

Abstract
Metabotropic glutamate receptors (mGluRs) have been implicated in a variety of cellular responses to glutamic acid. The work described in this manuscript extends the role of mGluRs to include protection from oxidative stress-induced programmed cell death. Glutamate analogs regulate inositol-1,4,5 triphosphate mass accumulation in accordance with their ability to protect cells from oxidative glutamate toxicity, and protection appears to take place at the level of glutathione metabolism. Short-term exposure of cells to low concentrations of glutamate desensitizes cells to a subsequent challenge from glutamate. Glutamate exposure upregulates the expression of mGluR5 in hippocampal HT-22 cells and mGluR1 in cortical primary cultures. Finally, group I mGluR agonists also protect cells from death programs initiated by glucose starvation and cystine deprivation.
Keywords: metabotropic receptors, oxidative stress, glutamate, toxicity, cell death, cystine
The identification of the metabotropic glutamate receptor (mGluR) family has greatly expanded the potential cellular responses to glutamate within the nervous system (Nakanishi, 1994). Experiments based primarily on the use of selective mGluR agonists and antagonists have shown that these receptors play roles in synaptic plasticity (Bashir et al., 1993; Manzoni et al., 1994; Riedel and Reymann, 1996), seizure activity (Thomsen et al., 1994), and excitotoxicity (Bruno et al., 1995a,b). The eight mGluRs are G-protein-coupled proteins that have been divided into three subgroups on the basis of sequence homology and pharmacological properties. Group II (mGluRs 2 and 3) and group III (mGluRs 4, 6, 7, and 8) receptors are coupled with the Gi and Go family of G-proteins, whereas group I (mGluRs 1 and 5) receptors couple to Gq/11. Group I mGluRs activate phospholipase C (PLC) to generate inositol-1,4,5 triphosphate (IP3) and diacylglycerol, which have multiple “second messenger” roles (Nakanishi, 1994; Joly et al., 1995; Pin and Duvoisin, 1995). In contrast, groups II and III receptors inhibit adenylyl cyclase and modify ion channel activity (Nakanishi, 1994; Buisson and Choi, 1995;Pin and Duvoisin, 1995). It is likely, however, that many of the roles of mGluRs in the nervous system remain to be discovered.
In addition to the activation of ionotropic and metabotropic glutamate receptors, a third way that glutamate can influence cellular metabolism is via its interaction with the cystine–glutamate antiporter, resulting in the depletion of intracellular cystine–cysteine and the lowering of the cysteine-containing tripeptide glutathione (GSH) (Murphy et al., 1989). The glutamate-induced depletion of GSH leads to oxidative stress and ultimately to cell death. Because glutamate-induced cell death can be blocked by a variety of antioxidants, the phenomenon is known as oxidative glutamate toxicity (Murphy et al., 1989) .
An excellent model for oxidative glutamate toxicity is the hippocampal cell line, HT-22. This immortalized mouse cell line lacks ionotropic glutamate receptors (Maher and Davis, 1996) and responds to oxidative glutamate toxicity with a form of programmed cell death that is distinct from classical apoptosis (Tan et al., 1998a). The signaling pathway that leads to cell death involves the lowering of GSH (Davis and Maher, 1994), the activation of 12-lipoxygenase (Li et al., 1997a), the accumulation of intracellular peroxides (Tan et al., 1998b), and the activation of a cGMP-dependent Ca2+ channel at a point near the end of the death cascade (Li et al., 1997b). A number of conditions protect cells from oxidative glutamate toxicity, including antioxidants (Murphy et al., 1989; Davis and Maher, 1994), growth factors such as epidermal growth factor (Schubert et al., 1992), and the activation of protein kinase C (Davis and Maher, 1994). Because at least one group of the mGluRs is coupled to phospholipase C to generate the second messengers IP3 and diacylglycerol (Nakanishi, 1994), and because diacylglycerol activates protein kinase C, it follows that the mGluRs have the potential to interfere with the signal transduction pathway leading to glutamate-induced cell death. The following experiments show that the activation of mGluRs in HT-22 cells and rat cortical neuron cultures protects cells from glutamate toxicity and other forms of stress. These data outline a novel neuroprotective role for mGluRs that may help to maintain cell viability within the CNS in the presence of excess glutamate.
MATERIALS AND METHODS
Cell culture and toxicity studies. The HT-22 hippocampal nerve cell line is a subclone of HT4 (Morimoto and Koshland, 1990). The HT-22 clone was selected for its sensitivity to glutamate toxicity. The cells do not possess active ionotropic glutamate receptors and are not subject to excitotoxicity (Maher and Davis, 1996). HT-22 cells are propagated in DMEM (Vogt and Dulbecco, 1963) supplemented with 10% fetal bovine serum. Cell survival was determined by the MTT [3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay as described (Schubert et al., 1992), which in this cell system, correlates with cell death as determined by trypan blue exclusion and a colony-forming assay (Davis and Maher, 1994). Briefly, HT-22 cells are dissociated with pancreatin (Life Technologies, Gaithersburg, MD) and seeded onto 96-well microtiter plates in 5% dialyzed fetal bovine serum at a density of 2.5 × 103 cells per well in 100 μl of medium. The next day cells are treated with various reagents according to the experimental design. Twenty hours after the addition of glutamate, 10 μl of the MTT solution (2.5 mg/ml) is added to each well, and the cells are incubated for 4 hr at 37°C. Solubilization solution (100 μl; 50% dimethylfomamide and 20% SDS, pH 4.8) is then added to the wells, and the next day the absorption values at 570 nm are measured. The results are expressed relative to the controls specified in each experiment and were subjected to statistical analysis (Student’s t test).
Primary cortical neurons were prepared from embryonic day 17 Sprague Dawley rats as described (Abe et al., 1990). Cells were dissociated from the cortex and maintained in MEM supplemented with 30 mm glucose, 2 mm glutamine, 1 mmpyruvate, and 10% fetal calf serum. For toxicity studies, cells were plated on polylysine-coated 96-well microtiter dishes at 50,000 cells/100 μl in each well and subjected to the treatments described 24 hr after the initial plating. The effect of various reagents on glutamate toxicity was assessed visually through cell counting and trypan blue exclusion. Results are expressed relative to the untreated controls.
cAMP determination. HT-22 cells were seeded at 2 × 105 cells/60 mm tissue culture dish in DMEM and 5% DFC. Twenty hours later the original medium was replaced with prewarmed DMEM, 5% DFC, and 1 mm IBMX. The test reagents were then added. After 10 min the cells were washed with cold PBS and lysed with 0.1N HCl in ethanol. The cells were scraped from the dish, centrifuged at 20,000 × g for 10 min, and the cAMP content was measured with a radioimmune assay kit (Amersham, Cleveland, OH) and the cAMP normalized to cellular protein.
Phosphoinositol determination. Cells were seeded at 2 × 105/60 mm tissue culture dish. Twenty hours later the test reagents were added for 15 sec at 37°C, after which the cells were washed twice with ice-cold PBS containing 10 mm LiCl. Ten percent trichloroacetic acid (TCA) was then added to the cultures, and the dishes were placed on ice for 15 min. Following high-speed centrifugation, the TCA supernatant was extracted with two volumes of cold Freon; octylamine (4:1 v/v) and the aqueous upper phase were retained. IP3 mass assays were performed using the bovine IP3-binding protein assay (Challiss et al., 1993) supplied in kit form by Amersham.
Cystine uptake. Cystine uptake was measured according toMurphy et al. (1989). Cells were washed once with prewarmed (37°C) HBSS and incubated with 0.5 ml of the same solution for 5 min. Then, the cells were washed as before and replaced with 250 μl of HBSS containing 70 μml-[35S]cystine (40–250mCi/mmol; Amersham, Cleveland, OH). After incubation at 37°C for 45 min, the uptake medium was aspirated, and the cells were washed rapidly three times with cold HBSS and lysed with 200 μl of 0.5 m NaOH. One hundred microliters was used for radioactivity determination, and the remaining was used for protein determination. The rate of uptake was linear up to 60 min. Results were first expressed as the amount of35S incorporated per milligram of protein and then converted to the percentage of the mock-treated sample in each trial.
Total intracellular GSH–GSH disulfide. Cells were washed twice with ice-cold PBS, collected by scraping, and lysed with 10% sulfosalicylic acid. Lysates were incubated on ice for 10 min, and supernatants were collected after centrifugation in an Eppendorf microfuge. On neutralization of supernatants with triethanolamine, total glutathione (reduced and oxidized) concentration was determined by the method described originally by Tietze (1969) and modified byGriffith (1980). Pure GSH was used to obtain the standard curve.
Analysis of mGluRs. Cells were collected directly in 1× Laemmli buffer (Laemmli, 1970). Cell lysates were resolved in 10% polyacrylamide gels containing SDS and electrophoretically transferred to polyvinylidene difluoride hybridization membranes (Micron Separations Inc., Westboro, MA). The membrane was first probed with a rabbit antiserum at a dilution of 1:2000 and then with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody at a dilution of 1:20,000. The antibody conjugates were detected using a chemiluminescence Western blot kit (Amersham, Buckinghamshire, England).
The measurement of reactive oxygen species. Intracellular accumulation of reactive oxygen species (ROS) was determined with 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) (Bass et al., 1983). Briefly, HT-22 cells were seeded at 2.5 × 105/60 mm dish and, 12 hr later, were either mock-treated or treated with the indicated concentrations of glutamate and other agents. After another 12 hr, samples were dissociated from culture dishes with pancreatin in DMEM containing 10 μmH2DCF-DA for 10 min at 37°C and washed once with room temperature DMEM (without phenol red) supplemented with 2% dialyzed FCS. The use of pancreatin did not affect the outcome of flow cytometric experiments as confirmed by fluorescence microscopy. The final cell suspension contained 10 μg/ml propidium iodide (PI). Flow cytometric analysis was performed using a FACScan instrument (Becton Dickinson, San Jose, CA) with 488 nm for the excitation and 525 nm for the emission wavelengths. Data were collected in list mode on 10,000 cells after gating for characteristic forward versus orthogonal light scatter and low PI fluorescence to exclude dead cells. Median fluorescence intensities of control and test samples were determined with CellQuest software (Becton Dickinson).
Reagents. Tissue culture reagents were purchased from Life Technologies, and the MEM used for cortical neurons was from Sigma (St. Louis, MO). The fluorescent dye 2′, 7′ H2DCF-DA was from Molecular Probes (Eugene, OR). The mGluR agonists and antagonists were all from Tocris Cookston, and the mGluR 1 and 2/3 antisera were from Chemicon (Temecula, CA). Anti-mGluR5 was a gift from Dr. R. Gereau (The Salk Institute, La Jolla, CA). The remaining reagents were from Sigma.
RESULTS
Group I mGluR antagonists potentiate glutamate toxicity
If mGluRs are involved in nerve cell toxicity, they may have either a protective role in glutamate toxicity or their activation may cause or potentiate toxicity. To examine the possible role of mGluRs in oxidative glutamate toxicity and other forms of stress, both short-term cortical primary cultures and the mouse hippocampal cell line HT-22 were studied. HT-22 cells lack ionotropic glutamate receptors (Maher and Davis, 1996) and are readily killed by exogenous glutamate via the oxidative pathway (Davis and Maher, 1994; Li et al., 1997a,b; Maher and Davis, 1996; Tan et al., 1998a,b). Cortical neurons in culture for <1 week also lack ionotropic glutamate receptors and are killed by glutamate via the oxidative pathway (Murphy and Baraban, 1990; Ratan et al., 1996). Because glutamate itself is the toxic agent in these experiments, mGluR antagonists are required to distinguish between the potential toxic and protective roles of these receptors. In the presence of marginally toxic levels of glutamate, mGluR antagonists would be predicted to potentiate glutamate toxicity if the activation of mGluRs by glutamate has a protective role and reduce glutamate toxicity if their activation potentiates toxicity. Figure1A shows a typical dose–response relationship between glutamate and cell viability. Half-maximal toxicity is ∼1.5 mm glutamate. Like glutamate, the mGluR agonists (±)-1-aminocyclopentane-trans-1,3, dicarboxylic acid (ACPD) and quisqualate are both directly toxic to cells, at least in part by virtue of inhibiting cystine uptake (see below). A newly identified group I agonist, (R,S)-3,5-dihydroxyphenylglycine (DHPG), is, however, nontoxic. To determine whether mGluR antagonists potentiate toxicity and to identify the pharmacological properties of the receptors involved, cells were preincubated for 30 min with a variety of mGluR antagonists, followed by the addition of 1 mm glutamate. In 1 mm glutamate alone, ∼90% of the cells survive. Figure 1B and Table1 show that two antagonists of phospholipase C linked group I mGluR activity, (R,S)-1-aminoindan-1,5-dicarboxylic acid (AIDA) and dl-2-amino-3-phosphonopropionic acid (AP-3), potentiate toxicity at relatively low concentrations, whereas at least 10-fold higher concentrations of group II and III antagonists are required for a similar effect. AP-3 is a noncompetitive and apparently irreversible inhibitor of group I mGluR-mediated IP3 hydrolysis (Schoepp et al., 1990) whereas AIDA is a potent competitive antagonist of group I receptors with a preference for mGluR1 (Moroni et al., 1997).
Fig. 1.

mGluR agonists and antagonists modify glutamate toxicity. Exponentially dividing cells were plated in 96-well plates as described in Materials and Methods, and 20 hr later the indicated reagents were added. Cell viability was determined by the MTT assay 24 hr later and verified by visual counting. All experiments were repeated at least three times with similar results. The error bars are the mean of triplicate assay points ± SEM. A, HT-22: ACPD, ×–×; glutamate, •–•; DHPG, ○–○; quisqualate, ▵–▵; U-73122, ▴–▴; AP-3, ■–■; and AIDA, ▪–▪.B, HT-22: AIDA, ×–×; AP-3, •–•; DHPG, ○–○; or U-73122 ▵–▵ were added to HT-22 cells 20 hr after plating at the indicated concentrations. Glutamate was added 30 min later at 1 mm to AIDA, U-73122, and AP-3 and at 1.5 mm to DHPG-containing cultures, and cell viability was determined by the MTT assay 24 hr later. Eighty-six percent of the cells survived in the presence of 1 mm glutamate alone. Because potentiation of toxicity was being assayed, the data were plotted with the 86% survival normalized to 100% survival for the sake of comparison with other data throughout the text. In the presence of the higher 1.5 mm glutamate concentration, only 51% of the cells survived. Because protection by DHPG was being assayed, 51% was normalized to zero (baseline) survival, and maximum protection (51–100%) was plotted as 100%. C, Cortical primary cultures: 2 d after plating, the culture medium was removed and replaced with fresh medium. To one set of cultures, AP-3 or AIDA was added at different concentrations followed by the addition of 2 mm glutamate. Viable cell number was determined 24 hr later by the MTT assay and verified by visual counting. In the absence of AIDA or AP-3, 81% of the cells survived. Cell death was potentiated by both AIDA (×–×) and AP-3 (•–•). Data are normalized to 81% survival, which is represented as 100% in the Figure. In another experiment, cells were exposed to increasing concentrations of DHPG (○–○) followed by 5 mm glutamate. In 5 mmglutamate alone, 47% of the cells survived. These data are normalized to 47% as 0% survival.
Table 1.
Effects of mGluR antagonists
| Reagent | Antagonist | HT22 | Primary |
|---|---|---|---|
| AIDA | Group I | 110 μm | 200 μm |
| AP-3 | Group I | 300 μm | 1 mm |
| E4CPG | Group I and II | 1 mm | 1.5 mm |
| EGLU | Group II | 2.5 mm | >5 mm |
| CPPG | Group III | 3 mm | >5 mm |
| MAP4 | Group II and III | >3 mm | >3 mm |
The above reagents were tested for their ability to potentiate the toxicity of 1 mm glutamate, as described in Figure 1. Antagonist concentrations between 1 μm and 5 mm were tested, and the data are the concentrations required for half-maximal potentiation. The dose–response curves were repeated at least three times with similar results.
DHPG is an agonist of group I mGluRs with a potency several fold higher than glutamate (Ito et al., 1992; Schoepp et al., 1994; Conn and Pin, 1997). Because DHPG is nontoxic at concentrations up to 1 mm and activates group I receptors more efficiently than glutamate, it should be possible to activate the protective mechanism against a low toxic dose of glutamate by preincubation with DHPG. Figure 1B shows that DHPG does indeed protect cells to a limited extent from low-dose glutamate toxicity. The activation of group I mGluRs also protects cells from cystine deprivation, a form of oxidative stress that mimics glutamate toxicity (see Fig. 5). These pharmacological data suggest that HT-22 cells express group I mGluRs.
Fig. 5.

DHPG blocks cell death by cystine or glucose starvation and desensitizes cells to glutamate. A, HT-22 or primary cortical cells were placed in culture medium containing 2% of the normal amount of cystine, followed by increasing amounts of DHPG. In the absence of DHPG, 72% of the HT-22 (■–■) cells survived, as did 57% of the primary cortical neurons (▵–▵). The data are normalized to 72 or 57% as 100% toxicity for the sake of comparison (see Fig. 1 legend). In another experiment, cells were changed to 0.5 mm glucose medium, and increasing amounts of DHPG were added. Under these conditions, 0% of the HT-22 (○–○) and 40% of the primary cortical cells (×–×) survived. The data are normalized as above. The data are the means of triplicate determinations ± SEM. The experiments were done at least three times. B, Exponentially dividing HT-22 cells were plated out in 96-well microtiter dishes at 2 × 103cells per well. Twenty-four hours later 100 μm glutamate or ACPD was added for a period of 2 hr. The plates were then washed, and fresh medium was added. Increasing amounts of glutamate or ACPD were added both to wells preincubated with glutamate (×–×) or ACPD (▵–▵), and glutamate (•–•) or ACPD (○–○) was also added to cultures with no previous exposure to glutamate or ACPD. The data are normalized to 100% for 100 μm glutamate and 100 μm ACPD alone producing 92 and 89% survival, respectively (Fig. 1). The data are the means of triplicate determinations ± SEM. The experiments were repeated at least four times with similar results.
To determine whether the pharmacological aspects of mGluR activation were similar between the clonal hippocampal cell line HT-22 and cortical neurons, primary cultures of rat cortical neurons were examined with respect to the promotion of glutamate toxicity by AIDA and AP-3 and its inhibition by DHPG. Figure 1C and Table 1show that, as with HT-22 cells, the group I mGluR antagonists AIDA and AP-3 greatly potentiate the minimally toxic levels of glutamate. In contrast, the group I mGluR agonist DHPG protects cells from glutamate.
HT-22 cells express mGluR1 and mGluR5
The above experiments suggest that HT-22 cells express functional group I mGluRs. To determine whether the receptor proteins are indeed expressed, cell lysates were immunoblotted with rabbit antisera to the C-terminal peptides of mGluR1, 2 and 3, and 5. Figure2A shows that HT-22 cells express both mGluR1 (lane 2) and mGluR5 (lane 6), but not 2 or 3 (lane 5). The microglial cell line N9 expresses none of these receptors and serves as a negative control (lanes 3and 7). The positive control for mGluR5 was the protein expressed in oocytes (lane 8), whereas the positive control for mGluRs 1 and 2/3 was a mouse brain lysate (lanes 1 and 4). The mGluRs extracted from brain frequently aggregate and migrate on SDS gels at a higher molecular weight (Alaluf et al., 1995).
Fig. 2.

Stress-induced changes in mGluR expression.A, HT-22 or glutamate-resistant clones were plated at 2 × 105/60 mm dish and the next day lysed, run on SDS acrylamide gels at 10 μg protein/lane, and immunoblotted with the indicated antisera. Antisera: Lanes 1–3, anti-mGluR1; lanes 4 and 5, anti-mGluR2/3; lanes 6–8, anti- mGluR5; lanes 9–12, anti-mGluR1; and lanes 13–16, anti-mGluR5. Lysates: Lane 1, mouse brain; lane 2, HT-22; lane 3, N9; lane 4, mouse brain; lane 5, HT-22; lane 6, HT-22; lane 7, N9; lane 8, oocyte-expressed mGluR5; lane 9, HT-22 wild-type;lane 10, HT-22 r2; lane 11, HT-22 r9; lane 12, HT-22 r10; lane 13, HT-22 wild-type; lane 14, HT-22 r2; lane 15, HT-22 r9; and lane 16, HT-22 r10. The immunoblots inlanes 9-16 were developed for a shorter period of time to emphasize the increased mGluR protein concentrations in the resistant clones. Scanning of gels generated the following relative increases in the resistant cells. Anti-mGluR1: control, 1; r2, 1.5 ± 0.01; r9, 12.1 ± 3.6; r10, and 24.4 ± 4.2. mGluR5: control, 1; r2, 5.7 ± 1.0; r9, 16.2 ± 2.4; r10, 70 ± 11.0; n = 4. B, HT-22 cells (2 × 105 cells/60 mm dish) or primary cortical neurons (1 × 106 cells/60 mm dish) were plated as described above, and 24 hr later the medium was changed to cystine-free or complete DMEM containing 5 mm glutamate. Cell lysates were prepared every 2 hr and immunoblotted with anti-mGluR1 or anti-mGluR5. I, −Cys, anti-mGluR1;II, +Glu, anti- mGluR1; III, −Cys, anti-mGluR5; and IV, +Glu, anti-mGluR5. The experiment was repeated three times with similar results.
mGluR activation modifies glutathione metabolism
The functional basis for the initiation of oxidative glutamate toxicity is the inhibition of cystine uptake from the medium by glutamate (Murphy et al., 1989). Because the mGluR agonists and antagonists are structural analogs of glutamate, it is possible that they alter glutamate toxicity by directly modifying the rate of cystine uptake. Figure 3 shows that the two most widely used mGluR agonists, ACPD and quisqualate, both strongly inhibit cystine uptake. Quisqualate (ID50, 40 μm) is slightly more effective than glutamate (ID50, 80 μm), whereas ACPD is less effective (ID50, 600 μm). The group I mGluR agonist DHPG and antagonist AIDA also inhibit cystine uptake at high concentrations (ID50, 1.2 mm), whereas the group II antagonist 2S-ethylglutamate (EGLU) and the group I antagonist AP-3 are ineffective. It cannot, therefore, be assumed that the mGluRs are the sole target in the nervous system for these glutamate analogs.
Fig. 3.

Effects of mGluR agonists and antagonists on cystine uptake. HT-22 cells were washed with prewarmed HBSS and incubated with 70 μml-[35S]cystine with or without the reagents at the indicated concentrations. Uptake was terminated by washing with cold HBSS, the cells were solubilized, and the isotope was determined. The data are presented as the mean percent of control uptake in the absence of the drugs. AIDA, ○–○; DHPG, ▵–▵; ACPD, ■–■; quisqualate, ▿–▿; glutamate, •–•; EGLU, ▴–▴; and AP-3, ▪–▪. The data from triplicate determinations are presented with the mean ± SE. The experiment was repeated twice with indistinguishable results.
Because AIDA or AP-3 and DHPG have the opposite effects on glutamate toxicity (Fig. 1B,C), it is possible that they have opposite effects on cystine uptake in the presence of marginally toxic 1 mm glutamate. To formally rule out this possibility, cells were incubated in 1 mmglutamate with the half-maximal effective concentrations of AIDA, AP-3, or DHPG. EGLU was included as a negative control. Figure4A shows that there is a negligible effect of AIDA on cystine uptake at concentrations in which dramatic potentiation of toxicity occurs (Fig.1B,C). DHPG and AP-3 are also inactive. These results show that the effects of AIDA, DHPG, and AP-3 on glutamate toxicity are not caused by their modification of the initial rates of cystine uptake.
Fig. 4.
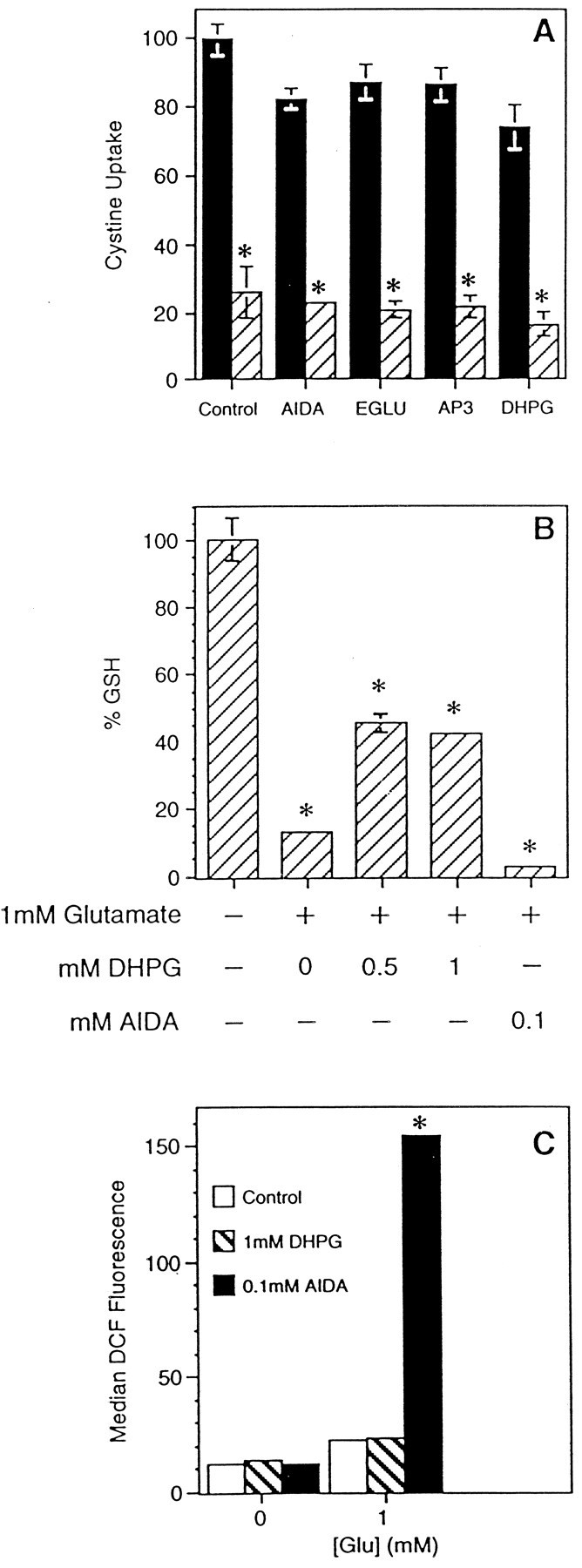
Effects of mGluR agonists and antagonists on cystine uptake, glutathione, and reactive oxygen species.A, Initial rates of cystine uptake were measured in the presence or absence of the indicated reagents and in the presence or absence of 1 mm glutamate (AIDA, 100 μm; AP-3, 500 μm; or EGLU, 500 μm) or 1.5 mm glutamate (DPHG, 600 μm). Solid bars, No glutamate; striped bars, +Glu. The cystine uptake of the control sample was 17,063 ± 831 cpm/mg protein, which was taken as 100%, and other values were derived accordingly. B, HT-22 cells were treated with the indicated reagents for 12 hr, and levels of glutathione were measured as described in Materials and Methods. The control value (80.2 ± 4.8 nmol GSH/mg protein) was taken as 100%, and the rest was expressed accordingly. C, Levels of reactive oxygen species were measured in HT-22 cells treated similarly to B in the presence of H2DCF-DA, and median channels of the fluorescence intensity of dichlorofluorescein (DCF) were reported. *Significantly different from control (p ≤ 0.001). The experiments were repeated at least two times with similar results.
The inhibition of cystine uptake by glutamate causes the loss of cellular GSH (Murphy et al., 1989) followed by the accumulation of ROS (Tan et al., 1998b). To understand where in this pathway the activation of mGluRs modifies glutamate toxicity, the effects of the mGluR agonist DHPG and the antagonist AIDA on these parameters were examined. HT-22 cells were treated with 1 mm glutamate in the presence or absence of mGluR ligands for 12 hr, harvested, and cellular GSH was determined. GSH decreases to ∼20% of the control value in the presence of 1 mm glutamate (Fig. 4B), and the cells do not die (Fig. 1A). In contrast, DHPG-treated cells retain 50% of the control GSH level under this condition, whereas AIDA-treated cells have ∼8% of the control level (Fig. 4B), and the cells die (Fig.1B). Next, cells were treated with glutamate along with the group I mGluR ligands, and the extent of ROS accumulation was determined using dichlorodihydrofluorescein diacetate. This membrane-permeant, nonfluorescent dye becomes fluorescent on deesterification and reaction with ROS in the cytoplasm (Bass et al., 1983). It was previously shown that the loss of GSH up to 85% of the control level causes only a 5- to 10-fold increase in the ROS level (Tan et al., 1998b). However, a greater loss of GSH leads to a several hundredfold increase in ROS, resulting in cell death (Tan et al., 1998b). In the presence of 1 mm glutamate, control and DHPG-treated cells have similar levels of ROS, but AIDA-treated cells accumulated 10-fold more ROS (Fig. 4C). From the above data, it can be concluded that the activation of the mGluR reduces the loss of cellular GSH. The mGluR antagonists interfere with this protective mechanism, resulting in the accumulation of excess ROS and, ultimately, cell death in the presence of nontoxic levels of glutamate.
mGluR agonists protect from other forms of neuronal toxicity
If oxidative glutamate toxicity leads to a form of free radical-mediated stress, then agonists for the group I mGluRs should protect cells from other forms of stress-induced cell death. Perhaps the least complex form of oxidative stress is caused by the depletion of cystine from the growth medium (Yonezawa et al., 1996). The depletion of cystine leads to a decrease in GSH, peroxide production, and cell death. Under these conditions it would be predicted that low, nontoxic levels of mGluR agonists would protect cells, whereas mGluR antagonists would do nothing. Figure5A shows that the group I agonist DHPG indeed protects cells. The group I antagonist AIDA is without effect (data not shown). Like cystine deprivation, glucose starvation (hypoglycemia) leads to cell death, which involves GSH depletion and the production of reactive oxygen species (Ikeda et al., 1994; Papadopoulos et al., 1997). It was therefore asked if the activation of group I mGluR also protects cells from hypoglycemia. HT-22 and primary cortical cultures were plated into microtiter dishes and 2 d later the medium glucose concentration was reduced from 25 to 0.5 mm. The mGluR agonist DHPG was added, and 24 hr later cell viability was determined by MTT and visual counting. Figure5A shows that DHPG protects both primary and HT-22 neurons from glucose starvation. As with cystine starvation, glucose deprivation is not potentiated by the group I antagonists (data not shown).
The activation of mGluRs is correlated with IP3 accumulation
A characteristic that distinguishes group I mGluRs from the other classes of mGluRs is that they are coupled to the phospholipase C pathway. In contrast, the activation of group II and III receptors leads to a decrease in cAMP (Nakanishi, 1994). Therefore the accumulation of IP3 mass was measured in response to glutamate and mGluR agonists. In addition, the cAMP response was assayed. Table 2 shows that glutamate and the group I mGluR agonist DHPG stimulate phosphoinositide accumulation, whereas the levels of cAMP are unchanged. The group I antagonist AIDA greatly reduces IP3 accumulation stimulated by glutamate. The phospholipase C and A2 inhibitor U-73122 inhibits phospholipase C activation (Table 2; Bleasdale et al., 1990). At a half-maximal concentration of 2.5 μm, U-73122 inhibits the accumulation of IP3 induced by DHPG (Table 2) and potentiates the toxicity of 1 mm glutamate (Fig.1B). These data support the antagonist and agonist data and indicate that IP3-linked group I metabotropic receptors are activated by glutamate in HT-22 cells.
Table 2.
Modulation of IP3 and cAMP by glutamate
| Reagent | Concentration | IP3 | cAMP |
|---|---|---|---|
| None | 100 | 100 | |
| Glutamate | 5 mm | 227 ± 15* | 110 ± 23 |
| Glutamate | 1 mm | 385 ± 32* | 130 ± 25 |
| AIDA | 1 mm | 118 ± 21 | 98 ± 15 |
| Glu (1 mm) + AIDA | 1 mm | 130 ± 20 | 118 ± 44 |
| U-73122 | 2.5 μm | 85 ± 10 | |
| DHPG | 1 mm | 410 ± 54* | 133 ± 38 |
| DHPG + U-73122 | 1 mm + 2.5 μm | 128 ± 15 | |
| Forskolin | 1 μm | 218 ± 11* | >2,000* |
| Glutamate2-a | 1 mm | 125 ± 12 | 92 ± 14 |
HT22 cells were exposed to the different reagents for 30 sec or 30 min and IP3 or cAMP determined, respectively, as described in Materials and Methods. The values are changes relative to control (100) ± SEM (n = 6, triplicate determinations, 2 independent experiments).
*Significantly different from controls (p ≤ 0.001).
Cells were exposed to 100 μmglutamate for 2 hr, followed by exposure to 1 mm glutamate. The basal concentrations were the following: IP3, 32 pmol/mg protein; cAMP, 4.3 pmol/mg protein.
The expression of mGluRs is modified by oxidative stress
If the expression and activation of group I mGluRs has a protective effect against glutamate toxicity, then it is possible that their synthesis is upregulated on exposure to glutamate and other forms of oxidative stress. In addition, cells that are selected for their resistance to glutamate may have higher levels of receptor. To determine whether the mGluRs are upregulated by glutamate, HT-22 cells were exposed to 5 mm glutamate for various lengths of time, and the cell lysates were run on SDS acrylamide gels and blotted with antisera against mGluR1, 5, or 2/3. There was no detectable mGluR 2/3 protein at any time point, but Figure 2B shows that there was an upregulation of mGluR5 (III), but not mGluR1 protein (II). The full time course is quantitated in Table 3. Although HT-22 cells are sensitive to short-term exposure to glutamate, cells have been selected and characterized that are not killed by 10 mm glutamate (Sagara et al., 1998). When cell lysates from these resistant cells were blotted with antisera to the mGluRs, there was a 70-fold increase in the expression of mGluR1 and a 24-fold increase in mGluR5 (Fig. 2A, lanes 9-16).
Table 3.
Oxidative stress induces metabotropic glutamate receptors
| Cells | Antibody | Condition | Time (hr) | ||||
|---|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | |||
| Primary | mGluR1 | −Cys | 1 ± 0.18 | 2.98 ± 0.35 | 8.79 ± 0.45 | 7.66 ± 0.26 | 13.2 ± 1.42 |
| Primary | mGluR1 | +Glu | 1 ± 0.15 | 3.81 ± 0.47 | 2.81 ± 0.34 | 15.6 ± 1.68 | 10.9 ± 1.17 |
| HT22 | mGluR5 | −Cys | 1 ± 0.21 | 2.64 ± 0.11 | 9.05 ± 1.42 | 15.8 ± 2.56 | 14.4 ± 2.26 |
| HT22 | mGluR5 | +Glu | 1 ± 0.17 | 2.61 ± 0.14 | 9.34 ± 0.56 | 12.6 ± 1.15 | 11.6 ± 1.18 |
The mGluRs were assayed after glutamate addition or growth in low cystine medium as described in Figure 2B. The density of the exposed film was quantitated and expressed as the relative increase (0 time = 1) ± SEM (n = 4).
To determine whether cortical neurons respond in a manner similar to HT-22 cells, primary cultures were exposed to glutamate, and the synthesis of mGluR 1 and 5 followed as a function of time by Western blotting. Figure 2B shows that in contrast to HT-22, cultured cortical neurons respond to glutamate by the upregulation of mGluR1 (II), whereas the expression of mGluR5 is not reproducibly changed (IV). The quantitative data in Table 3 show that the level of induced expression of mGluR5 in primary cultures is similar to the level of mGluR1 in HT-22 cells.
mGluR expression may be upregulated by virtue of the direct interaction of glutamate with its receptor or as the result of oxidative stress caused by glutamate. To distinguish between these alternatives, cells were exposed to media depleted of cystine, a condition that leads to a form of oxidative stress similar to oxidative glutamate toxicity (Yonezawa et al., 1996). The expression of mGluR1 and mGluR5 was then followed every 2 hr by Western blotting. Figure 5B shows that as with glutamate toxicity, mGluR5 is upregulated in HT-22 cells (III), and mGluR1 was upregulated in primary cortical neurons (I). These data are quantitated in Table3.
The protection from glutamate toxicity is rapidly desensitized
mGluRs can be rapidly desensitized on exposure to low concentrations of glutamate, because, in primary cortical cultures, the IP3 response is lost after 1 hr exposure to 100 μm glutamate (Catania et al., 1991). To determine whether the protective effect of mGluR activation is lost with glutamate exposure, HT-22 cells were exposed to 100 μm glutamate or 100 μm concentrations of the group I and II agonist ACPD for various periods of time, the agonist was washed out, and then the cells were continually exposed to increasing concentrations of glutamate. Cell viability was determined after 20 hr. The time for half-maximal desensitization to glutamate exposure was 45 min, and after 2 hr the cells preincubated with glutamate were much more sensitive to glutamate than the cells exposed directly to glutamate (Fig. 5B). This is attributable to the loss of the protective effect of mGluR activation and is similar to the effects of the mGluR antagonists AIDA and AP-3. If this desensitization is working through the inositol pathway as described by Catania et al., (1991), then preexposure of cells to glutamate should block a subsequent increase in IP3 by exposure to glutamate. Table 2 shows that cells preexposed to 100 μm glutamate for 2 hr no longer respond to 1 mm glutamate with an increase in IP3 mass.
DISCUSSION
The above data show that the activation of group I metabotropic glutamate receptors generates a cellular response that is protective to oxidative glutamate toxicity, cystine deprivation, and hypoglycemia. This conclusion is based on the observations that group I mGluR agonists protect cells and stimulate the accumulation of IP3. In contrast, group I antagonists potentiate glutamate toxicity. Finally, exposure of cells to glutamate both downregulates the protective response (desensitization) and increases the accumulation of mGluR1 in cortical neurons and mGluR5 HT-22 cells. These results expand the known functions of group I mGluRs within the CNS to include a role in the protection from several forms of oxidative stress.
mGluRs and nerve cell toxicity
There have been a number of apparently contradictory studies that examined the role of mGluRs in various forms of neurotoxicity. All of these studies were, however, done with heterogeneous populations of cells and, in some cases, very high doses of mGluR agonists were used, which can directly kill cells by the oxidative glutamate toxicity pathway (Figs. 1A, 3). Direct injection of the group I agonist ACPD into the hippocampus (Sacaan and Schoepp, 1992) or the systemic administration of ACPD (McDonald et al., 1993) induces seizures and cell death. These effects were not blocked by NMDA antagonists, which frequently protect cells from glutamate excitotoxicity. It has also been argued that the activation of mGluR1 contributes to trauma-induced cell death (Mukhin et al., 1996). In agreement with the in vivo data, some studies with cultured cells have shown that the activation of group I mGluRs potentiates NMDA receptor-mediated excitotoxicity, presumably because these receptors mediate an increase in IP3, resulting in an additional Ca2+ load in cells that is synergistic with the NMDA-gated Ca2+ influx (Bruno et al., 1995b). However, other groups have shown that the activation of group I receptors protects against excitotoxicity (Pizzi et al., 1993), enhances the survival of cultured Purkinje cells (Mount et al., 1993), and that group I antagonists cause retinal degeneration in developing rodents (Price et al., 1995). In contrast with conflicting reports with group I mGluRs, it is generally agreed that the activation of the cAMP-linked group II receptors leads to protection from excitotoxicity and other neuronal insults (Buisson and Choi, 1995; Nicoletti et al., 1996).
HT-22 cells lack ionotropic glutamate receptors (Maher and Davis, 1996) and do not express group II mGluRs (Fig. 2A). Therefore, examining the role of group I receptors in the protection from toxic conditions is less ambiguous than when dealing with mixed nerve cell populations. The above data show that group I mGluR1 receptor activation leads to the protection of cells from several toxic insults. This effect appears to be mediated by receptor coupling to PI hydrolysis, because protective group I receptor agonists stimulate IP3 accumulation, whereas group I receptor antagonists, which block IP3 accumulation, potentiate glutamate toxicity (Table 2). These data agree with the observations that the activation of PKC protects HT-22 cells from oxidative glutamate toxicity (Davis and Maher, 1994), because PI turnover leads to increased levels of diacylglycerol which, in turn, activate PKC.
The mechanism of protection
One of the initial events in oxidative glutamate toxicity and other forms of oxidative stress is the rapid decline in intracellular GSH (Murphy et al., 1989). This leads to a series of downstream events including the activation of lipoxygenase, peroxide formation, and the massive influx of calcium via cGMP-regulated channels immediately before cell lysis (Li et al., 1997a,b). The data in Figure 4 show that although mGluR group I agonists or antagonists do not modify cystine uptake, they do have a significant effect on GSH levels. Agonists increase GSH relative to glutamate alone, whereas antagonists decrease GSH. The extent of this GSH modification is sufficient to either inhibit or trigger all of the downstream events leading to cell death (Tan et al., 1998b). Although the exact mechanism by which mGluR activation influences GSH metabolism remains to be defined, several alternatives exist. mGluR group I activation may affect the expression or the activity of proteins required for the maintenance of cellular GSH under stressful conditions. For example, the expression of the rate-limiting GSH synthetic enzyme γ-glutamylcysteine synthetase (γ-GCS) can be upregulated by various stimuli (Morales et al., 1997;Mulcahy et al., 1997; Sekhar et al., 1997). Furthermore, the enzyme activity of γ-GCS can be elevated post-translationally (Ochi, 1995,1996). The gene expression and enzyme activity of these proteins may also be modulated by the transient increase of Ca2+(Bading et al., 1997), because mGluR group I activation increases intracellular IP3 (Table 2), and IP3 triggers Ca2+ release from ER via IP3 receptors (Dowson, 1997). Finally, instead of directly modulating GSH synthesis, mGluR group I activation may decrease GSH consumption, for example by reducing the production of ROS from mitochondria. These alternatives are currently being examined.
Glutamate leads to mGluR desensitization
Desensitization is defined as the tendency of the receptor response to decrease with time after exposure to an agonist. HT-22 cells are desensitized with respect to the ability of glutamate to kill cells by previous exposure to glutamate. Primary cultures of cerebellar granule cells are also desensitized by glutamate when polyphosphoinositide hydrolysis is measured after a second exposure to glutamate (Catania et al., 1991), probably by the PKC-dependent phosphorylation of the receptor (Alaluf et al., 1995; Gereau and Heinemann, 1998). A similar observation was made with HT-22 cells (Table 2). Because one of the functions of mGluRs appears to be the protection of cells from toxic insults, it is curious that a mechanism would evolve to terminate this response in a rather rapid manner. The desensitization may be necessary to allow a return of function of shared downstream receptor pathways that are required to maintain viability and normal physiology. For example, a large group of cell surface receptors is linked to IP3 turnover, and both desensitization and signaling can be mutually affected at various points along the signal transduction pathways, depending on the specific receptor (Wojcikiewicz et al., 1993; Fischer, 1995). Alternatively, the initial exposure to glutamate may be sufficient to induce changes in gene expression that lead to additional protection, because cells can become resistant to glutamate by the upregulation of antioxidant enzymes and the enzymes involved in glutathione metabolism (Sagara et al., 1998). Glutamate-resistant cells also upregulate mGluR5 expression up to 70-fold (Fig. 2A).
Glutamate and other forms of stress upregulate mGluR accumulation
The exposure of HT-22 cells to glutamate or oxidative stress caused by cystine deprivation increases the rate of accumulation of mGluR5 as defined by Western blotting; mGluR1 protein is not changed. In contrast, the concentration of mGluR1 is increased under the same conditions in primary cortical cultures, whereas mGluR5 expression is unchanged. Therefore, there are different coupling mechanisms between the stress signal transduction pathways and mGluR subtypes in cortical neurons and HT-22 cells. These data indicate, however, that the response to stress is similar and are consistent with in vivo results that show that group I mGluRs are upregulated in the hippocampus after transient global ischemia (Chen et al., 1988; Seren et al., 1989). The expression of mGluR5 is also modified by protein growth factors in cultured cortical astrocytes. Basic fibroblast growth factor and epidermal growth factor both upregulate mGluR5 expression (Miller et al., 1995), whereas thrombin downregulates mGluR5 expression (Miller et al., 1996). If IP3 signaling is involved in protection from cell injury or death, then the enhanced expression of group I mGluRs may make the cells more responsive to glutamate or allow for a higher basal level of IP3.
Conclusion
Finally, it must be asked what is the functional significance of the apparently short-term protective response elicited by the activation of group I mGluRs? Glutamate is the most abundant neurotransmitter in the mammalian CNS, with an intracellular concentration approaching several millimolar (Coyle and Puttfarcken, 1993). Because elevated exposure to glutamate can be toxic to neurons via both receptor-mediated and the oxidative glutamate toxicity pathways, and because transient excess glutamate may be a frequent occurrence in areas dense in glutaminergic terminals, the short-term activation of group I mGluRs may be required to prevent nerve cell and synaptic damage. Because the activation of group II mGluRs is also protective (Nicoletti et al., 1996), the combined effect of these two most abundant CNS mGluRs would lead to a potent protective response to excess glutamate. This mechanism could also be involved in minor trauma in which there is minimal cellular damage, and it may bridge the gap between the time of initial insult and the induction of a more long-term transcription-mediated stress response.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS09658 and PO1 NS-28121 to D.S. and a postdoctoral fellowship to Y.S. (2F32NS 10032). We thank Drs. P. Maher, H. Kimura, and Y. Liu for critically reading this manuscript, Dr. D. Chambers for assistance in the fluorescence-activated cell-sorting analysis, and Dr. R. Gereau for the oocyte expressed mGluR5.
Correspondence should be addressed to David R. Schubert, The Salk Institute for Biological Studies, La Jolla, CA 92037.
REFERENCES
- 1.Abe K, Takayanagi M, Saito H. Effects of recombinant human basic FGF and its modified protein CS23 on survival of primary cultured neurons from various regions of fetal rat brain. Jpn J Pharmacol. 1990;53:221–227. doi: 10.1254/jjp.53.221. [DOI] [PubMed] [Google Scholar]
- 2.Alaluf S, Mulvihill ER, McIlhinney RAJ. Rapid agonist mediated phosphorylation of the metabotropic glutamate receptor 1α by protein kinase C in permanently transfected BHK cells. FEBS Lett. 1995;367:301–305. doi: 10.1016/0014-5793(95)00575-t. [DOI] [PubMed] [Google Scholar]
- 3.Bading B, Hardingham GE, Johnson CM, Chawla S. Gene regulation by nuclear and cytoplasmic calcium signals. Biochem Biophys Res Commun. 1997;236:541–543. doi: 10.1006/bbrc.1997.7037. [DOI] [PubMed] [Google Scholar]
- 4.Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 5.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol. 1983;130:1910–1917. [PubMed] [Google Scholar]
- 6.Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human neutrophils. J Pharmacol Exp Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- 7.Bruno V, Battaglia G, Copani A, Giffard RG, Raciti G, Raffaele R, Shinozaki H, Nicoletti F. Activation of class II or III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur J Neurosci. 1995a;7:1906–1913. doi: 10.1111/j.1460-9568.1995.tb00712.x. [DOI] [PubMed] [Google Scholar]
- 8.Bruno V, Copani A, Knopfel T, Kuhn R, Casabona G, Dell’ Albani P, Condorelli DF, Nicoletti F. Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells. Neuropharmacology. 1995b;34:1089–1098. doi: 10.1016/0028-3908(95)00077-j. [DOI] [PubMed] [Google Scholar]
- 9.Buisson A, Choi DW. The inhibitory mGluR agonist, S-4-carboxy-3-hydroxy-phenylglycine selectively attenuates NMDA neurotoxicity and oxygen-glucose deprivation-induced neuronal death. Neuropharmacology. 1995;34:1081–1087. doi: 10.1016/0028-3908(95)00073-f. [DOI] [PubMed] [Google Scholar]
- 10.Catania MV, Aronica E, Sortino MA, Canocio PL, Nicoletti F. Desensitization of metabotrophic glutamate receptors in neuronal cultures. J Neurochem. 1991;56:1329–1335. doi: 10.1111/j.1471-4159.1991.tb11429.x. [DOI] [PubMed] [Google Scholar]
- 11.Challiss RAJ, Jenkinson S, Mistry R, Batty IH, Nahorski SR. Assessment of neuronal phosphoinositide turnover and its disruption by lithium. Neuroprotocols. 1993;3:135–144. [Google Scholar]
- 12.Chen CK, Silverstein FS, Fisher SK, Statman D, Johnston MV. Perinatal hypoxic-ischemic brain injury enhances quisqualic acid-stimulated phosphoinositide turnover. J Neurochem. 1988;51:353–359. doi: 10.1111/j.1471-4159.1988.tb01046.x. [DOI] [PubMed] [Google Scholar]
- 13.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 14.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 15.Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell line. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 16.Dowson AP. Calcium signalling: how do IP3 receptors work? Curr Biol. 1997;7:R544–R547. doi: 10.1016/s0960-9822(06)00277-6. [DOI] [PubMed] [Google Scholar]
- 17.Fisher SK. Homologous and heterologous regulation of receptor-stimulated phosphoinositide hydrolysis. Eur J Pharmacol. 1995;288:231–250. doi: 10.1016/0922-4106(95)90035-7. [DOI] [PubMed] [Google Scholar]
- 18.Gereau RW, Heinemann SF., IV Role of protein kinase C phosphorylation in rapid desensitization of metabotropic glutamate receptor 5. Neuron. 1998;20:143–151. doi: 10.1016/s0896-6273(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 19.Griffith OW. Determination of glutathione and glutathione disulfide using glucose reductase and 2-vinyl pyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda J, Morita L, Murota S. Involvement of nitric oxide and free radical (02-) in neuronal injury induced by deprivation of oxygen and glucose in vitro. Acta Neurochir Suppl. 1994;60:94–97. doi: 10.1007/978-3-7091-9334-1_24. [DOI] [PubMed] [Google Scholar]
- 21.Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H. 3,5-Dihydroxyphenyl-glycine: a potent agonist of metabotropic glutamate receptors. NeuroReport. 1992;3:1013–1016. [PubMed] [Google Scholar]
- 22.Joly C, Gomeza J, Brabet I, Curry K, Bockaert J, Pin J-P. Molecular, functional, and pharmacological characterization of the metabotropic glutamate receptor type 5 splice variants: comparison with mGluR1. J Neurosci. 1995;15:3970–3981. doi: 10.1523/JNEUROSCI.15-05-03970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997a;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Maher P, Schubert D. Requirement for cGMP in nerve cell death caused by glutathione depletion. J Cell Biol. 1997b;139:1317–1324. doi: 10.1083/jcb.139.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maher P, Davis J. The role of monoamine metabolism in oxidative glutamate toxicity. J Neurosci. 1996;16:6394–6401. doi: 10.1523/JNEUROSCI.16-20-06394.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manzoni OJ, Weisskopf MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 28.McDonald JW, Fix AS, Tizzano JP, Schoepp DD. Seizures and brain injury in neonatal rats induced by 1S,3R-ACPD, a metabotropic glutamate receptor agonist. J Neurosci. 1993;13:4445–4455. doi: 10.1523/JNEUROSCI.13-10-04445.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller S, Romano C, Cotman CW. Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. J Neurosci. 1995;15:6103–6109. doi: 10.1523/JNEUROSCI.15-09-06103.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller S, Sehati N, Romano C, Cotman CW. Exposure of astrocytes to thrombin reduces levels of the metabotropic glutamate receptor mGluR5. J Neurochem. 1996;67:1435–1447. doi: 10.1046/j.1471-4159.1996.67041435.x. [DOI] [PubMed] [Google Scholar]
- 31.Morales A, Garcia-Ruiz C, Miranda M, Mari M, Colell A, Ardite E, Fernandez-Checa JC. Tumor necrosis factor increases hepatocellular glutathione by transcriptional regulation of the heavy subunit chain of gamma-glutamylcysteine synthetase. J Biol Chem. 1997;272:30371–30379. doi: 10.1074/jbc.272.48.30371. [DOI] [PubMed] [Google Scholar]
- 32.Morimoto BH, Koshland DE., Jr Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron. 1990;5:875–880. doi: 10.1016/0896-6273(90)90347-i. [DOI] [PubMed] [Google Scholar]
- 33.Moroni F, Lombardi G, Thomsen C, Leonardi P, Attucci S, Peruginelli F, Torregrossa SA, DE P-G, Luneia R, Pellicciari R. Pharmacological characterization of 1-aminoindan-1,5-dicarboxylic acid, a potent mGluR1 antagonist. J Pharmacol Exp Ther. 1997;281:721–729. [PubMed] [Google Scholar]
- 34.Mount HTJ, Dreyfus CF, Black IB. Purkinje cell survival is differentially regulated by metabotropic and ionotropic excitatory amino acid receptors. J Neurosci. 1993;13:3173–3179. doi: 10.1523/JNEUROSCI.13-07-03173.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukhin A, Fan L, Faden AI. Activation of metabotropic glutamate receptor subtype mGluR1 contributes to post-traumatic neuronal injury. J Neurosci. 1996;16:6012–6020. doi: 10.1523/JNEUROSCI.16-19-06012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and beta-naphthoflavone-induced expression of the human gamma-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 37.Murphy TH, Baraban JM. Glutamate toxicity in immature cortical neurons precedes development of glutamate receptor currents. Brain Res. 1990;57:146–150. doi: 10.1016/0165-3806(90)90195-5. [DOI] [PubMed] [Google Scholar]
- 38.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 39.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 40.Nicoletti F, Bruno V, Copani A, Casabona G, Knopfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 41.Ochi T. Hydrogen peroxide increases the activity of γglutamylcysteine synthetase in cultured Chinese hamster V79 cells. Arch Toxicol. 1995;70:96–103. doi: 10.1007/BF02733669. [DOI] [PubMed] [Google Scholar]
- 42.Ochi T. Menadione causes increases in the level of glutathione and in the activity of γ-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Toxicology. 1996;112:45–55. doi: 10.1016/0300-483x(96)03348-3. [DOI] [PubMed] [Google Scholar]
- 43.Papadopoulos MC, Koumenis IL, Dugan LL, Giffard RG. Vulnerability to glucose deprivation injury correlates with glutathione levels in astrocytes. Brain Res. 1997;748:151–156. doi: 10.1016/s0006-8993(96)01293-0. [DOI] [PubMed] [Google Scholar]
- 44.Pin J-P, Duvoisin R. Metabotropic glutamate receptors: Structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 45.Pizzi M, Fallacara C, Arrighi V, Memo M, Spano PF. Attenuation of excitatory amino acid toxicity by metabotropic glutamate receptor agonists and aniracetam in primary cultures of cerebellar granule cells. J Neurochem. 1993;61:683–689. doi: 10.1111/j.1471-4159.1993.tb02173.x. [DOI] [PubMed] [Google Scholar]
- 46.Price MT, Romano C, Fix AS, Tizzano JP, Olney JW. Blockade of the second messenger functions of the glutamate metabotropic receptor is associated with degenerative changes in the retina and brain of immature rodents. Neuropharmacology. 1995;34:1069–1079. doi: 10.1016/0028-3908(95)00065-e. [DOI] [PubMed] [Google Scholar]
- 47.Ratan RR, Lee PJ, Baraban JM. Serum deprivation inhibits glutathione depletion-induced death in embryonic cortical neurons: evidence against oxidative stress as a final common mediator of neuronal apoptosis. Neurochem Int. 1996;29:153–157. doi: 10.1016/0197-0186(95)00115-8. [DOI] [PubMed] [Google Scholar]
- 48.Riedel G, Reymann KG. Metabotropic glutamate receptors in hippocampal long-term potentiation and learning and memory. Acta Physiol Scand. 1996;157:1–19. doi: 10.1046/j.1365-201X.1996.484231000.x. [DOI] [PubMed] [Google Scholar]
- 49.Sacaan AI, Schoepp DD. Activation of hippocampal metabotropic excitatory amino acid receptors leads to seizures and neuronal damage. Neurosci Lett. 1992;139:77–82. doi: 10.1016/0304-3940(92)90862-2. [DOI] [PubMed] [Google Scholar]
- 50.Sagara Y, Dargusch R, Chambers D, Davis J, Schubert D, Maher P. Cellular mechanisms of resistance to chronic oxidative stress. J Free Radic Res. 1998;24:1375–1389. doi: 10.1016/s0891-5849(97)00457-7. [DOI] [PubMed] [Google Scholar]
- 51.Schoepp DD, Johnson BG, Smith ECR, McQuaid LA. Stereoselectivity and mode of inhibition of phosphoinositide-coupled excitatory amino acid receptors by 2-amino-3-phosphonopropionic acid. Mol Pharmacol. 1990;38:222–228. [PubMed] [Google Scholar]
- 52.Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR. 3,5-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- 53.Schubert D, Kimura H, Maher P. Growth factors and Vitamin E modify neuronal glutamate toxicity. Proc Natl Acad Sci USA. 1992;89:8264–8267. doi: 10.1073/pnas.89.17.8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sekhar KR, Meredith MJ, Kerr LD, Soltaninassab SR, Spitz DR, Xu ZQ, Freeman ML. Expression of glutathione and γglutamylcysteine synthetase mRNA is Jun dependent. Biochem Biophys Res Commun. 1997;234:588–593. doi: 10.1006/bbrc.1997.6697. [DOI] [PubMed] [Google Scholar]
- 55.Seren MS, Alkinio C, Zanoni R, Leon A, Nicoletti F. Stimulation of inositol phospholipid hydrolysis by excitatory amino acids is enhanced in brain slices from vulnerable regions after transient global ischemia. J Neurochem. 1989;53:1700–1705. doi: 10.1111/j.1471-4159.1989.tb09233.x. [DOI] [PubMed] [Google Scholar]
- 56.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of peroxide production during programmed cell death. J Cell Biol. 1998a;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan S, Wood M, Maher P. Oxidative stress in nerve cells induces a form of cell death with characteristics of both apoptosis and necrosis. J Neurochem. 1998b;71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x. [DOI] [PubMed] [Google Scholar]
- 58.Thomsen C, Klitgaard H, Sheardown M, Jackson HC, Eskesen K, Jacobsen P, Treppendahl S, Suzdak PD. (S)-4-Carboxy-3-hydroxyphenylglycine, and antagonist of metabotropic glutamate receptor (mGluR) 1α and an agonist of mGluR2, protects against audiogenic seizures in DBA/2 mice. J Neurochem. 1994;62:2492–2495. doi: 10.1046/j.1471-4159.1994.62062492.x. [DOI] [PubMed] [Google Scholar]
- 59.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 60.Vogt M, Dulbecco R. Steps in the neoplastic transformation of hamster embryo cells by polyoma virus. Proc Natl Acad Sci USA. 1963;49:171–179. doi: 10.1073/pnas.49.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wojcikiewicz RJH, Tobin AB, Nahorski SR. Desensitization of cell signalling mediated by phosphoinositidase C. Trends Pharmacol Sci. 1993;14:279–285. doi: 10.1016/0165-6147(93)90131-3. [DOI] [PubMed] [Google Scholar]
- 62.Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine deprivation induces oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem. 1996;67:566–573. doi: 10.1046/j.1471-4159.1996.67020566.x. [DOI] [PubMed] [Google Scholar]