

Abstract
After peripheral nerve injury, macrophages infiltrate the degenerating nerve and participate in the removal of myelin and axonal debris, in Schwann cell proliferation, and in axonal regeneration.In vitro studies have demonstrated the role serum complement plays in both macrophage invasion and activation during Wallerian degeneration of peripheral nerve. To determine its rolein vivo, we depleted serum complement for 1 week in adult Lewis rats, using intravenously administered cobra venom factor. At 1 d after complement depletion the right sciatic nerve was crushed, and the animals were sacrificed 4 and 7 d later. Macrophage identification with ED-1 and CD11a monoclonal antibodies revealed a significant reduction in their recruitment into distal degenerating nerve in complement-depleted animals. Complement depletion also decreased macrophage activation, as indicated by their failure to become large and multivacuolated and their reduced capacity to clear myelin, which was evident at both light and electron microscopic levels. Axonal regeneration was delayed in complement-depleted animals. These findings support a role for serum complement in both the recruitment and activation of macrophages during peripheral nerve degeneration as well as a role for macrophages in promoting axonal regeneration.
Keywords: complement, macrophage, peripheral nerve, Wallerian degeneration, regeneration, axon
The transection of axons in the peripheral nervous system (PNS) leads to a pattern of distal axonal degeneration, followed by myelin degradation and Schwann cell and fibroblast proliferation (Ramon y Cajál, 1928; Griffin and Hoffman, 1993; Fu and Gordon, 1997). One of the most striking cellular responses during Wallerian degeneration (WD) in the PNS is the proliferation and infiltration of macrophages (Griffin et al., 1993;Perry, 1994; Bruck, 1997). Although a group of resident tissue macrophages from the PNS may contribute (Griffin et al., 1993; Vass et al., 1993), macrophages are recruited predominantly from the circulating pool of hematogenous monocytes (Ramon y Cajál, 1928;Beuche and Friede, 1984). Examination of the temporal course of the macrophage response found that it begins within 24 hr of axonal injury and reaches its peak by 14–21 d (Monaco et al., 1992; Avellino et al., 1995).
Macrophages participate in a wide array of cellular responses during WD. Once activated, they release factors that are mitogenic for Schwann cells (Baichwal et al., 1988). Although it appears that Schwann cells can initiate myelin phagocytosis (Bigbee et al., 1987; Stoll et al., 1989), the completion of WD relies on the phagocytic ability of macrophages to degrade myelin and axonal debris (Beuche and Friede, 1984; Hann Bonnekoh et al., 1989; Lunn et al., 1989; Griffin et al., 1992). In addition, macrophages can degrade molecules inhibitory to axonal regeneration (David et al., 1990; Bedi et al., 1992) as well as release factors, such as interleukin-1 (IL-1), which can promote axonal growth via the induction of neurotrophic factors such as nerve growth factor (NGF) (Heumann et al., 1987; Lindholm et al., 1987).
The precise mechanisms responsible for macrophage recruitment during WD are not completely understood. One group of factors that may play a role in macrophage recruitment and activation is the serum complement proteins. The importance of complement proteins in both traumatic and immune-mediated peripheral nerve injury has been demonstrated previously in vitro. In mixed cultures containing macrophages and peripheral nerve segments, blocking the complement type 3 receptor with monoclonal antibodies prevented the phagocytosis of opsonized myelin (Bruck and Friede, 1990b). Macrophages were unable to invade degenerating nerves when cocultured in C3-deficient serum, and C3-deficient serum applied to nerve cultures after the macrophages had successfully invaded was able to abolish the myelin phagocytic ability of the macrophages (Bruck and Friede, 1991). In experimental allergic neuritis, complement depletion diminished myelin breakdown and macrophage recruitment in vivo. (Feasby et al., 1987; Vriesendorp et al., 1995).
The importance of complement products during WD in vivo remains controversial (Lobato and Griffin, 1993). This study investigated the ability of macrophages to invade a degenerating sciatic nerve and become activated after the depletion of serum complement in vivo. By reducing macrophage infiltration and activation, we had the opportunity to examine the role of macrophages in axonal regeneration. The results provide evidence for the importance of complement and macrophages in both WD and axonal regenerationin vivo.
MATERIALS AND METHODS
Surgery. Adult male Lewis rats (300 gm; Charles River, Wilmington, MA) were anesthetized with intraperitoneal pentobarbital (45 mg/kg), and their depth of anesthesia was assessed via corneal blink responses and respiratory pattern. An indwelling central venous catheter was placed (Remie et al., 1990). The right neck was shaved and swabbed with Betadine solution. The right internal jugular vein was identified and isolated with two 4-0 silk ligatures, and a venotomy was performed. A silicone catheter (0.31 mm inner diameter × 0.57 mm outer diameter, length 42 mm; Dow-Corning, Midland, MI) was inserted into the right atrium, and both ligatures were tightened. The distal port of the catheter was tunneled through the neck to the posterior scalp, attached to a right angle metal connector, and then rigidly fixed to each animal’s skull with methylmethacrylate and microscrews. Wounds were closed with interrupted 3-0 nonabsorbable sutures, and the animals were allowed to recover. Before sciatic crush the catheter was flushed twice weekly with saline and 40% polyvinylpyrrolidine (Sigma, St. Louis, MO) in heparinized saline, 1000 U/ml (w/v). The catheters were monitored for a period of 2–3 weeks before complement was depleted to ensure proper functioning throughout the experiment.
For the crush injury the animals were anesthetized with intravenous pentobarbital (30–45 mg/kg) and again monitored for depth of anesthesia. The lateral thigh was shaved and prepped with Betadine solution. A longitudinal incision was made along the lateral thigh, and the hamstring and gluteal muscles were exposed. The sciatic nerve was identified by dissecting the plane separating these two muscles, and a 15–20 mm length of the nerve was identified. At a point ∼3–5 mm distal to the insertion of the obturator tendon, the nerve was crushed with a jeweler’s forceps (number 5 Taxal) for 30 sec, and the crush site was tagged with a 10-0 Dermalon suture. Then the wound was closed by using 4-0 absorbable sutures for the muscle fascia and surgical clips to approximate the skin. The left sciatic nerve served as the intact control specimen.
Complement depletion. The animals were separated into two groups, depending on whether they underwent depletion of complement (depleted, n = 11) or were treated with saline alone (control, n = 11). Animals were complement-depleted with cobra venom factor (CVF, Diamedix, Miami, FL), 200 U/kg intravenously on the day before crush and 100 U/kg intravenously daily until their deaths. Control animals received equal volumes of sterile normal saline. Blood samples were drawn through the indwelling catheter before complement depletion, on the day of surgery (day 0), and on days 4 and 7 after surgery. The serum was collected and stored at −80°C until serum complement values could be measured.
Complement depletion was checked with a standard CH50 assay for hemolysis of sheep erythrocytes (Kabat and Mayer, 1971). Sera from animals as well as a standard rat serum (Diamedix) with a known CH50 value were incubated with sensitized sheep erythrocytes, and the absorbance of the supernatants was read at 415 nm. Complement values were expressed as both CH50 units and as a percentage of the standard serum CH50 values.
Histology. Animals were killed at 4 d (n = 3) or 7 d (n = 8) after sciatic nerve crush with an overdose of pentobarbital. The intact (left) and crushed (right) sciatic nerves were removed and blocked to include the nerve 3 mm proximal to and at least 15 mm distal to the crush site. The nerves were frozen in a block of Tissue Tek OCT (Miles, Elkhart, IN), using isopentane cooled with dry ice. Serial 8 μm longitudinal cryostat sections were cut at −20°C, collected on 3-aminopropyltriethoxy-silane (ASA)-coated slides, and stored at −80°C.
Immunohistochemistry. Sections for immunohistochemistry were reacted by using the avidin–biotin–peroxidase complex (ABC) method (Hsu et al., 1981). Slides were removed from the freezer, air-dried at room temperature for 1 hr, and then fixed in cold acetone for 10 min. Slides were preincubated in serum blocking solutions depending on the antibody reaction (ED-1 and CD11a: 50% rat and 2% horse serum in 0.1m PBS; neurofilament: 5% horse in 0.1 m PBS) for 1 hr. Then the slides were reacted with primary antibody for 2 hr at room temperature. The following antibodies were used: (1) ED-1 (1:1000; Serotec, Oxford, UK), (2) neurofilament, 2F11 clone against 200 and 70 kDa epitomes (1:200; Dako, Copenhagen, Denmark), and (3) CD11a (1:500; a gift of ICOS, Seattle, WA). After serial washes in PBS-Tween, the sections were incubated for 30 min at room temperature with secondary antibody (1:200 to 1:400; rat-absorbed biotinylated horse, anti-mouse IgG, Vector Laboratories, Burlingame, CA). After being washed, the ABC reagents (Vectastain-Elite kit, Vector) were added to each slide for 1 hr at room temperature. Visualization of the antibodies was achieved by using 3,3-diaminobenzidine (DAB; Sigma), with 3% hydrogen peroxide added for 10 min. In the neurofilament reaction, 1% (w/v) nickel sulfate was added to the DAB as a chromogen. Sections were washed in distilled water, followed by dehydration in a series of graded ethanols to xylene, and coverslips were mounted with Permount (Fisher Scientific, Pittsburgh, PA).
Electron microscopy. Three animals from each of the 7 d groups had tissue harvested for electron microscopy (EM). The sciatic nerves were harvested, and a 3 mm section of the distal tibial nerve was removed and immersion-fixed in Rager’s solution for 48 hr. Sections were stored at 4°C in 4% (w/v) formaldehyde in 0.1m sodium cacodylate solution before being processed for EM. Then the tissues were rinsed in 0.1 m sodium cacodylate buffer for 1 hr and osmicated in 1% osmium tetroxide for 2 hr at room temperature. The tissue was rinsed in buffer and sequentially dehydrated in a series of ethyl alcohols (EtOH) until reaching 100% EtOH and then infiltrated in a graded series of EtOH/Spurr’s resin (Electron Microscopy Sciences, Fort Washington, PA) from 50 to 100%. After infiltration in the 100% Spurr’s resin overnight, the tissue was embedded in fresh Spurr’s and polymerized at 70°C for 12 hr. Ultrathin sections were cut on a Reichert–Jung Ultracut E microtome and viewed on a JEOL 100 electron microscope.
Quantification. All counts were performed by an observer blinded to the conditions of the experiment. Macrophage counts were performed at 200× magnification by counting all positively stained cells that contained a visible nucleus in three randomly chosen fields in each degenerating nerve segment. The fields were all chosen at least 3 mm distal to the crush site, and all counts were performed on sections cut at 8 μm thickness. The area of each field was measured with a digitizing tablet (Microcomputer Imaging Device, IBM; software version 4.20, Imaging Research, St. Catherine’s, Ontario, Canada), which converted screen pixels to surface area; a standard area was chosen so that identical volumes from each nerve were examined. Then the counts were normalized to an area of 0.1 mm2, and a mean value was determined for each nerve.
Because of the difference in macrophage size between complement-depleted and control groups, it is possible that small macrophages could be missed at a higher frequency than large macrophages. We addressed this possibility by counting only ED-1-labeled cells that had a visible nucleus. The size of the nucleus did not differ when macrophages from the two treatment groups were compared (0.58 × 10−4 mm2for the control group vs 0.59 × 10−4mm2 for the complement-depleted group). If no nucleus was observed, the stained area was assumed to be a fragment of macrophage cytoplasm and was not counted. By counting only the ED-1-labeled cells with a nucleus, we should have removed any bias introduced by differences in cell size.
Cell surface area measurements were performed on slides immunostained with ED-1 on animals 7 d after sciatic nerve crush. A standard area of 0.1 mm2 was identified at least 3 mm distal to the crush site, and every immunostained cell within the chosen area was outlined with the digitizing tablet. Then the resulting pixel area was converted to area in square millimeters. Three 0.1 mm2 areas were examined per nerve so that identical volumes were examined from each nerve, and a mean macrophage surface area was determined for each nerve.
Axon counts were performed at 400× magnification by counting axons stained with neurofilament antibody at points 3 and 10 mm from the crush site. Only axons crossing a 25 μm line placed perpendicular to the longitudinal axis of the nerve were counted, and counts were expressed as the number of stained axons crossing this line. All counts were performed in triplicate, and a mean value was determined for each nerve.
Quantification of myelin morphology was performed at 25× magnification by measuring the optical density of sections stained with Erichrome Cyanine R. The section measured was 8–10 mm distal to the crush site, and only sections 7 d after crush without cutting artifact were chosen for analysis (n = 7 for each group). The camera was set to capture all stained tissue within a standard pixel area. A proportional area of stained tissue was generated by measuring the number of pixels above the optical density threshold and dividing by the total pixel area of the section analyzed (stained tissue/total tissue). An identical pixel area was examined for each nerve.
Statistical analysis. Means and SEs were determined for each time point and each experimental group. The statistical significance of count differences between experimental and control groups at the same time point was tested with a paired Student’s t test. Differences between time points were tested with the one-way ANOVA. Statistical significance was set at p < 0.05.
RESULTS
Complement depletion
In all of the animals treated with CVF, the serum complement values had fallen to <5% of control values by the time of surgery (day 0, mean CH50 2.1 ± 0.3 vs 46.9 ± 8.9 for control) and remained at <12% of control values at 4 and 7 d when they were killed. An attempt to deplete complement over a 14 d time course showed that values had returned to levels >60% of control values (data not shown).
Cell counts
All counts were expressed as the number of positively stained cells per 0.1 mm2 of tissue area. Staining with the ED-1 antibody revealed that in the intact nerve there was a small number of macrophages primarily in the perineurium and perivascular spaces with an elongated ramified appearance. There were no differences in ED-1 counts in intact nerves of control and complement-depleted animals (8.2 ± 0.8 vs 8.4 ± 1.6). At 4 d there was a significant increase in the number of macrophages in the crushed nerves in both groups. Although there was a trend toward fewer macrophages in the complement-depleted animals, the difference did not reach statistical significance (23.4 ± 5.3 vs 27.2 ± 2.3). There was a further increase in macrophage number at 7 d after crush, and now the difference between complement-depleted and control animals was significant (31.2 ± 2.1 vs 38.9 ± 2.1; Figs.1A,2A–D).
Fig. 1.
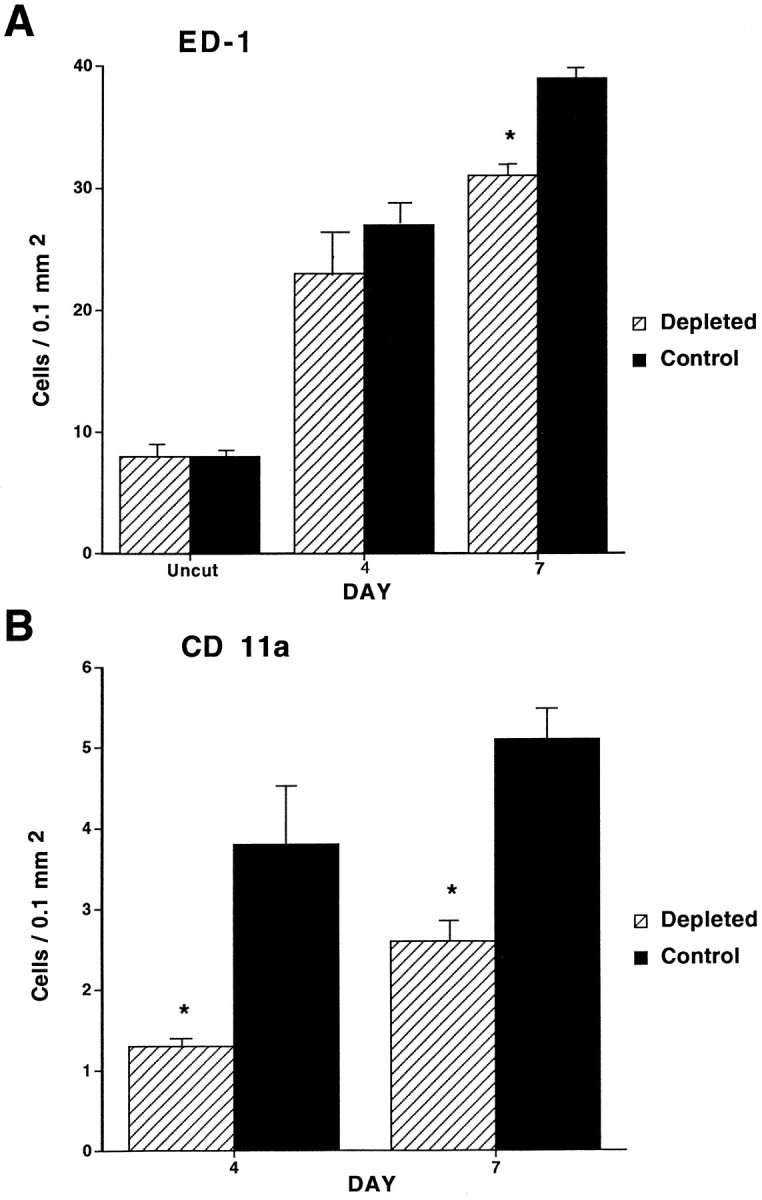
Graphs depicting the macrophage response in control and complement-depleted animals. Counts represent the number of macrophages per 0.1 mm2 stained with either ED-1 (A) or CD11a (B) antibodies. A, In intact nerves there is no difference in macrophage counts between control and complement-depleted groups. At 4 d after crush there is a trend toward fewer macrophages in the complement-depleted animals (23.4 ± 5.3 vs 27.2 ± 2.3;p = 0.1). At 7 d after crush the difference in macrophage counts between the two groups has reached statistical significance (31.2 ± 2.1 vs 38.9 ± 2.1; *p < 0.05). B, In intact nerves there is no staining with the CD11a antibody. At both 4 d (1.3 ± 0.1 vs 3.8 ± 1.3; *p < 0.05) and 7 d (2.6 ± 0.3 vs 5.1 ± 0.5; *p < 0.05) after crush the complement-depleted group has a statistically significant reduction in the number of stained cells when compared with the control group.
Fig. 2.
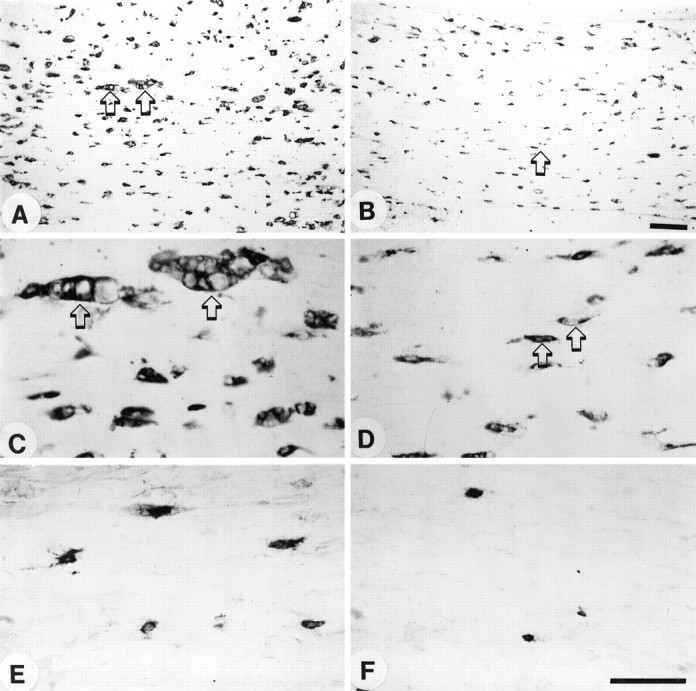
Longitudinal sections of degenerating sciatic nerve stained with either ED-1 or CD11a antibodies at 7 d after crush injury. A–D, Staining with ED-1 antibody.E, F, Staining with CD11a antibody.A, C, E, Control animal.B, D, F, A complement-depleted animal. Note the reduced number of macrophages in the complement-depleted animal (B) in comparison to the control animal (A). C, Shown at higher power are large multivacuolated macrophages (arrows) in a control animal as compared with the much smaller macrophages in the complement-depleted animal (D, arrows). E,F, Shown are small CD11a antibody-staining cells only, presumably representing recently recruited macrophages. Many more CD11a-staining cells are seen in the control (E) than in the complement-depleted (F) animal. Scale bars: in A, B, 0.1 mm; inC–F, 0.05 mm.
Intact nerves had no CD11a-staining cells. At 4 d after crush there was a population of small round cells, most likely representing newly recruited monocytes, which were immunostained with CD11a. More CD11a-stained cells were found in control animals than in complement-depleted animals (1.3 ± 0.1 vs 3.8 ± 1.3). At 7 d after crush, both groups had more stained cells than at 4 d, with complement-depleted animals having fewer macrophages than control animals (2.6 ± 0.3 vs 5.1 ± 0.5; Figs.1B, 2E,F).
Macrophage morphology
One of the most striking differences between control and complement-depleted animals lies in the morphology of ED-1-stained macrophages 7 d after crush. Control animals had numerous large multivacuolated macrophages that appeared to be actively engaging in phagocytosis (Fig. 3B). Far fewer of these large multivacuolated cells were seen in complement-depleted animals, with most of the macrophages more closely resembling the elongated ramified macrophages seen in intact nerves (Figs. 3A,C). Macrophages from complement-depleted animals had a smaller mean cell surface area than those from control animals (1.31 ± 0.04 vs 2.50 ± 0.23 × 10−4 mm2), a difference that was statistically significant (p < 0.05). In addition, there were none of the very large macrophages (>7 × 10−4 mm2) in complement-depleted animals (Fig. 4). However, as pointed out in Materials and Methods, there was no difference in nuclear size of the macrophages between the complement-depleted and control groups (0.59 ± 0.02 vs 0.58 ± 0.06 × 10−4 mm2).
Fig. 3.
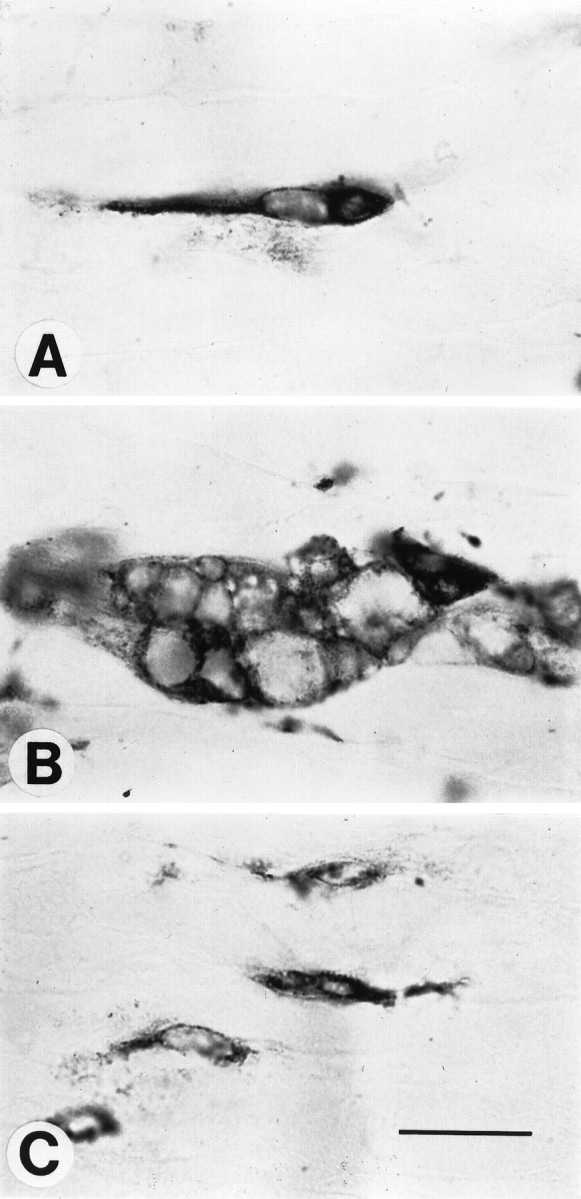
Macrophages in different states of activation.A, Shown is a ramified resident macrophage from an intact nerve in a control animal. B, Shown is an example from a control animal of a large multivacuolated foamy macrophage in a degenerating nerve crushed 7 d previously. C, Shown are macrophages in a degenerating nerve crushed 7 d previously from a complement-depleted animal. These macrophages resemble the ramified endogenous macrophages seen in intact nerves. Scale bar, 0.02 mm.
Fig. 4.

Histogram depicting macrophage cell size, expressed in surface area, in control (n = 8) and complement-depleted (n = 8) degenerating nerves 7 d after crush. Note that the complement-depleted group has a greater proportion of macrophages that are <2 × 10−4 mm2, very few cells that are >3 × 10−4 mm2, and no cells that are >7 × 10−4mm2. The mean cell area was significantly smaller for the complement-depleted group (1.31 ± 0.04 × 10−4 mm2) than for the control group (2.50 ± 0.23 × 10−4mm2; p < 0.05).
Myelin breakdown
Myelin was examined by light and electron microscopy. At the light microscopic level, staining with Erichrome Cyanine R allowed for the comparison of the gross morphology of intact and degenerating nerve. In the intact nerves the myelin profiles extend as regular tube-like structures along the longitudinal axis of the nerve. Each myelin profile appears to have a fairly consistent diameter extending along the section of the nerve. After 7 d following crush, there is a tendency for the nerves from the control animals to lose the pattern of myelin staining so that areas within the nerve now have abundant myelin ovoids, many clear spaces between myelin sheaths, and a much greater variance in the diameters of the myelin profiles. In the nerves of complement-depleted animals the clear spaces between profiles are not so prominent and numerous, and the diameter along myelin profiles does not vary so significantly as in the control animals (Fig.5). Quantification of the loss of myelin staining shows a greater area of stained tissue (and thus fewer cleared areas) in the complement-depleted group at 7 d after crush than in the control group (proportional area 0.91 for the complement-depleted group vs 0.81 for the control group;p < 0.05). No difference was noted between the two groups at 4 d after crush.
Fig. 5.
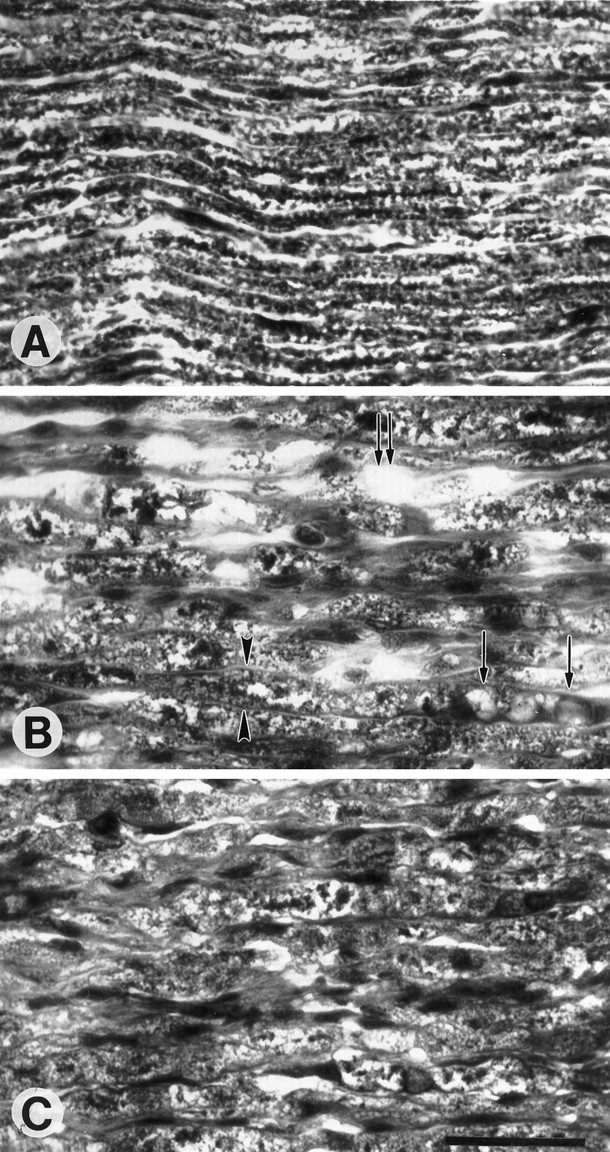
Longitudinal sections of degenerating peripheral nerve stained with Erichrome Cyanine R to depict myelin profiles.A, Represented is an intact nerve from a control animal. Note the regular elongated tube-like myelin profiles. B, Shown is a degenerating nerve from a control animal 7 d after crush in which myelin ovoids (single arrows) and clear spaces (double arrows) have replaced the normal myelin sheaths. Persistent myelin sheaths have varying diameters, with some exhibiting markedly increased space between the basal laminae (arrowheads). C, A degenerating nerve from a complement-depleted animal shows greater preservation of myelin. Scale bar, 0.05 mm.
EM displayed the difference in myelin clearance between the two groups more clearly. In the control animals the macrophages with large irregular nuclei containing a ring of dark heterochromatin were prominent. The cytoplasm formed pseudopodia, often appearing to make contact with the degenerating myelin sheaths as the cells phagocytosed the debris. In addition, the cytoplasm contained fragments of ingested myelin as well as lipid droplets. Some sections revealed myelin debris and lipid droplets packed within a basement membrane; these structures represented the axons that had degenerated and left the bands of Bungner (Fig. 6A,B). In contrast, EM sections taken from complement-depleted animals showed many remaining myelin sheaths. However, the myelin sheaths had begun to collapse and separate from the surrounding Schwann cell cytoplasm, forming concentric circles within the basement membrane. Occasional cells with dark irregular nuclei were observed and represented infiltrating monocytes that had not yet engaged in phagocytosis (Fig.6C). Thus, although there is evidence of WD in complement-depleted animals, it does not appear to have progressed to the same degree as in the control animals.
Fig. 6.
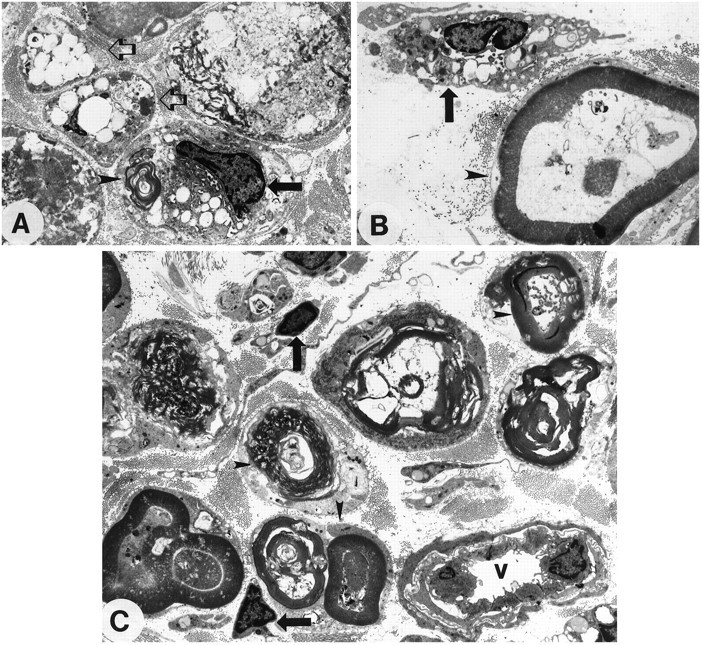
Representative electron micrographs from degenerating nerves 7 d after crush from control (A, B) and complement-depleted (C) animals. In A, an intratubal macrophage (black arrow) is associated with lipid vacuoles and myelin fragments (arrowhead) within a degenerating myelin sheath. Two adjacent structures (clear arrows) depict basement membrane containing lipid vacuoles and cellular remnants, but no intact myelin. B, Shown is a macrophage (arrow) with an irregular nucleus, dark heterochromatin, and pseudopodia engaging a myelin sheath (arrowhead). In a complement-depleted animal (C) the myelin remains visible within the sheath (arrowheads), although it may have collapsed and separated from the surrounding Schwann cell cytoplasm. There are some small macrophages (arrows) that have not yet engaged in the phagocytosis of myelin. In the bottom right corneris a blood vessel (v) containing monocytes that have not yet entered the degenerating nerve segment. All pictures are displayed at 800× original magnification.
Axonal regeneration
Neurofilament staining with the pan-neurofilament antibody 2F11 was used as a marker for axonal regeneration. At 4 and 7 d after crush there was less axon staining in the complement-depleted animals than in the control animals (Fig. 7). At 4 d after crush the control animals showed a twofold increase in stained axons 3 mm from the crush site (5.7 ± 0.7 vs 10.0 ± 1.0). No animals had any axon staining at 10 mm from the crush site 4 d after the crush. The difference between complement-depleted and control animals persisted at 7 d after crush both 3 mm (15.0 ± 0.9 vs 21.8 ± 0.9) and 10 mm (10.7 ± 0.6 vs 15.9 ± 0.5) from the crush site (Fig.8). In addition, the front of regenerating axons in the complement-depleted animals lagged behind the front in the control animals, with the maximal point of regeneration at 12–13 mm in the complement-depleted nerves, as compared with 15–17 mm in the control animals at 7 d after crush. Several animals within the control group had axons that extended up to the limits of the harvested tissue, suggesting that the point of maximal regeneration may have been farther than 17 mm in the control animals.
Fig. 7.

Longitudinal sections of nerves stained for regenerating axons with a neurofilament antibody at 7 d after crush injury. These sections were taken 10 mm distal to the crush site.A and B are low-power whereasC and D are high-power photomicrographs. Sections from a control animal (A, C) show a greater number of regenerated axons than in a complement-depleted animal (B, D). In addition, the complement-depleted animal has a greater amount of residual neurofilament debris (arrowheads,D) as compared with the control animal (C). Scale bar, 0.05 mm.
Fig. 8.

Graph showing the counts of neurofilament-stained regenerating axons at different times and different points from the crush site. At both 4 and 7 d after crush, complement-depleted animals have fewer regenerating axons than control animals. At 4 d after crush, only the 3 mm point was examined (5.7 ± 0.7 vs 10.0 ± 1.0; *p< 0.05). At 7 d after crush, both 3 mm (15.0 ± 0.9 vs 21.8 ± 0.9; **p < 0.001) and 10 mm points (10.7 ± 0.6 vs 15.9 ± 0.5; ***p < 0.001) were examined.
DISCUSSION
The role of complement in macrophage infiltration
Intact peripheral nerves contain a resident population of macrophages that comprise from 2 to 9% of the cells (Oldfors, 1980;Monaco et al., 1992; Griffin and George, 1993). Our results show equal numbers of elongated and ramified resident macrophages in intact nerves from both complement-depleted and control animals. The importance of infiltrating macrophages during WD was suggested initially by Ramon y Cajál (1928) when he described cells of hematogenous origin that had invaded the basement membrane of Schwann cells and then phagocytosed myelin debris. Although Schwann cells have been proposed as the primary cell of myelin phagocytosis (Nathaniel and Pease, 1963), the experiments of Beuche and Friede (1984) firmly established the role of macrophages in WD. By preventing nonresident cells from infiltrating a degenerating nerve isolated intraperitoneally with a Millipore chamber, a substantial reduction in the intracellular ingestion of myelin resulted. Subsequent investigations using immunohistochemical markers have shown that the recruited cells are indeed macrophages (Perry et al., 1987; Scheidt and Friede, 1987; Hann Bonnekoh et al., 1989). Macrophage infiltration and proliferation during WD of peripheral nerve in vivo may begin as early as day 1 and reaches a maximum response between 14 and 21 d after a nerve transection injury (Stoll et al., 1989; Monaco et al., 1992; Perry, 1994; Avellino et al., 1995).
The signals modulating macrophage recruitment during WD of peripheral nerve remain incompletely understood. One group of molecules that have been implicated in this process is the complement proteins. When degenerating sciatic nerves were cocultured with peritoneal macrophages, the use of C3 deficient serum prevented the infiltration of macrophages (Bruck and Friede, 1991). During the T-cell-mediated process of experimental allergic neuritis, depletion of complement reduced the number of macrophages infiltrating the PNS (Feasby et al., 1987; Vriesendorp et al., 1995). Reduced macrophage recruitment might be the result of a decrease in C3 cleavage products, particularly the chemoattractant C5a, which has been implicated in monocyte locomotion and adhesion (Rollins and Springer, 1985; Springer, 1994).
In our experiments depletion of complement reduced the number of macrophages at both 4 and 7 d after sciatic nerve crush injury when compared with control animals. However, the number of macrophages in complement-depleted animals did rise above baseline levels, suggesting that other factors are involved in macrophage recruitment during WD. We could not deplete complement beyond 7 d, because animals then begin to make antibodies to CVF, which makes the treatment ineffective (Feasby et al., 1987).
The role of complement in macrophage activation
Our experiments allowed us to investigate the role of complement in the activation of infiltrating macrophages that participate in the clearance of myelin and axonal debris. One marker of macrophage activation is cell size (Papadimitriou and Ashman, 1989). In crushed nerves undergoing WD we found a significant difference in macrophage morphology and size between control and complement-depleted animals (see Fig. 3). The mean surface area of macrophages in control nerves 7 d after crush was significantly larger than in complement-depleted animals (2.50 ± 0.23 × 10−4 mm2 vs 1.31 ± 0.04 × 10−4 mm2). Control nerves contained a population of very large and multivacuolated macrophages that appeared to be ingesting myelin and axonal debris. Most macrophages in complement-depleted nerves had an elongated and ramified appearance consistent with a reduced state of activation.
Macrophage infiltration involves the expression of cell adhesion molecules (CAMs), which mediate their transendothelial migration into the degenerating nerve (Yong and Khwaja, 1990; Springer, 1994). The CAM CD11a is the α-subunit of the integrin LFA-1 that binds ICAM-1, present on endothelial cells, and mediates the diapedesis of circulating monocytes into areas of injury. In vitro studies have shown increased expression of CD11a when macrophages become activated (Stent et al., 1995). Blocking CD11a with monoclonal antibodies prevented macrophages from migrating across the blood–brain barrier and thus improved the outcome in a model of experimental allergic encephalitis (Gordon et al., 1995). We found a markedly reduced number of cells staining with CD11a in complement-depleted nerves in comparison to control nerves at both 4 and 7 d after crush injury (see Fig. 1B). This result suggests that complement depletion reduces the ability of circulating monocytes to become activated and then to infiltrate a degenerating peripheral nerve.
Phagocytosis of myelin is regulated by complement proteins after traumatic nerve injury (Beuche and Friede, 1986; Bruck and Friede, 1990b, 1991; Bruck, 1997). Although immunoglobulin depletion in sciatic nerve cultures did not interfere with myelin phagocytosis (Hann et al., 1988), depletion of C3 components, blocking of the type 3 complement receptor, and inhibition of the type 3 complement receptor withl-fucosidase all greatly reduced the phagocytosis and clearance of myelin debris in vitro (Bruck and Friede, 1990a,b, 1991). Complement-mediated clearance of myelin proceeds by both classic and alternative pathways, both of which have C3 as an intermediate component (Koski et al., 1985, 1996). Our results providein vivo evidence that serum complement is involved in myelin clearance during WD.
In a number of studies the suppression of macrophage recruitment during WD has been correlated with the preservation of myelin profiles. Silica injection (Beuche and Friede, 1986; Tanaka et al., 1992), whole body irradiation (Perry et al., 1995), and depletion of monocytes with liposomes containing dichloromethylene diphosphonate (Bruck et al., 1996) have all been used to reduce macrophage recruitment in vivo during WD of peripheral nerve. Although Schwann cells may initiate myelin breakdown in the absence of macrophages (Stoll et al., 1989; Reichert et al., 1994; Fernandez-Valle et al., 1995), macrophages serve to complete the process of myelin breakdown in the degenerating nerve segment (Stoll et al., 1989; Bruck, 1997). The preservation of myelin profiles in complement-depleted animals observed at both the light and electron microscopic levels in the current study demonstrates the importance of complement-mediated phagocytosis by macrophages during WD.
The role of macrophages in axonal regeneration
The reduced macrophage response seen in complement-depleted animals provided us with the opportunity to examine the role of macrophages on peripheral nerve regeneration in vivo. Our experimental findings demonstrate a reduced number of regenerating axons in complement-depleted animals at both 4 and 7 d after crush nerve injury. Previous experiments using the C57BL/Ola mouse showed how delayed degeneration of axons could lead to a delay in both macrophage recruitment and the clearance of myelin debris (Lunn et al., 1989;Perry et al., 1990; Glass et al., 1993; Bruck et al., 1995). This slow WD response was noted initially to produce a delay in the regeneration of sensory axons (Lunn et al., 1989; Brown et al., 1992). Further investigations showed a delay in the regeneration of motor axons as well (Chen and Bisby, 1993; Brown et al., 1994).
Other experiments have shown that axons can grow on predegenerated, but not intact, peripheral nerve sections in vitro (Bedi et al., 1992). Several studies provide evidence for the important role of macrophages in promoting axonal regeneration in degenerating peripheral nerve. Regeneration of dorsal root ganglia axons was enhanced by providing a substrate rich in macrophages (Lu and Richardson, 1991;Hikawa et al., 1993; Miyauchi et al., 1997). Conversely, reducing the macrophage response in an injured peripheral nerve reduced axonal regeneration (Tanaka et al., 1992; Calcutt et al., 1994; Dahlin, 1995;Miyauchi et al., 1997).
One explanation for reduced peripheral nerve regeneration in the setting of a reduced macrophage response is that myelinated Schwann cells express molecules, such as chondroitin sulfate proteoglycan, which inhibit regeneration and must be broken down to allow for axonal regeneration (Braunewell et al., 1995; Zuo and Muir, 1997). Matrix metalloproteinases, which are products of neurons and activated macrophages (La Fleur et al., 1996), have been shown to degrade chondroitin sulfate proteoglycan and eliminate the inhibitory effects of these molecules on axonal regeneration in vitro(Ferguson et al., 1997).
Another potential explanation is that macrophages modulate the production of neurotrophic and/or neurotropic molecules that promote axonal regeneration (Nathan, 1987; Perry and Brown, 1992). For example, the increase in NGF found in degenerating peripheral nerve appears to be mediated, at least in part, by IL-1, which is a secretory product of macrophages (Heumann et al., 1987; Lindholm et al., 1987; Taniuchi et al., 1988). In the C57BL/Ola mouse a reduced macrophage recruitment has been correlated with low levels of NGF mRNA and poor axonal regeneration (Brown et al., 1991a,b).
In summary, we have shown that complement depletion reduces the number of infiltrating macrophages during WD and markedly decreases their state of activation. In addition, serum complement-depleted animals showed a delay in the regeneration of axons after a peripheral nerve crush injury. The reduced macrophage response not only diminishes the clearance of cellular debris but also may curtail the production of trophic/tropic factors that promote axonal regeneration. Therefore, the macrophage response during Wallerian degeneration of peripheral nerve appears to be an important determinant of axonal regeneration in vivo.
Footnotes
This work was supported by funds from the Spinal Cord Research Foundation (A.D. and M.K.), a Clinical Investigator Development Award from National Institutes of Health (M.K.), and Program Project Grant NS 70144 to the Department of Neurological Surgery, University of Washington (A.D., A.A., and M.K.). We thank Kate Andrus for her excellent technical advice and assistance, Paul Schwartz and Janet McClardy for their photographic expertise, and Dr. H. R. Winn for his generous support.
Correspondence should be addressed to Dr. Andrew T. Dailey at his present address: Department of Neurosurgery, University of Utah, 3B-409 SOM, 50 North Medical Drive, Salt Lake City, UT 84132.
REFERENCES
- 1.Avellino AM, Hart D, Dailey AT, MacKinnon M, Ellegala D, Kliot M. Differential macrophage responses in the peripheral and central nervous system during Wallerian degeneration of axons. Exp Neurol. 1995;136:183–198. doi: 10.1006/exnr.1995.1095. [DOI] [PubMed] [Google Scholar]
- 2.Baichwal RR, Bigbee JW, DeVries GH. Macrophage-mediated myelin-related mitogenic factor for cultured Schwann cells. Proc Natl Acad Sci USA. 1988;85:1701–1705. doi: 10.1073/pnas.85.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bedi KS, Winter J, Berry M, Cohen J. Adult rat dorsal root ganglion neurons extend neurites on predegenerated but not on normal peripheral nerves in vitro. Eur J Neurosci. 1992;4:193–200. doi: 10.1111/j.1460-9568.1992.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 4.Beuche W, Friede RL. The role of non-resident cells in Wallerian degeneration. J Neurocytol. 1984;13:767–796. doi: 10.1007/BF01148493. [DOI] [PubMed] [Google Scholar]
- 5.Beuche W, Friede RL. Myelin phagocytosis in Wallerian degeneration depends on silica-sensitive, bg/bg-negative and Fc-positive monocytes. Brain Res. 1986;378:97–106. doi: 10.1016/0006-8993(86)90289-1. [DOI] [PubMed] [Google Scholar]
- 6.Bigbee JW, Yoshino JE, Devries GH. Morphological and proliferative responses of cultured Schwann cells following rapid phagocytosis of a myelin-enriched fraction. J Neurocytol. 1987;16:487–496. doi: 10.1007/BF01668503. [DOI] [PubMed] [Google Scholar]
- 7.Braunewell KH, Martini R, LeBaron R, Kresse H, Faissner A, Schmitz B, Schachner M. Upregulation of a chondroitin sulphate epitope during regeneration of mouse sciatic nerve: evidence that the immunoreactive molecules are related to the chondroitin sulphate proteoglycans decorin and versican. Eur J Neurosci. 1995;7:792–804. doi: 10.1111/j.1460-9568.1995.tb00682.x. [DOI] [PubMed] [Google Scholar]
- 8.Brown MC, Lunn ER, Perry VH. Poor growth of mammalian motor and sensory axons into intact proximal nerve stumps. Eur J Neurosci. 1991a;3:1366–1369. doi: 10.1111/j.1460-9568.1991.tb00069.x. [DOI] [PubMed] [Google Scholar]
- 9.Brown MC, Perry VH, Lunn ER, Gordon S, Heumann R. Macrophage dependence of peripheral sensory nerve regeneration: possible involvement of nerve growth factor. Neuron. 1991b;6:359–370. doi: 10.1016/0896-6273(91)90245-u. [DOI] [PubMed] [Google Scholar]
- 10.Brown MC, Lunn ER, Perry VH. Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. J Neurobiol. 1992;23:521–536. doi: 10.1002/neu.480230507. [DOI] [PubMed] [Google Scholar]
- 11.Brown MC, Perry VH, Hunt SP, Lapper SR. Further studies on motor and sensory nerve regeneration in mice with delayed Wallerian degeneration. Eur J Neurosci. 1994;6:420–428. doi: 10.1111/j.1460-9568.1994.tb00285.x. [DOI] [PubMed] [Google Scholar]
- 12.Bruck W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997;7:741–752. doi: 10.1111/j.1750-3639.1997.tb01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruck W, Friede RL. l-Fucosidase treatment blocks myelin phagocytosis by macrophages in vitro. J Neuroimmunol. 1990a;27:217–227. doi: 10.1016/0165-5728(90)90072-u. [DOI] [PubMed] [Google Scholar]
- 14.Bruck W, Friede RL. Anti-macrophage CR3 antibody blocks myelin phagocytosis by macrophages in vitro. Acta Neuropathol (Berl) 1990b;80:415–418. doi: 10.1007/BF00307696. [DOI] [PubMed] [Google Scholar]
- 15.Bruck W, Friede RL. The role of complement in myelin phagocytosis during PNS Wallerian degeneration. J Neurol Sci. 1991;103:182–187. doi: 10.1016/0022-510x(91)90162-z. [DOI] [PubMed] [Google Scholar]
- 16.Bruck W, Bruck Y, Maruschak B, Friede RL. Mechanisms of macrophage recruitment in Wallerian degeneration. Acta Neuropathol (Berl) 1995;89:363–367. doi: 10.1007/BF00309630. [DOI] [PubMed] [Google Scholar]
- 17.Bruck W, Huitanga I, Dijkstra CD. Liposome-mediated monocyte depletion during Wallerian degeneration defines the role of hematogenous phagocytes in myelin removal. J Neurosci Res. 1996;46:477–484. doi: 10.1002/(SICI)1097-4547(19961115)46:4<477::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 18.Calcutt NA, McMurray HF, Moorhouse DF, Bache M, Parthasarathy S, Powell HC, Mizisin AP. Inhibition of macrophage chemotaxis and peripheral nerve regeneration in normal and hyperglycemic rats by the aldose reductase inhibitor Tolrestat. Exp Neurol. 1994;128:226–232. doi: 10.1006/exnr.1994.1131. [DOI] [PubMed] [Google Scholar]
- 19.Chen S, Bisby MA. Impaired motor axon regeneration in the C57BL/Ola mouse. J Comp Neurol. 1993;333:449–454. doi: 10.1002/cne.903330310. [DOI] [PubMed] [Google Scholar]
- 20.Dahlin LB. Prevention of macrophage invasion impairs regeneration in nerve grafts. Brain Res. 1995;679:274–280. doi: 10.1016/0006-8993(95)00249-p. [DOI] [PubMed] [Google Scholar]
- 21.David S, Bouchard C, Tsatas O, Giftochristos N. Macrophages can modify the nonpermissive nature of the adult mammalian central nervous system. Neuron. 1990;5:463–469. doi: 10.1016/0896-6273(90)90085-t. [DOI] [PubMed] [Google Scholar]
- 22.Feasby TE, Gilbert JJ, Hahn AF, Neilson M. Complement depletion suppresses Lewis rat experimental allergic neuritis. Brain Res. 1987;419:97–103. doi: 10.1016/0006-8993(87)90572-5. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson T, Hernandez J, Maria B, Muir D. Neuronal matrix metalloproteinase-2 degrades and inactivates neurite-inhibiting chondroitin sulfate proteoglycan. Soc Neurosci Abstr. 1997;23:21.13. doi: 10.1523/JNEUROSCI.18-14-05203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez-Valle C, Bunge RP, Bunge BM. Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. J Neurocytol. 1995;24:667–679. doi: 10.1007/BF01179817. [DOI] [PubMed] [Google Scholar]
- 25.Fu SY, Gordon T. The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol. 1997;14:67–116. doi: 10.1007/BF02740621. [DOI] [PubMed] [Google Scholar]
- 26.Glass JD, Brushart TM, George EB, Griffin JW. Prolonged survival of transected nerve fibers in C57BL/Ola mice is an intrinsic characteristic of the axon. J Neurocytol. 1993;22:311–321. doi: 10.1007/BF01195555. [DOI] [PubMed] [Google Scholar]
- 27.Gordon EJ, Myers KJ, Dougherty JP, Rosen H, Ron Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;62:153–160. doi: 10.1016/0165-5728(95)00120-2. [DOI] [PubMed] [Google Scholar]
- 28.Griffin JW, George R. The resident macrophages in the peripheral nervous system are renewed from the bone marrow: new variations on an old theme. Lab Invest. 1993;69:257–260. [PubMed] [Google Scholar]
- 29.Griffin JW, Hoffman PN. Degeneration and regeneration in the peripheral nervous system. In: Dyck P, Thomas P, editors. Peripheral neuropathy. Saunders; Philadelphia: 1993. pp. 361–376. [Google Scholar]
- 30.Griffin JW, George R, Lobato C, Tyor WR, Yan LC, Glass JD. Macrophage responses and myelin clearance during Wallerian degeneration: relevance to immune-mediated demyelination. J Neuroimmunol. 1992;40:153–166. doi: 10.1016/0165-5728(92)90129-9. [DOI] [PubMed] [Google Scholar]
- 31.Griffin JW, George R, Ho T. Macrophage systems in peripheral nerves: a review. J Neuropathol Exp Neurol. 1993;52:553–560. doi: 10.1097/00005072-199311000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Hann PG, Beuche W, Neumann U, Friede RL. The rate of Wallerian degeneration in the absence of immunoglobulins. A study in chick and mouse peripheral nerve. Brain Res. 1988;451:126–132. doi: 10.1016/0006-8993(88)90756-1. [DOI] [PubMed] [Google Scholar]
- 33.Hann Bonnekoh PG, Scheidt P, Friede RL. Myelin phagocytosis by peritoneal macrophages in organ cultures of mouse peripheral nerve. A new model for studying myelin phagocytosis in vitro. J Neuropathol Exp Neurol. 1989;48:140–153. doi: 10.1097/00005072-198903000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Heumann R, Korsching S, Bandtlow C, Thoenen H. Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. J Cell Biol. 1987;104:1623–1631. doi: 10.1083/jcb.104.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hikawa N, Horie H, Takenaka T. Macrophage-enhanced neurite regeneration of adult dorsal root ganglia neurones in culture. NeuroReport. 1993;5:41–44. doi: 10.1097/00001756-199310000-00010. [DOI] [PubMed] [Google Scholar]
- 36.Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–580. doi: 10.1177/29.4.6166661. [DOI] [PubMed] [Google Scholar]
- 37.Kabat EA, Mayer MM. Experimental immunochemistry. Thomas; Springfield, IL: 1971. [Google Scholar]
- 38.Koski CL, Vanguri P, Shin ML. Activation of the alternative pathway of complement by human peripheral nerve myelin. J Immunol. 1985;134:1810–1814. [PubMed] [Google Scholar]
- 39.Koski CL, Estep AE, Sawant-Mane S, Shin ML, Highbarger L, Hansch GM. Complement regulatory molecules on human myelin and glial cells: differential expression affects the deposition of activated complement proteins. J Neurochem. 1996;66:303–312. doi: 10.1046/j.1471-4159.1996.66010303.x. [DOI] [PubMed] [Google Scholar]
- 40.La Fleur M, Underwood JL, Rappolee DA, Werb Z. Basement membrane and repair of injury to peripheral nerve: defining a potential role for macrophages, matrix metalloproteinases, and tissue inhibitor of metalloproteinases-1. J Exp Med. 1996;184:2311–2326. doi: 10.1084/jem.184.6.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lindholm D, Heumann R, Meyer M, Thoenen H. Interleukin-1 regulates synthesis of nerve growth factor in non-neuronal cells of rat sciatic nerve. Nature. 1987;330:658–659. doi: 10.1038/330658a0. [DOI] [PubMed] [Google Scholar]
- 42.Lobato GC, Griffin JW. Macrophage response in mast cell-deficient and complement 5-deficient mice. Soc Neurosci Abstr. 1993;19:419. [Google Scholar]
- 43.Lu X, Richardson PM. Inflammation near the nerve cell body enhances axonal regeneration. J Neurosci. 1991;11:972–978. doi: 10.1523/JNEUROSCI.11-04-00972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 45.Miyauchi A, Kanje M, Danielsen N, Dahlin LB. Role of macrophages in the stimulation and regeneration of sensory nerves by transposed granulation tissue and temporal aspects of the response. Scand J Plast Reconstr Hand Surg. 1997;31:17–23. doi: 10.3109/02844319709010501. [DOI] [PubMed] [Google Scholar]
- 46.Monaco S, Gehrmann J, Raivich G, Kreutzberg GW. MHC-positive, ramified macrophages in the normal and injured rat peripheral nervous system. J Neurocytol. 1992;21:623–634. doi: 10.1007/BF01191724. [DOI] [PubMed] [Google Scholar]
- 47.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–326. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nathaniel EJH, Pease DC. Degenerative changes in rat dorsal roots during Wallerian degeneration. J Ultrastruct Res. 1963;9:511–532. doi: 10.1016/s0022-5320(63)80082-9. [DOI] [PubMed] [Google Scholar]
- 49.Oldfors A. Macrophages in peripheral nerves: an ultrastructural and enzyme histochemical study on rats. Acta Neuropathol (Berl) 1980;49:43–49. doi: 10.1007/BF00692218. [DOI] [PubMed] [Google Scholar]
- 50.Papadimitriou JM, Ashman RB. Macrophages: current views on their differentiation, structure, and function. Ultrastruct Pathol. 1989;13:343–372. doi: 10.3109/01913128909048488. [DOI] [PubMed] [Google Scholar]
- 51.Perry HV. Macrophages and the nervous system. Landes; Austin, TX: 1994. [Google Scholar]
- 52.Perry VH, Brown MC. Role of macrophages in peripheral nerve degeneration and repair. BioEssays. 1992;14:401–406. doi: 10.1002/bies.950140610. [DOI] [PubMed] [Google Scholar]
- 53.Perry VH, Brown MC, Gordon S. The macrophage response to central and peripheral nerve injury: a possible role for macrophages in regeneration. J Exp Med. 1987;166:1685–1701. doi: 10.1084/jem.165.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perry VH, Brown MC, Lunn ER, Tree P, Gordon S. Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur J Neurosci. 1990;2:802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x. [DOI] [PubMed] [Google Scholar]
- 55.Perry VH, Tsao JW, Fearn S, Brown MC. Radiation-induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur J Neurosci. 1995;7:271–280. doi: 10.1111/j.1460-9568.1995.tb01063.x. [DOI] [PubMed] [Google Scholar]
- 56.Ramon y Cajál S. Degeneration and regeneration of the nervous system. Oxford UP; Oxford: 1928. [Google Scholar]
- 57.Reichert F, Saada A, Rotshenker S. Peripheral nerve injury induces Schwann cells to express two macrophage phenotypes: phagocytosis and the galactose-specific lectin MAC-2. J Neurosci. 1994;14:3231–3245. doi: 10.1523/JNEUROSCI.14-05-03231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Remie R, Van Dongen JJ, Rensema JW. Permanent cannulation of the jugular vein (acc. to Steffens). In: Van Dongen J, Remie R, Wunnik V, editors. Manual of microsurgery on the laboratory rat, Pt I. Elsevier; New York: 1990. pp. 159–169. [Google Scholar]
- 59.Rollins TE, Springer MS. Identification of the polymorphonuclear leukocyte C5a receptor. J Biol Chem. 1985;260:7157–7160. [PubMed] [Google Scholar]
- 60.Scheidt P, Friede RL. Myelin phagocytosis in Wallerian degeneration. Properties of millipore diffusion chambers and immunohistochemical identification of cell populations. Acta Neuropathol (Berl) 1987;75:77–84. doi: 10.1007/BF00686796. [DOI] [PubMed] [Google Scholar]
- 61.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 62.Stent G, Irving L, Lewin S, Crowe SM. The kinetics of surface expression of CD11/CD18 integrins and CD54 on monocytes and macrophages. Clin Exp Immunol. 1995;100:366–376. doi: 10.1111/j.1365-2249.1995.tb03678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoll G, Griffin JW, Li CW, Trapp BD. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophage in myelin degradation. J Neurocytol. 1989;18:671–683. doi: 10.1007/BF01187086. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka K, Zhang QL, Webster HD. Myelinated fiber regeneration after sciatic nerve crush: morphometric observations in young adult and aging mice and the effects of macrophage suppression and conditioning lesions. Exp Neurol. 1992;118:53–61. doi: 10.1016/0014-4886(92)90022-i. [DOI] [PubMed] [Google Scholar]
- 65.Taniuchi M, Clark HB, Schweitzer JB, Johnson EM., Jr Expression of nerve growth factor receptors by Schwann cells of axotomized peripheral nerves: ultrastructural location, suppression by axonal contact, and binding properties. J Neurosci. 1988;8:664–681. doi: 10.1523/JNEUROSCI.08-02-00664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vass K, Hickey WF, Schmidt RE, Lassman H. Bone marrow-derived elements in the peripheral nervous system: an immunohistochemical and ultrastructural investigation in chimeric rats. Lab Invest. 1993;19:275–282. [PubMed] [Google Scholar]
- 67.Vriesendorp FJ, Flynn RE, Pappolla MA, Koski CL. Complement depletion affects demyelination and inflammation in experimental allergic neuritis. J Neuroimmunol. 1995;58:157–165. doi: 10.1016/0165-5728(95)00006-n. [DOI] [PubMed] [Google Scholar]
- 68.Yong K, Khwaja A. Leucocyte cellular adhesion molecules. Blood Rev. 1990;4:211–225. doi: 10.1016/0268-960x(90)90001-9. [DOI] [PubMed] [Google Scholar]
- 69.Zuo J, Muir D. Neurite-inhibition by chondroitin sulfate proteoglycan in the PNS. Soc Neurosci Abstr. 1997;23:21.10. [Google Scholar]