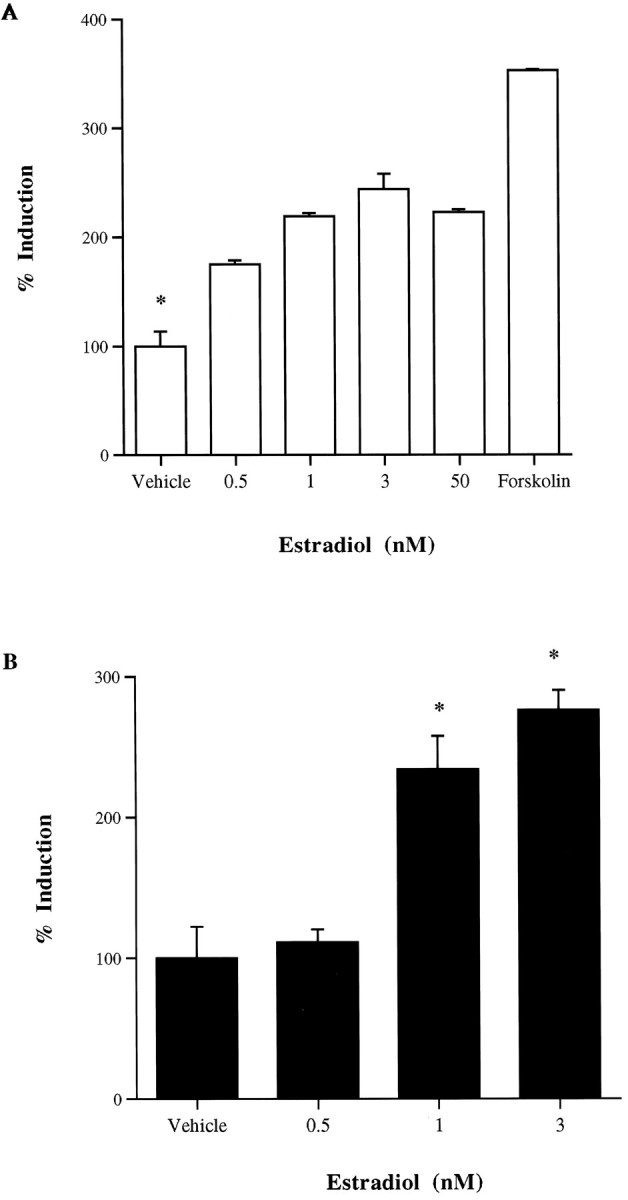
Dose–response curves depicting the effect of 17β-estradiol (E2) treatment on cAMP-responsive reporter gene constructs transiently transfected into SK-N-SH cells.A, NT/N promoter luciferase construct; B, the α168 luciferase construct. Forskolin was administered at a dose of 10 μm, and vehicle-treated wells received water vehicle.
The effects of estrogen on cAMP accumulation are independent of gene transcription in SK-N-SH cells
The accumulation of intracellular cAMP that was maximal at 60 min and returned to baseline within 90 min of treatment was induced by 3 nm 17β-estradiol in the presence of IBMX (Fig.3A). Given the protracted period of this effect, it seemed possible that estrogen could be acting on nuclear estrogen receptors to enhance the expression of estrogen-inducible genes that promote cAMP accumulation at 60 min. Therefore, we performed identical experiments in the presence of the transcription inhibitor actinomycin D. A dose of actinomycin D (1 μm), previously determined to block ERE-mediated transcription in this cell line, was added to cells 30 min before estrogen exposure and failed to block the increases in cAMP observed with estrogen treatment.
Fig. 3.

A, Effect of estrogen and actinomycin D on cAMP accumulation in SK-N-SH cells. Cells were treated with 3 nm estrogen alone, estrogen and 1 μmactinomycin D together, or water vehicle in the presence of 200 μm IBMX for the times indicated. Data are graphed as percent vehicle-induced cAMP accumulation for estrogen treatment alone or as vehicle-induced cAMP accumulation in the presence of actinomycin D. *p < 0.05 from vehicle-treated cAMP levels.B, Western blot depicting estrogen induction of PCREB immunoreactivity in SK-N-SH cells. Ten micrograms of nuclear extract protein were loaded per lane. PCREB immunoreactivity is denoted in thetop panel, and the bottom panel indicates CREB immunoreactivity in the same cells. C, Quantitative ratio of optical densities of PCREB to CREB immunoreactivities of Western blot bands.
Estrogen induces the phosphorylation of the cAMP response element-binding protein in SK-N-SH cells
Because the peak of estrogen-induced cAMP accumulation occurred at 60 min after estrogen treatment of these cells, we reasoned that the phosphorylation of the CREB protein should occur within 5–15 min after activation of PKA (Fig. 3B, top panel). Time points earlier and later were also evaluated, with no noticeable change in phosphorylation status of the CREB protein. In contrast, using an antibody to monitor total CREB nuclear protein, no change was evident in the amount of CREB itself (Fig. 3B, middle panel). Figure 3B, bottom panel, illustrates the quantitative depiction of the PCREB-to-CREB ratio of immunoreactivity as measured by optical density. A pronounced increase in PCREB-IR was noted between 65 and 75 min after estrogen treatment. The ratio of phosphorylated CREB to total CREB-IR increased dramatically over this period. This response waned by 90 min.
Effect of estrogen receptor antagonists and various protein kinase inhibitors on NT/N gene expression in SK-N-SH cells
The estrogen receptor antagonists tamoxifen and ICI 182,780 failed to block estrogen-induced NT/N gene transcription (Fig.4A). In fact, as observed in vivo with tamoxifen, it and ICI 182,780 acted as agonists of the NT/N gene response, and when coadministered elicited the same response as estradiol itself. Figure 4Billustrates the ability of H89, a potent and selective PKA inhibitor, to block the transcription of the NT/N reporter gene construct induced by estrogen. H89 was also able to inhibit the induction of the NT/N gene induced by tamoxifen. Other protein kinase inhibitors, BIM selective for several protein kinase C isoforms, and KN-62, an inhibitor of calmodulin kinase II, were incapable of blocking the effect of estrogen on transcription of the NT/N construct. Together these data suggest a selective role for PKA.
Fig. 4.

A, Effect of estrogen receptor antagonists on estrogen-induced NT/N gene transcription in SK-N-SH cells. NT/N gene expression is induced by 3 nm estrogen. Pretreatment with 1 μm tamoxifen (T) or 1 μm ICI 182,780 (I) fails to block the effects of estrogen. One micromolar tamoxifen and I alone and both together have agonistic activity on the NT/N gene. *p < 0.05 from all other treatments. B, Blockade of 3 nm estrogen and 1 μm T induced NT/N gene transcription by 5 μm H89 but not by 5 μmBIM or KN-62. *p < 0.05 from vehicle and E2 + H89 treatments. C, Effect of dominant negative PKA and CREB and overexpression of PKI on estrogen induction of NT/N gene transcription in SK-N-SH cells. KCREB, REVAB, and PKI were cotransfected into SK-N-SH cells with the wild-type NT/N luciferase reporter construct. Vehicle-treated cells received water, and estradiol-treated cells received 10 nm E2. Coexpression of the dominant negative PKA subunit REVAB or of the nonactivatable form of CREB (KCREB) inhibited the effects of estrogen on the NT/N gene. Overexpression of the PKA peptide inhibitor also blocked the effects of estrogen on NT/N transcription. *p < 0.05 from vehicle treatment of NT reporter construct alone.
Effect of dominant negative PKA and CREB and overexpression of PKI in SK-N-SH cells on NT/N gene transcription
A dominant negative PKA construct, REVAB (Clegg et al., 1987), a regulatory subunit that is unable to bind cAMP and therefore is incapable of releasing free catalytic subunit, was cotransfected along with the NT/N reporter construct into SK-N-SH cells. The presence of REVAB inhibited the ability of estrogen to increase NT/N gene transcription (Fig. 4C). Additionally, a dominant negative form of CREB, KCREB (Walton et al., 1992), in which a mutation in the DNA binding domain allows CREB dimerization but not transactivation, also abolished estrogenic induction of the NT/N gene. Last, overexpression of the native peptide inhibitor of PKA, PKI (Day et al., 1989), resulted in blockade of the effects of estrogen on the NT/N gene, further indicating the involvement of cAMP/PKA-mediated mechanisms in the effects of estrogen on NT/N gene expression.
Mice bearing a targeted disruption in PKA are unable to elicit estrogen-induced increases in NT/N mRNA in the MPON
Mice bearing targeted disrupted genes for either the regulatory type IIβ (Brandon et al., 1998) or catalytic β1 (Qi et al., 1996) subunit of the PKA holoenzyme were studied to assess the involvement of the PKA-dependent signaling pathway in mediating both the chronic and acute effects of estrogen on NT/N gene transcription in the brain. RIIβ knock-out mice exhibit region-specific reductions in PKA activity. The cAMP-stimulated PKA activity in the MPON of these knock-out mice was reduced to 50% of wild-type animals (Fig. 5A). This was not true of Cβ1 knock-out mice (Fig.5A). In the chronic paradigm, wild-type, Cβ1−/−, and RIIβ−/−mice were ovariectomized for 7 d and replaced with 10 μg of EB intraperitoneally for 3 d. In both wild-type and Cβ1−/− mice, significant increases in NT/N mRNA were observed after estrogen treatment, whereas in RIIβ knock-out mice, this response was completely absent (Fig.5B,C). However, detectable levels of NT/N mRNA were evident in the MPON of some intact RIIβ knock-out intact animals. Because it was possible that disruption of the RIIβ gene had in some way impaired ERE-dependent transcriptional effects involving the estrogen receptor, we performed experiments on the RIIβ mice to test the ability of estrogen to induce PR mRNA expression in the MPON. PR gene expression was examined because the role of ERE-dependent effects of estrogen on transcription of this gene have been well documented (Kraus et al., 1994). We did this to assure that the lack of effect we observed on NT/N mRNA was not a result of an improperly phosphorylated and functioning estrogen receptor. Mice were treated with 1 μg of EB, 100 μg of tamoxifen, or both for 3 hr, with tamoxifen being administered 30 min before EB. Previous studies had shown that maximal effects of estrogen on PR mRNA in wild-type animals are evident within 6 hr. We observed a significant increase in PR mRNA in response to EB treatment of the RIIβ−/− mice (Fig. 5E). Tamoxifen alone was without effect and when given together with EB reduced the induction elicited by EB alone by ∼50%. In these same animals, NT/N mRNA was unaltered by any of these treatments (Fig.5D), again indicating that the animals are capable of exhibiting an ERE-mediated transcriptional response in preoptic neurons but not one that appears to be PKA-dependent.
Fig. 5.

A, Graph depicting decreased PKA activity levels in MPON of mouse brain. cAMP-stimulated PKA activity is decreased by >50% in RIIβ knock-out mice when compared with Cβ1 knock-out and wild-type controls. Basal PKA activity levels are similar in both wild-type and knock-out animals.B, Composite of coronally sliced mouse brain sections depicting NT/N mRNA expression in the MPON of wild-type (top panel), Cβ1 knock-out (middle panel), and RIIβ knock-out (bottom panel) mouse brains in ovariectomized (OVX; sesame oil vehicle) and ovariectomized and estrogen (10 μg)-replaced animals (OVX +E) for 3 d. C, Graphical representation of NT/N mRNA optical density in the MPON of wild-type, RIIβ, and Cβ1 knock-out animals. *p< 0.05 from OVX animals. D, Graphical representation of NT/N mRNA optical density measured in the MPON of RIIβ knock-out animals ovariectomized and acutely treated with 1 μg of EB, 100 μg of T, or both together for 3 hr. E, Graphical representation of PR mRNA optical density measured in the MPON. EB elicited a significant increase in PR mRNA in the MPON (*p < 0.05), which was significantly reduced 50% by pretreatment of animals with T.
DISCUSSION
We have used a human neuroblastoma cell line, SK-N-SH, as a model in which to study the molecular events through which estrogen promotes expression of the neuropeptidergic gene NT/N. In vivo, estrogen alters the expression of neurotensin as it varies throughout the estrous cycle in various estrogen receptor-rich regions of the rat brain. The time course of estrogen-dependent induction of neurotensin gene expression in the MPON is very rapid and occurs within 2–3 hr after peripheral estrogen treatment.
In cultured neuroblastoma cells, estrogen drives the expression of a luciferase NT/N promoter–luciferase construct maximally at a concentration of 3 nm, well within the physiological range of estrogen concentrations encountered by the rodent hypothalamus, because the hormone varies throughout the estrous cycle (Bixo et al., 1986). Consensus ERE-like elements are not present in the promoter region fused to the luciferase reporter. We have shown previously that tamoxifen behaves as an antagonist of the effects of estrogen on ERE-mediated gene transcription in SK-N-SH cells both in the wild-type state and when the human estrogen receptor (ERα) is overexpressed in these cells (Watters et al., 1997). However, tamoxifen acts as a full agonist to induce NT/N–luciferase, which strongly suggests that the effects of both estrogen and tamoxifen on NT/N transcription in this cell line do not involve ERE-mediated mechanisms.
Estrogen increases NT/N–luciferase activity in these cells approximately twofold to threefold, an effect comparable with that of forskolin, a direct activator of adenylate cyclase. Estrogen also elicits an increase in intracellular cAMP in these neural cells, which peaks within 1 hr after treatment. It appears that this increase in cAMP allows estrogen to drive the expression of the CRE-containing promoter α168 and the NT/N promoter. The increase in cAMP induced by estrogen was not blocked by actinomycin D, a transcription inhibitor, indicating that although the period of estrogen activation of cAMP is delayed, gene transcription is likely not required for the effects of estrogen on cAMP. In MCF-7 cells, estrogen promotes an increase in cAMP that is maximal at 1 hr. Aronica et al. (1994) have reported that these increases in cAMP were caused not by inhibition of a phosphodiesterase but by the activation of adenylate cyclase. Additionally, we found that estrogen promotes the phosphorylation of the cAMP response element-binding protein in SK-N-SH cells, an event that is necessary for transcriptional activation of the CREB protein and subsequently of promoters containing CREs. The phosphorylation of CREB can be performed on the Ser-133 residue by several enzymes, including PKA, which appears to be activated in these cells, by virtue of the increases in cAMP elicited by estrogen. The time course of CREB phosphorylation we have observed is consistent with this hypothesis.
In our study, estrogen and tamoxifen action was blocked by addition of H89, a potent and selective PKA inhibitor, and by cotransfection of dominant negative CREB and PKA regulatory subunit, implying a central role for PKA and the phosphorylated form of CREB in the effects of estrogen on the NT/N promoter. Additionally, overexpression of the endogenous PKA peptide inhibitor PKI also blocked the effects of estrogen on NT/N transcription.
The estrogen receptor antagonists tamoxifen and ICI 182,780 failed to block the effects of estrogen on the NT/N reporter gene construct. In fact, both mimic the effects of estrogen itself. These antagonists have also been reported to increase cAMP in MCF-7 cells (Aronica et al., 1994). In vivo, tamoxifen was unable to reverse the effects of estrogen on NT/N gene transcription and, in addition, promoted both the phosphorylation of the CREB protein and subsequent activation of NT/N expression in the MPON. Tamoxifen is a partial estrogen receptor antagonist, and in certain tissues such as the uterus, it exerts agonistic effects (for review, see Kuo and Runowicz, 1995). The estrogenic effects of tamoxifen that have been noted in other tissues have been attributed to effects on estrogen receptor function that involve actions other than those mediated by estrogen receptor–ERE interactions (Webb et al., 1995) possibly via AP-1 sites. Tamoxifen has been shown to block the actions of estrogen in brain, both on estrogen-induced sexual behavior and induction of progesterone receptor mRNA (McKenna et al., 1992). ICI 182,780 interferes with activated hormone–receptor complex binding to DNA by preventing dimerization and nuclear translocation. Because ICI 182,780 was unable to block the effects of estrogen on NT/N transcription in this cell line, DNA binding of the activated estrogen receptor also appears not to be involved. Interestingly, overexpression of ERα in these cells along with the neurotensin–luciferase reporter construct failed to further augment the effects of estrogen on the NT/N gene (our unpublished observation). Taken together, these data suggest that ERα and classic estrogen receptor–ERE transactivation is not involved in the transcription of the NT/N gene.
An alternative explanation is suggested by studies in MCF-7 cells (Fujimoto and Katzenellenbogen, 1994). In these breast cancer cells, activation of PKA reduces the antagonist activity of tamoxifen as measured using ERE-containing reporter constructs, reducing the antagonist effects of tamoxifen in cells in proportion to the degree to which PKA was activated. Conversely, the agonist activity of tamoxifen was enhanced. This effect was also shown to be promoter-specific and was not noted in all ERE reporter constructs tested. Thus, it is possible that by virtue of the cAMP increases induced by estrogen in SK-N-SH cells (and perhaps in MPON neurons), PKA activation modulates the agonist and antagonist potency of tamoxifen on transcription of the NT/N gene. Alternatively, the estrogen receptor present in these cells may differ from the classical estrogen receptor. This potentially novel estrogen receptor appears to have the ability to transactivate an ERE to a small degree, and it appears to be capable of eliciting increases in intracellular levels of cAMP. Recently, a novel estrogen receptor, termed ERβ, has been cloned from the rat prostate, having significant homology in the DNA binding domain but not in the ligand binding domain of the estrogen receptor protein (Kuiper et al., 1996). Thus, there is evidence suggesting the existence of at least one alternate receptor for estrogen, potentially possessing different ligand specificities. A further possibility is that tamoxifen may be acting as an agonist through its previously documented ability to augment transactivation via AP-1 sites present in the promoters of certain genetic origin (Webb et al., 1995). This interaction has been described in uterine cells but does not appear to occur in cells of breast. In HeLa cells, Webb et al. (1995) showed that the estrogen receptor forms a protein complex with c-Jun and c-Fos proteins and acts to facilitate transcription of AP-1-containing reporter constructs but not of ERE-containing constructs. A raloxifene response element (RRE) has been elucidated recently that differs in nucleotide sequence from the ERE and appears to support transactivation induced by metabolites of estrogen and by the estrogen receptor antagonist raloxifene (Yang et al., 1996). Sequence analysis of the NT/N reporter used in our studies revealed that a segment of DNA is present that exhibits 75% identity to the RRE. It is therefore possible that some of the agonist effects of the estrogen receptor antagonists and of estrogen itself we observe on the NT/N gene might partly be mediated through this homologous site. Another potential explanation for how the estrogen receptor antagonists function as agonists of the NT/N response is given by a recent report that transactivation by ERβ at AP-1 sites occurs when the receptor is complexed with ER antagonists (Paech et al, 1997). Thus, there are several potential explanations for the agonist effect of the estrogen receptor antagonists used in our experiments. It will be necessary to further characterize the estrogen receptor present in SK-N-SH cells to determine which of these mechanisms is involved in the effects noted here. It is conceivable that the estrogen receptor present in these cells also exists in vivo and may be responsible for the estrogen-dependent modulation of cAMP reported in the literature for several decades.
A role for PKA in mediating the effects of estrogen in vivois implied by the fact that the knock-out mice deficient in the RIIβ subunit of the PKA holoenzyme, but not those for the Cβ1subunit, lack the ability to induce NT/N gene transcription. RIIβ−/− mice show a 50% decrease in cAMP-stimulated PKA activity in the hypothalamus, whereas PKA activity in the same region of CB1−/− animals are similar to the wild-type controls. Estrogen does, however, elicit increases in PR mRNA in the RIIβ−/− animals, an effect that is believed to be via ERE-mediated events (Savouret et al., 1991). This strongly suggests that although the knock-out animals retain the ability to mount a “classical” estrogen response, they are deficient in transcriptional responses involving PKA-dependent signaling.
Given these results, it is also possible that estrogen, or antiestrogens such as tamoxifen, may have an effect on NT/N mRNA stability in addition to the transcriptional effects we have observedin vivo. This mechanism has been shown to be involved in the pulsatility of luteinizing hormone-releasing hormone secretion in the rat hypothalamus (Maurer and Wray, 1997). Although post-transcriptional mechanisms appear not to be involved in the modulation of NT/N promoter–luciferase constructs in SK-N-SH cells, this mechanism might very well be used in addition to transcriptional mechanisms in the intact brain. It is also possible that PKA activation by estrogen in the brain might indirectly increase the stability of NT/N mRNA by activating another protein that might bind to and stabilize the mRNA. More experiments directed at determining the processes induced by estrogen in vivo, in relation to the mRNA for NT/N, would be needed to answer this question specifically. In a more general sense, estrogen might affect the expression of many neuropeptide genes in the brain using post-transcriptional modifications of their mRNAs either solely or in addition to other mechanisms such as transcription.
Our data represent the first report of transcriptional effects of estrogen involving cross-talk with another signal transduction pathway in neurons both in vitro and in vivo. They provide a possible mechanism by which estrogen is able to regulate the expression of several neuropeptidergic genes with promoters that are devoid of classical estrogen response elements. Both our in vitro findings on cAMP-dependent gene transcription and the lack of effect of estrogen in a mouse model deficient in PKA activity provide evidence that estrogen may exert effects on the expression of numerous target genes containing cAMP response elements. More importantly, these data illustrate the potential importance of cross-talk signaling as a relevant feature of steroid hormone action in the brain.
Footnotes
This work was supported by Pharmacological Sciences Training Grant GM670489 (J.J.W.) and United States Public Health Service Grants NS20311 and AG05136 (D.M.D.). We thank Cong Xu and Monique Adams for their laboratory help and assistance and helpful scientific discussions. We also thank Elena Chartoff and Sherry Neher for their help with preparation of this manuscript. Also, we are grateful to the following individuals for providing reagents: Dr. Paul Dobner for the NT–luciferase construct, Dr. Richard Goodman for the KCREB construct, Dr. Richard Maurer for the PKI construct, Dr. Gene Erwin for the mouse NT/N riboprobe plasmid, and Dr. David Ginty for the 8466 anti-PCREB antibody. Additionally, we thank Zeneca Ltd. for donating the ICI 182,780 compound used in our experiments and Dr. Daniel Storm for generously allowing us to perform the cAMP assays in his laboratory. Last, we thank Dr. Stanley McKnight, Rejean Idzerda, and Eugene Brandon for so generously providing the PKA knock-out mice that were used in our studies and for performing the PKA assay on the hypothalami of these mice.
Correspondence should be addressed to Dr. Daniel M. Dorsa, Department of Pharmacology and Psychiatry, University of Washington, Box 356560, Seattle, WA 98195.
REFERENCES
- 1.Adams MR, Brandon EP, Chartoff EH, Idzerda RL, Dorsa DM, McKnight GS. Loss of haloperidol induced gene expression and catalepsy in protein kinase A-deficient mice. Proc Natl Acad Sci USA. 1997;94:12157–12161. doi: 10.1073/pnas.94.22.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander MJ, Leeman SE. Estrogen-inducible neurotensin immunoreactivity in the preoptic area of the female rat. J Comp Neurol. 1994;345:496–509. doi: 10.1002/cne.903450403. [DOI] [PubMed] [Google Scholar]
- 3.Alexander MJ, Dobner PR, Miller MA, Bullock BP, Dorsa DM, Leeman SE. Estrogen induces neurotensin/neuromedin N messenger ribonucleic acid in a preoptic nucleus essential for the preovulatory surge of luteinizing hormone in the rat. Endocrinology. 1989a;125:2111–2117. doi: 10.1210/endo-125-4-2111. [DOI] [PubMed] [Google Scholar]
- 4.Alexander MJ, Miller MA, Dorsa DM, Bullock BP, Melloni RH, Jr, Dobner PR, Leeman SE. Distribution of neurotensin/neuromedin N mRNA in rat forebrain: unexpected abundance in hippocampus and subiculum. Proc Natl Acad Sci USA. 1989b;86:5202–5206. doi: 10.1073/pnas.86.13.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexander MJ, Kiraly ZJ, Leeman SE. Sexually dimorphic distribution of neurotensin/neuromedin N mRNA in the rat preoptic area. J Comp Neurol. 1991;311:84–96. doi: 10.1002/cne.903110107. [DOI] [PubMed] [Google Scholar]
- 6.Aronica SM, Kraus WL, Katzenellenbogen BS. Estrogen action via the cAMP signaling pathway: stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc Natl Acad Sci USA. 1994;91:8517–8521. doi: 10.1073/pnas.91.18.8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Axelson JF, Shannon W, Van Leeuwen FW (1992) Immunocytochemical localization of estrogen receptors within neurotensin cells in the rostral preoptic area of the rat hypothalamus Neurosci Lett [Erratum (1992) 139:141] 136:5–9. [DOI] [PubMed]
- 8.Bixo M, Backstrom T, Winblad B, Selstam G, Andersson A. Comparison between preovulatory and postovulatory distributions of oestradiol and progesterone in the brain of the PMSG-treated rat. Acta Physiol Scand. 1986;128:241–246. doi: 10.1111/j.1748-1716.1986.tb07972.x. [DOI] [PubMed] [Google Scholar]
- 9.Brandon EP, Logue SF, Adams MR, Qi M, Sullivan SP, Matsumoto HM, Dorsa DM, Wehner JM, McKnight GS, Idzerda RL. Detective motor behavior and neural gene expression in RIIβ-protein kinase A mutant mice. J Neurosci. 1998;18:3639–3649. doi: 10.1523/JNEUROSCI.18-10-03639.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brot MD, De Vries GJ, Dorsa DM. Local implants of testosterone metabolites regulate vasopressin mRNA in sexually dimorphic nuclei of the rat brain. Peptides. 1993;14:933–940. doi: 10.1016/0196-9781(93)90069-s. [DOI] [PubMed] [Google Scholar]
- 11.Clegg CH, Correll LA, Cadd GG, McKnight GS. Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. J Biol Chem. 1987;262:13111–13119. [PubMed] [Google Scholar]
- 12.Day RN, Walder JA, Maurer RA. A protein kinase inhibitor gene reduces both basal and multihormone-stimulated prolactin gene transcription. J Biol Chem. 1989;264:431–436. [PubMed] [Google Scholar]
- 13.Delegeane AM, Ferland LH, Mellon PL. Tissue-specific enhancer of the human glycoprotein hormone alpha-subunit gene: dependence on cyclic AMP-inducible elements. Mol Cell Biol. 1987;7:3994–4002. doi: 10.1128/mcb.7.11.3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franklin KBJ, Paxinos G. The mouse brain in sterotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 15.Fujimoto N, Katzenellenbogen BS. Alteration in the agonist/antagonist balance of antiestrogens by activation of protein kinase A signaling pathways in breast cancer cells: antiestrogen selectivity and promoter dependence. Mol Endocrinol. 1994;8:296–304. doi: 10.1210/mend.8.3.7517003. [DOI] [PubMed] [Google Scholar]
- 16.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 17.Harrison RJ, McNeil GP, Dobner PR. Synergistic activation of neurotensin/neuromedin N gene expression by c-jun and glucocorticoids: novel effects of fos family proteins. Mol Endocrinol. 1995;9:981–993. doi: 10.1210/mend.9.8.7476995. [DOI] [PubMed] [Google Scholar]
- 18.Kislauskis E, Dobner PR. Mutually dependent response elements in the cis-regulatory region of the neurotensin/neuromedin N gene integrate environmental stimuli in PC12 cells. Neuron. 1990;4:783–795. doi: 10.1016/0896-6273(90)90205-t. [DOI] [PubMed] [Google Scholar]
- 19.Kislauskis E, Bullock B, McNeil S, Dobner PR. The rat gene encoding neurotensin and neuromedin N. Structure, tissue-specific expression, and evolution of exon sequences. J Biol Chem. 1988;263:4963–4968. [PubMed] [Google Scholar]
- 20.Kraus WL, Montano MM, Katzenellenbogen BS. Identification of multiple, widely spaced estrogen responsive regions in the rat progesterone receptor gene. Mol Endocrinol. 1994;8:952–969. doi: 10.1210/mend.8.8.7997237. [DOI] [PubMed] [Google Scholar]
- 21.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustaffson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo DY, Runowicz CD. Gynaecologic effects of tamoxifen. Med Oncol (Lond) 1995;12:87–94. doi: 10.1007/BF01676708. [DOI] [PubMed] [Google Scholar]
- 23.Leeman SE, Aronin N, Ferris C. Substance P and neurotensin. Recent Prog Horm Res. 1982;38:93–132. doi: 10.1016/b978-0-12-571138-8.50008-1. [DOI] [PubMed] [Google Scholar]
- 24.Malayer JR, Gorski J. An integrated model of estrogen receptor action. Domest Anim Endocrinol. 1993;10:159–177. doi: 10.1016/0739-7240(93)90021-3. [DOI] [PubMed] [Google Scholar]
- 25.Maurer JA, Wray S. Luteinizing hormone-releasing hormone (LHRH) neurons maintained in hypothalamic slice explant cultures exhibit a rapid LHRH mRNA turnover rate. J Neurosci. 1997;17:9481–9491. doi: 10.1523/JNEUROSCI.17-24-09481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna SE, Simon NG, Cologer-Clifford A. An assessment of agonist/antagonist effects of tamoxifen in the female mouse brain. Horm Behav. 1992;26:536–544. doi: 10.1016/0018-506x(92)90020-v. [DOI] [PubMed] [Google Scholar]
- 27.Miller MA, Zoeller RT, Dorsa DM. Detection of vasopressin messenger RNA in cells within the bed nucleus of the stria terminalis by in situ hybridization histochemistry. Neurosci Lett. 1988;94:264–268. doi: 10.1016/0304-3940(88)90028-6. [DOI] [PubMed] [Google Scholar]
- 28.Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakhla AM, Khan MS, Romas NP, Rosner W. Estradiol causes the rapid accumulation of cAMP in human prostate. Proc Natl Acad Sci USA. 1994;91:5402–5405. doi: 10.1073/pnas.91.12.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP 1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 31.Park OK, Mayo KE. Transient expression of progesterone receptor messenger RNA in ovarian granulosa cells after the preovulatory leutenizing hormone surge. Mol Endocrinol. 1991;7:967–978. doi: 10.1210/mend-5-7-967. [DOI] [PubMed] [Google Scholar]
- 32.Qi M, Zhou M, Skalhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking C beta 1 catalytic subunit of cAMP-dependent kinase. Proc Natl Acad Sci USA. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 34.Savouret JF, Bailley A, Mishrahi M, Rauch C, Redeuilh G, Chauchereau A, Milgrom E. Characterization of the hormone responsive element involved in the regulation of the progesterone receptor gene. EMBO J. 1991;10:1875–1883. doi: 10.1002/j.1460-2075.1991.tb07713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szot P, Dorsa DM. Expression of cytoplasmic and nuclear vasopressin RNA after castration and testosterone replacement: evidence for transcriptional regulation. Mol Cell Neurosci. 1994;5:1–10. doi: 10.1006/mcne.1994.1001. [DOI] [PubMed] [Google Scholar]
- 36.Wade GN, Blaustein JD, Gray JM, Meredith JM. ICI 182,780: a pure antiestrogen that affects behaviors and energy balance in rats without acting in the brain. Am J Physiol. 1993a;265:R1392–R1398. doi: 10.1152/ajpregu.1993.265.6.R1392. [DOI] [PubMed] [Google Scholar]
- 37.Wade GN, Powers JB, Blaustein JD, Green DE. ICI 182,780 antagonizes the effects of estradiol on estrous behavior and energy balance in Syrian hamsters. Am J Physiol. 1993b;265:R1399–R1403. doi: 10.1152/ajpregu.1993.265.6.R1399. [DOI] [PubMed] [Google Scholar]
- 38.Walton KM, Rehfuss RP, Chrivia JC, Lochner JE, Goodman RH. A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- 39.Watters JJ, Wilkinson CW, Dorsa DM. Glucocorticoid regulation of vasopressin V1a receptors in rat forebrain. Mol Brain Res. 1996;38:276–284. doi: 10.1016/0169-328x(95)00345-s. [DOI] [PubMed] [Google Scholar]
- 40.Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM. Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen activated protein kinase signalling cascade and c-fos immediate early gene transcription. Endocrinology. 1997;138:4030–4033. doi: 10.1210/endo.138.9.5489. [DOI] [PubMed] [Google Scholar]
- 41.Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443–456. doi: 10.1210/mend.9.4.7659088. [DOI] [PubMed] [Google Scholar]
- 42.Wong YH, Federman A, Pace AM, Zachary I, Evans T, Pouyss’egur J, Bourne HR. Mutant alpha subunits of Gi2 inhibit cyclic AMP accumulation. Nature. 1991;351:63–65. doi: 10.1038/351063a0. [DOI] [PubMed] [Google Scholar]
- 43.Yang NY, Venugopalan M, Hardikar S, Glasebrook A. Identification of an estrogen response element activated by metabolites of 17-B estradiol and roloxifene. Science. 1996;273:1222–1225. doi: 10.1126/science.273.5279.1222. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Y, Watters JJ, Dorsa DM. Estrogen rapidly induces the phosphorylation of the cAMP response element binding protein in rat brain. Endocrinology. 1996;137:2163–2166. doi: 10.1210/endo.137.5.8612562. [DOI] [PubMed] [Google Scholar]