

Abstract
In addition to the regulation of neuronal survival and differentiation, neurotrophins may play a role in synapse development and plasticity. Application of brain-derived neurotrophic factor (BDNF) promotes long-term potentiation (LTP) in CA1 synapses of neonatal hippocampus, which otherwise exhibit only short-term potentiation. This is attributable, at least in part, to an attenuation of the synaptic fatigue induced by high-frequency stimulation (HFS). However, the prevention of synaptic fatigue by BDNF could be mediated by an attenuation of synaptic vesicle depletion from presynaptic terminals and/or a reduction of the desensitization of postsynaptic receptors. Here we provide evidence supporting a presynaptic effect of BDNF. The effect of BDNF on synaptic fatigue depended on the stimulation frequency, not on the stimulus duration nor on the number of stimulation pulses. BDNF was only effective when the synapses were stimulated at frequencies >50 Hz. Treatment with BDNF also potentiated paired-pulse facilitation (PPF), a parameter reflecting changes in the properties of presynaptic terminals. This effect of BDNF was restricted only to PPF elicited with interpulse intervals ≤20 msec. Changes in the extracellular calcium concentration altered the magnitude of the BDNF effect on PPF and synaptic responses to HFS, suggesting that BDNF regulates neurotransmitter release. When the desensitization of glutamate receptors was blocked by cyclothiazide or aniracetam, the BDNF potentiation of the synaptic responses to HFS was unaltered. Taken together, these results suggest that BDNF acts presynaptically. When two pathways in the same slice were monitored simultaneously, BDNF treatment potentiated the tetanized pathway without affecting the synaptic efficacy of the untetanized pathway. The selective potentiation of high-frequency transmission by BDNF appears to contribute directly to the effect of BDNF on LTP rather than indirectly by inducing the release of additional diffusible factors. The preferential potentiation of highly active synapses by BDNF may have implications in the Hebbian mechanism of synaptic plasticity.
Keywords: BDNF, presynaptic, hippocampus, LTP, synaptic fatigue, plasticity
Although the traditional view holds that neurotrophins are signaling proteins essential for neuronal survival and differentiation, a series of recent studies suggests a novel role of neurotrophins in synaptic transmission and plasticity (Lo, 1995; Thoenen, 1995; Bonhoeffer, 1996; Lu and Figurov, 1997). Brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) rapidly enhance synaptic transmission at the developing neuromuscular junction in culture (Lohof et al., 1993). Detailed analyses indicate that this effect was a result of an enhancement of transmitter release, most likely caused by increased calcium concentrations at the nerve terminal (Stoop and Poo, 1995, 1996). NT-3 also has been shown to have a long-term effect on the maturation of neuromuscular synapses (Wang et al., 1995; Liou et al., 1997). Moreover, the expression of NT-3 in postsynaptic muscle cells is regulated by nerve cell innervation and membrane depolarization, and this activity-dependent expression of NT-3 may serve as a retrograde signal for modulating synaptic efficacy and/or stabilizing the neuromuscular synapses (Xie et al., 1997). In the CNS neurotrophins appear to have both acute and long-term effects on the synaptic function and plasticity. Studies on long-term effects have focused primarily on the visual system (Maffei et al., 1992; Gu et al., 1994; Cabelli et al., 1995; Riddle et al., 1995). The acute effects of neurotrophins on neuronal activity and synaptic transmission have been observed in cultured neurons and in slices by a number of laboratories. Knipper et al. (1994) first reported that NGF and BDNF enhance high K+-induced release of acetylcholine from hippocampal synaptosomes. Then rapid and acute effects of neurotrophins were demonstrated electrophysiologically in cortical and hippocampal neurons in culture (Kim et al., 1994; Lessmann et al., 1994; Levine et al., 1995). Kang and Schuman (1995) showed that BDNF and NT-3 rapidly enhance basal excitatory synaptic transmission in CA1 synapses of adult hippocampal slices. Because long-term potentiation (LTP) and BDNF-induced enhancement of synaptic transmission do not occlude each other and the BDNF effect cannot be blocked by the NMDA receptor antagonists, these authors suggested that these two phenomena may involve independent mechanisms. They also showed that this effect requires protein synthesis in the dendrites and axons (Kang and Schuman, 1996). However, using a whole-cell recording technique, Tanaka et al. (1997) recently reported that BDNF does not affect basal excitatory synaptic transmission but rapidly reduces IPSCs at CA1 synapses in hippocampal slices.
Using the CA1 synapses in the hippocampus as a model system, a number of recent studies have addressed the role of BDNF in LTP. Korte and colleagues (1995) were the first to demonstrate a defect of CA1 synapses in BDNF knock-out mice. A severe defect in tetanus-induced LTP has been observed in these mice in the hippocampus despite normal brain morphology, basal synaptic transmission, and behavior in these mice. Our laboratory subsequently demonstrated that the application (3–4 hr) of BDNF to hippocampal slices from immature rats facilitates tetanus-induced LTP (Figurov et al., 1996). The defect in LTP in BDNF knock-out mice can be rescued after prolonged (2–4 hr) incubation with recombinant BDNF (Patterson et al., 1996) or by infection with BDNF-containing adenovirus (Korte et al., 1996), suggesting that the absence of BDNF per se, rather than cumulative developmental defects, is responsible for impaired LTP in the hippocampus of BDNF knock-out mice.
Our previous study indicated that a critical level of BDNF is important for LTP in the CA1 synapses of the hippocampus (Figurov et al., 1996). Application of exogenous BDNF promotes LTP induction in neonatal hippocampal slices, where the endogenous BDNF levels are low. In contrast, treatment with TrkB-IgG, a fusion protein that scavenges endogenous BDNF, reduces the magnitude of LTP in adult hippocampus, where the endogenous BDNF levels are high. We also demonstrated that this effect was attributable to an enhanced ability of hippocampal synapses to respond to high-frequency stimulation (HFS) rather than to a direct modulation of LTP-triggering mechanisms. In the present study we have investigated potential mechanisms by which BDNF regulates synaptic plasticity in the hippocampus. We have studied the effects of BDNF on paired-pulse facilitation (PPF) and the synaptic responses to HFS. We have examined the effects of varying the extracellular Ca2+ concentration ([Ca2+]o) as well as the effects of compounds that block the desensitization of postsynaptic glutamate receptors on these BDNF-induced changes. Our results suggest that BDNF acts presynaptically to facilitate high-frequency synaptic transmission at developing hippocampal synapses.
MATERIALS AND METHODS
The methods used in the present study were essentially the same as previously described (Figurov et al., 1996). Briefly, hippocampal slices (350–400 μm thick) were prepared from 12- to 13-d-old (P12–P13) Sprague Dawley rats (Taconics, Germantown, NY), maintained in an interface chamber for both recovery (2 hr) and recording, and exposed to an artificial atmosphere of 95% O2/5% CO2. Perfusion medium [artificial CSF (ACSF) at 34°C] contained (in mm): 124 NaCl, 3.0 KCl, 2.5 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.25 KH2PO4, 10 glucose, and 2 ascorbate, pH 7.4. Low- or high-calcium ACSF was identical to control ACSF, except that the calcium concentration was changed to 0.5 or 5.0 mm, respectively. The perfusion rate was 15 ml/hr. Human recombinant BDNF was added directly into the chamber and perfused in a closed circle of ∼3 ml at a final concentration of 2 nm. Other drugs were added to the perfusion medium at the concentrations indicated. Cyclothiazide and aniracetam (Sigma, St. Louis, MO) were dissolved first in DMSO and then diluted at least 1000 times into ACSF. An extracellular recording electrode was filled with ACSF and placed in stratum radiatum of CA1. Basal synaptic transmission was measured by stimulating the Schaffer collaterals of CA1 alternately at low frequency (1/min, constant current) through two stimulating electrodes (S1 and S2) positioned on either side of the recording electrode. Only slices exhibiting field EPSPs of 2–3 mV in amplitude without superimposed population spikes were used. The stimulus strength was adjusted to evoke field EPSPs of ∼1.3 mV. Repetitive stimulation at varying frequencies and paired-pulse stimulation at different interpulse intervals was used to examine synaptic fatigue and PPF, respectively. Theta burst stimulation, which consisted of 10 bursts at 5 Hz, each composed of four pulses at 100 Hz, was used as tetanic stimulation to induce LTP. Responses were digitized (10 kHz), analyzed on-line, and stored on magnetic media, using acquisition systems DaQSys 1.0 and Process 2.0 (University of Geneva) and custom-developed software (kindly provided by Dr. Takafumi Inoue, University of Tokyo). Statistical significance was determined by ANOVA, followed by Fisher’s Protected Least Significant Difference as a post hoc test. A nonparametric Mann–Whitney U test was applied alternatively when indicated. The asterisk denotes p < 0.05. Data are presented as the mean ± SEM.
RESULTS
In all experiments the extracellular recordings were performed on CA1 synapses of hippocampal slices taken from P12–P13 rats and maintained in an interface chamber at 34°C, continuously perfused with ACSF with or without BDNF (2 nm) for up to 3 hr at a flow rate of 15 ml/hr. Under these conditions the application of BDNF had no effect on basal synaptic transmission. The initial slope of the EPSPs evoked every 30 sec in the same pathway remained unchanged for up to 3 hr after the application of BDNF (Fig.1). Although this result is consistent with similar recordings at CA1 synapses of hippocampal slices from rats (Figurov et al., 1996; Tanaka et al., 1997) and mice (Korte et al., 1996; Patterson et al., 1996), others have observed a potentiation by BDNF on basal synaptic transmission (Kang and Schuman, 1995). This discrepancy was later attributed, at least in part, to technical differences (i.e., type of chamber used, temperature, flow rate, etc.) (Kang et al., 1996).
Fig. 1.

BDNF does not affect basal synaptic transmission in CA1 synapses of P12–P13 hippocampus. The slopes of the EPSPs were measured every 30 sec before and during continuous application of BDNF to the interface chamber (black bar, up to 3 hr). Eachpoint represents the average slope of 10 consecutive EPSPs (5 min interval) normalized to the average slope of the EPSPs acquired before BDNF application (20 min baseline;n = 5). Representative traces acquired before and after 3 hr of BDNF treatment are shown at the top(averages of 10 consecutive traces). In this and all of the following figures, slices from P12–P13 rats were used. BDNF was applied to the interface chamber at the final concentration of 2 nm for 3 hr.
Our previous experiments showed that in P12–P13 hippocampal slices a train of HFS pulses (1 sec, 100 Hz) induces a gradual decline of the evoked EPSPs during the train. BDNF rescues this synaptic depression or fatigue without affecting basal synaptic transmission (Figurov et al., 1996). It is unclear, however, whether this effect of BDNF is dependent on the stimulation frequency and, if so, whether BDNF only affects synapses that undergo synaptic fatigue. Because synaptic fatigue is thought to be attributable to a depletion of the releasable pool of quanta or synaptic vesicles (Zucker, 1989; Larkman et al., 1991;Dobrunz and Stevens, 1997), an attenuation of synaptic fatigue by BDNF would suggest a presynaptic mechanism. To test this hypothesis, we applied HFS to the Schaffer commissural pathway (Fig.2). A train of stimuli was delivered to one pathway (S1); then the slices were treated with BDNF (2 nm) for 3 hr, and the identical train was applied to a second pathway (S2). The contribution of three variables was examined independently: stimulus duration, number of pulses, and stimulus frequency. When stimulation trains with different frequencies but fixed duration (400 msec) were applied, slices treated with BDNF exhibited higher EPSP slopes at the end of the trains than control slices only when the stimulation frequency reached 100 Hz (Fig.3a). Similarly, when stimulation trains with different frequencies and with a fixed number of pulses (40 pulses) were applied, a significant difference was observed between the control and BDNF-treated slices only at the highest stimulation frequency (100 Hz; Fig. 3b). Finally, when stimulation frequencies were fixed at 100 Hz, BDNF enhanced synaptic responses at the 20th, 40th, 70th, and 100th pulses of the trains (Fig. 3c). Moreover, in all cases, BDNF prevented EPSP depression only during the stimulation trains when the synapses exhibited an obvious fatigue (when stimulated at 100 Hz). The effect of BDNF was not attributable to any time-dependent changes that may have occurred while the slices were incubating in the chamber. The synaptic responses to HFS remain unchanged for up to 4 hr (31% ± 2.88,n = 14 at time 0 vs 34% ± 2.01, n = 14 at 4 hr later). Therefore, the attenuation of synaptic fatigue by BDNF is dependent preferentially on the stimulation frequency rather than on the number of pulses or stimulus duration.
Fig. 2.

Synaptic responses to repetitive stimulation at different frequencies. Shown are representative field EPSPs elicited by trains of afferent stimulation at the indicated frequencies. Because no difference was seen between control and BDNF-treated slices of EPSPs elicited by 25 and 50 Hz stimulation, only control traces are presented. At 100 Hz, EPSPs from BDNF-treated slices are superimposed on those from control slices.
Fig. 3.

Effect of BDNF on synaptic responses to repetitive afferent stimulation is dependent on the stimulation frequency. Repetitive stimulation was delivered to afferent fibers. One of three variables (duration, number of pulses, or stimulation frequency) was held constant. The last EPSP slopes at different conditions were calculated as the percentage of the first EPSP slope. The data were analyzed by ANOVA, followed by Fisher’s post hoc test (*p < 0.05). In this and all of the other figures, the numbers associated with each columnrepresent the number of pathways recorded. a, BDNF effect on EPSPs elicited by stimulation at different frequencies with a fixed train duration (400 msec). BDNF affected only responses to stimulation at 100 Hz. b, BDNF effect on EPSPs elicited by stimulation at different frequencies with a fixed number of pulses (40 pulses). Only those elicited by 100 Hz stimulation exhibited a difference between control and BDNF-treated slices. c, BDNF effect on EPSPs elicited by stimulation with different numbers of pulses at a fixed frequency (100 Hz). Data on the 20th, 40th, 70th, and 100th pulse are shown. In all cases the EPSP slopes are higher in BDNF-treated slices as compared with the control.
One possible effect of BDNF is to recruit more presynaptic fibers when afferent pathways are stimulated at high frequency. Because the number of activated fibers is proportional to the amplitude of the fiber volley in extracellular recordings, we compared the amplitude of the fiber volley elicited by HFS in control and BDNF-treated pathways. To isolate the fiber volley from the field EPSP, we incubated slices in a low-calcium ACSF (0.5 mm[Ca2+]o plus 0.5 mmcadmium) to block the influx of calcium (Fig.4a). The ratio of the amplitudes of the 40th fiber volley relative to the first elicited by a train of 100 Hz stimulation remained unchanged in the same pathway before and 3 hr after BDNF treatment (Fig. 4b,c, Control: 89%, n = 10; BDNF: 80%, n = 10). Similar results were obtained by using a combination of the NMDA receptor antagonist 2-amino-5-phosphonovalerate (APV, 100 μm) and the non-NMDA receptor antagonist 6-cyano-7-nitroquinozaline-2,3-dione (CNQX, 50 μm; Control: 71% ± 2.7, n = 4; BDNF: 65% ± 5,n = 6) or by using the noncompetitive glutamate receptor antagonist kynurenic acid (10 mm; Control: 86% ± 5.9, n = 19; BDNF: 89% ± 9.4,n = 12). Thus, BDNF did not appear to recruit more active presynaptic fibers during HFS.
Fig. 4.

Effect of BDNF on fiber volley. To better visualize the fiber volley elicited by high-frequency stimulation, we treated slices with a low-calcium ACSF ([Ca2+]o = 0.5 mm) plus cadmium (0.5 mm) to block the influx of calcium.a, Representative traces of EPSPs before (left) and after (right) the application of cadmium/0.5 mm [Ca2+]oACSF. Note that this treatment virtually eliminated the EPSP, allowing for the isolation of the fiber volley (arrow), which is masked by the EPSP in the control trace (left).b, Representative traces of the first and 40th fiber volleys during a 100 Hz train of afferent stimulation before (left) and 3 hr after (right) BDNF treatment in the same slice. Vertical scale: 1 mV for aand 0.5 mV for b; horizontal scale: 100 msec fora and 70 msec for b. c, Summary of BDNF effect on fiber volley. A train of 100 Hz pulses was delivered to the slices for 1 sec, and the isolated fiber volley was recorded. The amplitude of the 40th fiber volley is presented as a percentage of the first fiber volley in control and BDNF-treated slices. No significant differences were found in the fiber volley amplitudes between control and BDNF-treated slices (Control: 89% ± 6.9, n = 10; BDNF: 80% ± 5.6,n = 10).
Another way to investigate a potential presynaptic effect is to determine whether BDNF alters PPF, the enhancement of a second EPSP relative to the first, elicited by two successive stimulation pulses. This facilitation is believed to reflect an enhancement of transmitter release from presynaptic terminals (Creager et al., 1980; Wigstrom and Gustafsson, 1981; Foster and McNaughton, 1991; Schultz et al., 1994). In control slices maximal PPF occurred when interpulse intervals were between 30 and 50 msec, and the facilitation declined when the two pulses were closer than 20 msec (Fig. 5) (see also Castro-Alamancos and Connors, 1997; Dobrunz et al., 1997). In slices treated with BDNF there was a significant increase in PPF, particularly in those at shorter intervals (7, 10, and 20 msec, Fig.5b). It is important to note that the enhancement in the PPF was attributable to an increase of the second EPSP rather than a decrease in the first EPSP of the pair (Fig. 5a). These data are consistent with the observation that BDNF does not alter synaptic responses to low-frequency stimulation (see Fig. 1).
Fig. 5.

BDNF enhances paired-pulse facilitation at short interpulse intervals. Each slice was tested for PPF in one pathway at different interpulse intervals from 7 to 200 msec (only intervals 7–75 msec are shown). BDNF was added to the perfusion medium; 3 hr later a second pathway was stimulated with paired stimuli at the same intervals. a, Representative examples of PPF recorded at 10 or 50 msec interpulse intervals before and after BDNF treatment. Traces of the first and second EPSPs are superimposed. Note that, at the 10 msec interval, BDNF significantly increased the size of the second EPSP without affecting the size of the first EPSP.b, PPF ratio curves showing facilitation at different interpulse intervals. BDNF significantly affected PPF only at interpulse intervals between 7 and 20 msec (*p < 0.05, ANOVA).
To test whether BDNF affects transmitter release at CA1 synapses, we varied the extracellular Ca2+ concentration ([Ca2+]o). It is widely accepted that changes in [Ca2+]omodify the probability of transmitter release (Pr) from presynaptic terminals (Katz and Miledi, 1968), having a direct influence on short-term synaptic plasticity (for review, see Zucker, 1989). Elevation of [Ca2+]o will increase thePr from individual stimuli, leading to a faster depletion of presynaptic vesicles during repetitive HFS and, therefore, more pronounced synaptic fatigue. A decrease in [Ca2+]o will have the opposite effect. Indeed, reducing the [Ca2+]o from 2.5 mm in normal ACSF to 0.5 mm led to significantly less synaptic fatigue (Fig.6a). Synaptic responses to HFS under different conditions were measured as a percentage of the 40th EPSP slope over the first EPSP slope (from here on referred to as “response to HFS”). The response to HFS improved in lower [Ca2+]o, increasing from 33% in 2.5 mm [Ca2+]o to 68% in 0.5 mm [Ca2+]o. In contrast, increasing [Ca2+]o to 5.0 mm resulted in much more severe synaptic fatigue: the response to HFS declined from 33% in 2.5 mm[Ca2+]o to 17% in 5 mm[Ca2+]o.
Fig. 6.

The effect of BDNF on synaptic fatigue at different [Ca2+]o. a, BDNF effect on EPSPs elicited by HFS (100 Hz, 1 sec) at different [Ca2+]o. Synaptic responses to HFS under different conditions are measured as a percentage of the 40th EPSP slope over the first EPSP slope. The data were analyzed by ANOVA, followed by Fisher’s post hoc test. Statistically significant differences between a control and BDNF pair at a particular [Ca2+]o are marked by anasterisk (p < 0.05). Thenumbers associated with each column are as follows. At 0.5 mm[Ca2+]o, Control: 68% ± 9.0,n = 3; BDNF: 91% ± 15.3, n = 4. At 2.5 mm[Ca2+]o, Control: 33% ± 3.1,n = 3; BDNF: 68% ± 6.1, n = 4. At 5.0 mm[Ca2+]o, Control: 17% ± 0.33,n = 3; BDNF: 39% ± 4.1, n = 4. Note that at 0.5 mm[Ca2+]o the difference between control and BDNF groups is not statistically significant. b, The percentage of change seen after the addition of BDNF in the various [Ca2+]o during HFS. All calculations are done with data from a. The percentage of BDNF effect under different [Ca2+]o = (BDNF − Control)/Control (0.5 mm, 33%; 2.5 mm, 106%; 5.0 mm, 125%).
Using the synaptic fatigue during HFS as a parameter of presynaptic function, we determined whether BDNF alters the effectiveness of changing [Ca2+]o. First, if both BDNF and lowering [Ca2+]o affect transmitter release during HFS in a similar manner, treatment of slices in normal [Ca2+]o with BDNF should mimic the effect of lowering [Ca2+]oin control slices, and the two manipulations should occlude each other. Indeed, treatment with BDNF and lowering [Ca2+]o had similar effects (Fig.6a). Both BDNF-treated slices in normal [Ca2+]o (response to HFS, 68%) and slices in lower [Ca2+]o (68%) exhibited much less fatigue than the control slices in normal [Ca2+]o (33%) (p < 0.05; Mann–Whitney U test). Moreover, treatment with BDNF became less effective when combined with lowering [Ca2+]o (Fig. 6a), and the percentage of BDNF effect in 0.5 mm[Ca2+]o was significantly smaller than that in 2.5 mm [Ca2+]o(Fig. 6b). Thus, BDNF was less effective in preventing synaptic fatigue when Pr was already reduced by lowering [Ca2+]o. It should be noted, however, that treatment with BDNF and lowering [Ca2+]o did not occlude each other completely, although the difference between control and BDNF groups in 0.5 mm [Ca2+]o was not statistically significant (Fig. 6a). Second, if the BDNF modulation of synaptic fatigue was purely postsynaptic, changes in [Ca2+]o should not affect the magnitude of the BDNF effect. However, whereas the magnitude (%) of BDNF effect was similar in normal and higher [Ca2+]o, it was much less in lower [Ca2+]o. The percentage of increase in synaptic responses to HFS elicited by BDNF treatment was 33, 106, and 125, respectively (Fig. 6b). The differential effects of changing [Ca2+]o on synaptic fatigue in control and BDNF-treated pathways suggest that BDNF modulates transmitter release when the synapses are stimulated at high frequency.
We then examined the effect of changing [Ca2+]o on PPF in control and BDNF-treated slices. We chose to examine the PPF at the 10 msec interpulse interval, which exhibited the largest effect of BDNF in normal [Ca2+]o (see Fig.5b). In control slices, lowering [Ca2+]o to 0.5 mmdecreased whereas elevating [Ca2+]o to 5 mm increased the first EPSP of the pair, leading to an increase and decrease in PPF, respectively (Fig.7a). BDNF increased PPF in all three [Ca2+]o. However, the effect of BDNF in lower [Ca2+]o was relatively small, although comparison between control and BDNF groups still showed statistically significant differences (Fig. 7a, 0.5 mm [Ca2+]o). In contrast, treatment with BDNF became more effective when combined with increasing [Ca2+]o (Fig.7b, 5 mm[Ca2+]o). Thus, the net effect of BDNF at enhancing PPF was slightly smaller whenPr was reduced by lowering [Ca2+]o (19%) and was significantly larger when Pr was increased by raising [Ca2+]o (70%), as compared with that in normal [Ca2+]o (23%) (Fig.7b). Since changing [Ca2+]ois known to alter transmitter release, modulating the effectiveness of changing [Ca2+]o with BDNF supports the notion that BDNF may modulate transmitter release mechanisms.
Fig. 7.

The effect of BDNF on PPF at different [Ca2+]o. a, The effect of BDNF on PPF at a 10 msec interpulse interval at different [Ca2+]o. PPF is measured as the ratio of facilitation (%). The data were analyzed by ANOVA, followed by Fisher’s post hoc test. Statistically significant differences between a control and BDNF pair at a particular [Ca2+]o are marked by anasterisk (p < 0.05). Thenumbers associated with each column are as follows. At 0.5 mm[Ca2+]o, Control: 188% ± 4.3,n = 6; BDNF: 224 ± 11.6,n = 10. At 2.5 mm[Ca2+]o, Control: 144% ± 8.2,n = 8; BDNF: 178% ± 7.3, n = 8. At 5.0 mm[Ca2+]o, Control: 89% ± 4.6,n = 6; BDNF: 151% ± 8.7, n = 12. b, The percentage change of PPF seen after the addition of BDNF in the various [Ca2+]o. All calculations are done with data from a. The percentage of BDNF effect under different [Ca2+]o = (BDNF − Control)/Control (0.5 mm, 19%; 2.5 mm, 23%; 5.0 mm, 70%).
Despite the evidence for a presynaptic effect of BDNF on high-frequency synaptic transmission, the possibility exists that HFS elicits a gradual desensitization of postsynaptic glutamate receptors and that BDNF reduces or prevents this receptor desensitization. Using selective antagonists of NMDA and non-NMDA glutamate receptors (APV and CNQX, respectively), we found that the field EPSPs elicited by HFS were mediated predominantly by non-NMDA receptors (data not shown). If BDNF reduces the desensitization of postsynaptic non-NMDA receptors during HFS, the blockade of non-NMDA receptor desensitization by specific inhibitors such as cyclothiazide (CTZ) or aniracetam (Ito et al., 1990;Tang et al., 1991; Yamada and Rothman, 1992) should prevent the BDNF effect. We found that incubation of the slices with CTZ (0.1 mm, 30 min) alone did not alter the synaptic depression during HFS (Fig. 8a) (see alsoDobrunz and Stevens, 1997). The responses to HFS were 43 and 44% in control and CTZ-treated slices, respectively. The response to HFS after the application of BDNF to the CTZ-treated slices was not altered in magnitude and was comparable to that observed in slices treated with BDNF alone (Fig. 8a, BDNF, 71%; BDNF/CTZ, 77%). Similar results were seen in slices treated with another inhibitor of non-NMDA receptor desensitization, aniracetam (5 mm) (Fig.8b, Control, 41%; Anirac., 39%; Anirac./BDNF, 86%). Thus, the attenuation of synaptic fatigue by BDNF is not attributable to a reduction of the desensitization of postsynaptic non-NMDA receptors.
Fig. 8.
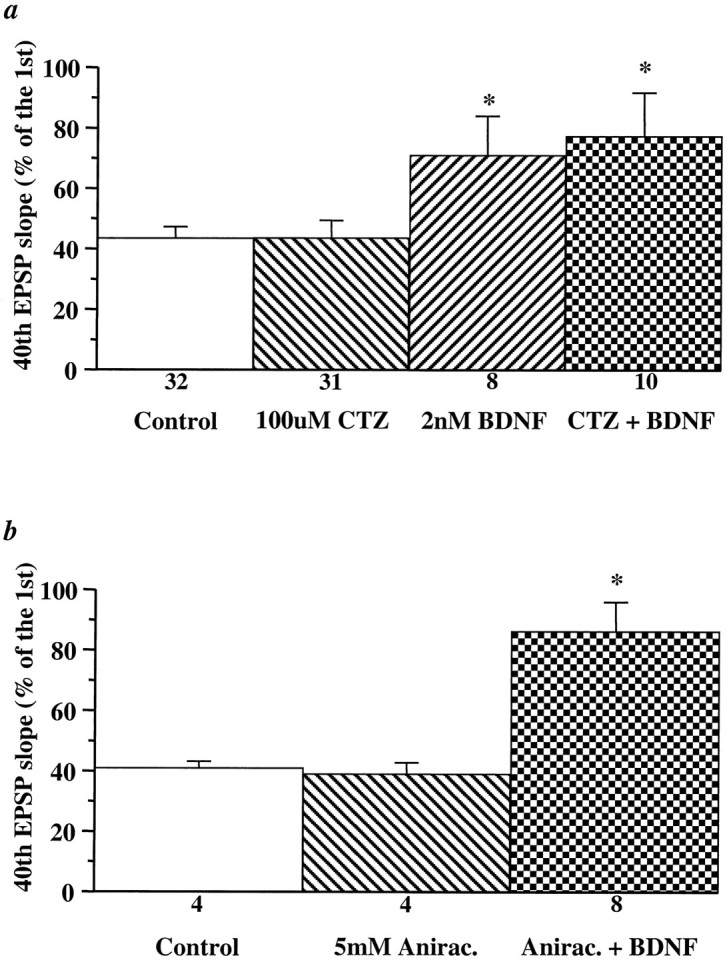
Blockade of desensitization of non-NMDA-type glutamate receptors does not alter the BDNF-induced increases in synaptic responses to HFS. Cyclothiazide (a,CTZ, 0.1 mm) or aniracetam (b, Anirac., 5 mm) was applied to the slices either alone or 30 min before BDNF application. EPSPs evoked by HFS were compared before and 3 hr after BDNF and CTZ or aniracetam application. The slopes of the 40th EPSP are presented as a percentage of the first EPSP slope. The magnitude of the BDNF attenuation of the synaptic fatigue during HFS is unaltered in the presence of CTZ or aniracetam (*p < 0.05, ANOVA).
It has been reported that BDNF could modulate NMDA channels directly by a mechanism not involving TrkB receptors (Jarvis et al., 1997). Our previous study showed that EPSPs elicited by low-frequency stimulation in the presence of CNQX and low magnesium remained unchanged up to 3 hr after BDNF application, suggesting that BDNF does not affect NMDA components of field EPSPs (Figurov et al., 1996). Unlike the direct effect of BDNF on NMDA channels reported by Jarvis et al. (1997), the BDNF effect on synaptic responses to HFS was blocked by K252A (0.2 μm), a specific inhibitor for Trk receptor tyrosine kinases (Control: 39% ± 4.7, n = 6; K252A: 43% ± 5.1, n = 5; K252A/BDNF: 41% ± 3.9, n= 12). To test the involvement of NMDA receptors further, we examined the effects of BDNF on synaptic responses during the HFS in the presence of the NMDA receptor blocker APV (100 μm). No significant difference was seen between control slices and those treated with APV (Control: 45% ± 5.6, n = 8; APV: 60% ± 5.9, n = 8; p > 0.05). In addition, BDNF still rescued synaptic fatigue in the presence of APV (APV/BDNF: 83% ± 3.9, n = 6). Thus, it is unlikely that directly regulation of NMDA channels by BDNF contributes to the BDNF effect on synaptic responses to HFS.
Finally, we examined the physiological significance of the presynaptic, selective enhancement of high-frequency synaptic transmission by BDNF. Our previous experiments indicated that exogenous BDNF promotes tetanus-induced LTP in neonatal hippocampal slices, which in the absence of BDNF exhibit only short-term potentiation (Figurov et al., 1996). This effect is thought to be attributable to, at least in part, the reduced synaptic depression during HFS after BDNF treatment, leading to prolonged postsynaptic depolarization. Although BDNF selectively regulates high-frequency transmission (see Fig. 3), it is unclear whether the enhanced responses to tetanus directly affect LTP or whether the effect of BDNF on LTP is mediated indirectly by the release of additional diffusible factors. To test this idea, we monitored synaptic efficacy in two independent Schaffer commissural pathways within CA1 stratum radiatum (∼300 μm apart). In slices treated with BDNF for 3 hr, only the tetanized pathway (S1) exhibited LTP (Fig. 9). The EPSPs recorded from the untetanized pathway (S2) remained unchanged throughout the experiment. Thus, the facilitating effect of BDNF on LTP in neonatal slices appears to be a direct consequence of selective potentiation of the tetanized synapses.
Fig. 9.

BDNF selectively facilitates the induction of LTP in the tetanized pathway without affecting the synaptic efficacy of the untetanized pathway. EPSPs were recorded in the CA1 area of BDNF-treated slices. Two stimulating electrodes were positioned on either side of a single recording electrode to stimulate two different groups of afferents converging in the same dendritic field in CA1. Stimulation was applied to Schaffer collaterals alternately at low frequency (1 per min). After a period of baseline recording, LTP was induced with a theta burst stimulation applied at time 0only to one pathway (S1, filled squares). Simultaneous recording of an independent pathway (S2,open circles) showed no change in its synaptic strength after the theta burst was delivered to S1. Synaptic efficacy (initial slope of field EPSPs) is expressed as a percentage of baseline value recorded during the 20 min before the tetanus. Representative traces of field EPSPs from S1 andS2 pathways were taken 10 min before and 40 min after the theta burst stimulation.
DISCUSSION
Pharmacological and genetic studies have demonstrated that BDNF plays a role in hippocampal LTP (Korte et al., 1995, 1996; Figurov et al., 1996; Patterson et al., 1996; Kang et al., 1997). In addition, the effect of BDNF on LTP appears to be mediated by an enhanced ability of hippocampal synapses to follow high-frequency afferent stimulation (Figurov et al., 1996). The goal of the present study was to determine whether BDNF regulates hippocampal plasticity via a presynaptic mechanism. We focused on phenomena known to alter presynaptic function, such as PPF, responses to HFS, and changes in [Ca2+]o. We also used pharmacological agents that block the desensitization of non-NMDA postsynaptic glutamate receptors. Our results support a presynaptic mechanism for the effects of BDNF observed in hippocampal CA1 synapses. These data may provide further insight into the mechanisms of the neurotrophin modulation of CNS synaptic maturation and plasticity.
Although we are not excluding a postsynaptic effect, we have provided three pieces of evidence for a presynaptic action of BDNF.
(1) The effect of BDNF on repetitive excitatory synaptic responses is dependent on the stimulation frequency. A wide variety of experiments suggests that HFS could rapidly deplete the releasable pool of quanta or synaptic vesicles in presynaptic terminals, leading to synaptic fatigue or depression (Zucker, 1989; Larkman et al., 1991; Dobrunz and Stevens, 1997). It is logical to propose that a modulation that could prevent synaptic fatigue is presynaptic in nature as well. However, during HFS the synapses also receive more stimulation pulses. To exclude the possibility that the effect of BDNF is dependent on the number of pulses, we performed experiments in which the same number of pulses was delivered to CA1 synapses at different frequencies. We showed that BDNF is effective only when the synapses were stimulated at 100 Hz or higher, which induced severe synaptic fatigue. Thus, BDNF may act via a presynaptic mechanism that prevents synaptic fatigue. Considering that hippocampal synaptic fatigue often is associated with immature synapses (Figurov et al., 1996), these data not only support a presynaptic action of BDNF but also raise the possibility that BDNF is involved in synapse maturation during hippocampal development.
(2) We analyzed BDNF modulation of PPF, a simple and reliable measure of presynaptic properties with very few assumptions, especially in hippocampal CA1 synapses (Foster and McNaughton, 1991; Schultz et al., 1994; Dobrunz et al., 1997). A modulation of the PPF strongly indicates a presynaptic change, although an additional postsynaptic component cannot be excluded. We showed that BDNF-treated slices exhibited higher PPF at interpulse intervals ≤20 msec. Because PPF is thought to result from higher Pr to the second pulse attributable to residual presynaptic [Ca2+]i left from the first pulse, an increase in the PPF after BDNF application suggests that BDNF may regulate [Ca2+]i dynamics within presynaptic terminals, leading to an enhancement in transmitter release (Zucker, 1989).
(3) We also examined PPF and synaptic responses to HFS under different [Ca2+]o. Elevating [Ca2+]o increases thePr to a single stimulus, leading to more rapid depletion of synaptic vesicles during HFS and, thus, more severe synaptic fatigue. Increasing Pr also decreases the PPF, presumably because fewer releasable vesicles are available after an initial successful release. The Pr in CA1 excitatory synapses from neonatal hippocampus is high (0.9), and it gradually decreases as the animals mature (<0.5) (Bolshakov and Siegelbaum, 1995). Thus, immature CA1 synapses have a lower PPF and more pronounced fatigue during HFS. We showed that BDNF mimicked the effect of lowering [Ca2+]o in PPF and responses to HFS. Moreover, BDNF became much less effective when combined with lowering [Ca2+]o, although the two manipulations did not occlude each other completely. Finally, changing [Ca2+]o clearly altered the magnitude of BDNF effects: in lower [Ca2+]o BDNF attenuated synaptic fatigue less effectively, whereas in higher [Ca2+]o BDNF potentiated PPF more effectively. Taken together, our data support the notion that BDNF regulates transmitter release mechanisms in neonatal hippocampus, leading to conditions of reduced synaptic fatigue and enhanced synaptic facilitation. However, because lowering [Ca2+]o did not occlude the BDNF effect completely, we could not exclude the possibility that BDNF also acts on sites different from that modulated by Ca2+.
This study also provides evidence that the reduced synaptic fatigue in BDNF-treated slices is not likely to be attributable to an attenuation of non-NMDA receptor desensitization. We showed that the magnitude of the BDNF effect on synaptic fatigue was unaltered in the presence of CTZ, a compound that blocks the desensitization of non-NMDA glutamate receptors. Non-NMDA receptors in hippocampal CA1 synapses have relatively fast deactivation (4.5 msec) and slow desensitization time constants (10 msec) (Trussell and Otis, 1996), and the waveform of the EPSP is dominated mostly by receptor deactivation (Jonas and Spruston, 1994). Assuming that the glutamate released in the synaptic cleft has a lifetime of ∼2 msec (Clements, 1996), the majority of non-NMDA receptors probably has not entered the desensitized state, even when synapses are stimulated at 100 Hz. Consistent with this point, CTZ alone did not have a significant effect on the HFS response. In addition to its postsynaptic effect, CTZ has been shown to enhance presynaptic transmitter release (Diamond and Jahr, 1995). However, when we used aniracetam, which also blocks non-NMDA receptor desensitization with little known presynaptic effects, we observe similar attenuation of synaptic fatigue by BDNF.
Although we have demonstrated a lack of BDNF effect on non-NMDA receptor desensitization, possibilities exist that BDNF may still have other postsynaptic effects. HFS causes postsynaptic membrane depolarization, decreasing the driving force for Na+and K+ ion flow, and thus reducing the EPSP amplitude. BDNF may regulate ion channel properties, allowing for faster repolarization of postsynaptic membranes. Indeed, BDNF has been shown to enhance the expression of K+ channels that are critical in membrane repolarization (Du and McBain, 1997). Another possibility is that BDNF induces morphological changes in postsynaptic dendrites and/or spines. In slices derived from neonatal ferret visual cortex, long-term treatment with BDNF elicited a dramatic change in dendritic arborizations (McAllister et al., 1995, 1996). In hippocampal neuronal cultures the BDNF-induced increase in the amplitudes of EPSPs could be blocked by loading postsynaptic cells with K252A (Levine et al., 1995). BDNF also could modulate NMDA channels directly via mechanisms not involving TrkB receptor tyrosine kinases (Jarvis et al., 1997). Although NMDA receptors did not contribute significantly to the field EPSP (however, see Muller et al., 1990) and BDNF still enhances synaptic responses to HFS in the presence of APV, our present study did not rule out completely the possibility that recruitment of NMDA receptor activity during HFS may be altered by BDNF. Neurotrophic modulation of postsynaptic structure and function represents an important new area of research that requires further investigation.
The present study demonstrated that BDNF preferentially potentiates synaptic responses to HFS. An intuitive interpretation of the link between the effect of BDNF on HFS responses and on LTP would be that an enhanced response to the tetanus could lead to more sustained postsynaptic depolarization and larger Ca2+ influx, an essential step in LTP induction (Bliss and Collingridge, 1993). Indeed, our previous study showed that BDNF facilitates hippocampal LTP by potentiating synaptic responses to a tetanus without affecting the magnitude of TEA-induced LTP, suggesting that the LTP-triggering mechanism or mechanisms are not affected by BDNF (Figurov et al., 1996). However, this notion is not firmly established because others have shown an impairment in pairing-induced LTP in BDNF knock-out mice (Korte et al., 1995). Alternatively, one could imagine that an enhanced synaptic activity during tetanus could induce the release of additional diffusible factors, which in turn may contribute to LTP. If the latter were the case, one would predict a potentiation of other untetanized synapses within the same slice as well. On the contrary, we showed that BDNF treatment potentiated the tetanized pathway without affecting the synaptic efficacy of the untetanized pathway in the same P12–P13 slices. However, we cannot exclude the possibility that some diffusible factors may affect synapses immediately adjacent to the tetanized synapses. With the use of an elegant local superfusion technique, it has been shown recently that even the input specificity of LTP breaks down at short distances (Engert and Bonhoeffer, 1997). LTP can spread to naive synapses as far as 70 μm from the tetanized synapses. Although the spread of LTP and the potential spread of the effect of BDNF are two separate issues, they share similar conceptual constraints. Nevertheless, our data suggest that the effect of BDNF is relatively spatially restricted within CA1 stratum radiatum (∼300 μm).
Our finding that the presynaptic effect of BDNF is dependent on stimulation frequency suggests a model that may help to understand how neurotrophins may serve as retrograde signals for activity-dependent processes, such as synaptic competition during development and synaptic plasticity in the adult (Fig. 10). First, our model provides a different way to look at the involvement of neurotrophins in the Hebbian-type synaptic plasticity. The widely accepted Hebb’s hypothesis predicts that more active synapses are favored during synaptic competition (Stent, 1973). It has been postulated that activity-dependent synthesis and/or release of neurotrophins may mediate this positive reinforcement (Thoenen, 1995). However, because BDNF is a soluble protein, it probably will diffuse once it is released and act on neighboring synapses. Our present findings indicate that BDNF preferentially enhances highly active synapses, and this effect may contribute directly to the BDNF effect on LTP rather than indirectly by some additional diffusible factors. The physiological consequence of the BDNF regulation of the responses to tetanus is relatively confined and will not spread to distant synapses. Thus, even if the release of BDNF is not restricted to one active synapse, it still could favor more active synapses (Fig. 10). Second, the selective pairing of high-frequency neuronal activity and BDNF concentration potentially could provide the signal for coincidence detection, analogous to presynaptic activity and postsynaptic membrane depolarization leading to NMDA receptor activation in LTP induction. Finally, the presynaptic action of BDNF supports the hypothesis that this neurotrophin may serve as a retrograde factor that regulates synaptic plasticity.
Fig. 10.

Schematic model for the selective potentiation of active synapses by BDNF. High-frequency stimulation elicits synthesis and/or release of BDNF, increasing its concentration surrounding the hippocampal neurons. BDNF may potentiate transmitter release only at synapses that undergo high-frequency transmission (red) without affecting less active synapses (blue).
Footnotes
We express our gratitude to Drs. Chris McBain [National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH)], Elis Stanley (National Institute of Neurological Diseases and Stroke, NIH), and Serena Dudek (NICHD, NIH) for helpful discussions and critical comments on this manuscript; Takafumi Inoue (University of Tokyo) for acquisition and analysis software; and Dr. Colombe Chappey (National Center for Biotechnology Information, NIH) for discussions on statistical analyses.
Correspondence should be addressed to Dr. Bai Lu, Unit on Synapse Development and Plasticity, National Institute of Child Health and Human Development, National Institutes of Health, Building 49, Room 5A38, 49 Convent Drive, Bethesda, MD 20892-4480.
REFERENCES
- 1.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 3.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 4.Cabelli RJ, Horn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 5.Castro-Alamancos MA, Connors BW. Distinct forms of short-term plasticity at excitatory synapses of hippocampus and neocortex. Proc Natl Acad Sci USA. 1997;94:4161–4166. doi: 10.1073/pnas.94.8.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clements JD. Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 7.Creager R, Dunwiddie T, Lynch G. Paired pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 9.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1018. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 10.Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci USA. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du J, Tao-Cheng J-H, Zerfas P, McBain CJ. The subcellular distribution of the K+ channel subunit kVZ.1 in hippocampal and cortical neurons and the regulation of K+ channel expression of BDNF and NGF. Soc Neurosci Abstr. 1997;23:680.13. [Google Scholar]
- 12.Engert F, Bonhoeffer T. Synapse specificity of long-term potentiation breaks down at short distances. Nature. 1997;388:279–284. doi: 10.1038/40870. [DOI] [PubMed] [Google Scholar]
- 13.Figurov A, Pozzo-Miller L, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 14.Foster TC, McNaughton BL. Long-term synaptic enhancement in CA1 is due to increased quantal size, not quantal content. Hippocampus. 1991;1:79–91. doi: 10.1002/hipo.450010108. [DOI] [PubMed] [Google Scholar]
- 15.Gu Q, Liu Y, Cynader MS. Nerve growth factor-induced ocular dominance plasticity in adult cat visual cortex. Proc Natl Acad Sci USA. 1994;91:8408–8412. doi: 10.1073/pnas.91.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito I, Tanabe S, Kohda A, Sugiyama H. Allosteric potentiation of quisqualate receptors by a nootropic drug aniracetam. J Physiol (Lond) 1990;424:533–543. doi: 10.1113/jphysiol.1990.sp018081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- 18.Jonas P, Spruston N. Mechanisms shaping glutamate-mediated excitatory postsynaptic currents in the CNS. Curr Opin Neurobiol. 1994;4:366–372. doi: 10.1016/0959-4388(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 19.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 20.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 21.Kang H, Jia LZ, Suh KY, Tang L, Schuman EM. Determinants of BDNF-induced hippocampal synaptic plasticity: role of the TrkB receptor and the kinetics of neurotrophin delivery. Learn Memory. 1996;3:188–196. doi: 10.1101/lm.3.2-3.188. [DOI] [PubMed] [Google Scholar]
- 22.Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 23.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits γ-aminobutyrateric synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knipper M, da Penha Berzaghi M, Blochl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur J Neurosci. 1994;6:668–671. doi: 10.1111/j.1460-9568.1994.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 26.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkman A, Stratford K, Jack J. Quantal analysis of excitatory synaptic action and depression in hippocampal slices. Nature. 1991;350:344–347. doi: 10.1038/350344a0. [DOI] [PubMed] [Google Scholar]
- 29.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 30.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liou JC, Yang RS, Fu WM. Regulation of quantal secretion by neurotrophic factors at developing motoneurons in Xenopus cell cultures. J Physiol (Lond) 1997;503:129–139. doi: 10.1111/j.1469-7793.1997.129bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 33.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 34.Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Maffei L, Berardi N, Domenici L, Parisi V, Pizzorusso T. Nerve growth factor (NGF) prevents the shift in ocular dominance distribution of visual cortical neurons in monocularly deprived rats. J Neurosci. 1992;12:4651–4662. doi: 10.1523/JNEUROSCI.12-12-04651.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 37.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 38.Muller D, Lynch G. Synaptic modulation of N-methyl-d-aspartate receptor-mediated responses in hippocampus. Synapse. 1990;5:94–103. doi: 10.1002/syn.890050203. [DOI] [PubMed] [Google Scholar]
- 39.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knock-out mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 40.Riddle DR, Lo DC, Katz LC. NT-4-mediated rescue of lateral geniculate neurons from effects of monocular deprivation. Nature. 1995;378:189–191. doi: 10.1038/378189a0. [DOI] [PubMed] [Google Scholar]
- 41.Schultz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stent G. A physiological mechanism for Hebb’s postulate of learning. Proc Natl Acad Sci USA. 1973;70:997–1001. doi: 10.1073/pnas.70.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stoop R, Poo MM. Potentiation of transmitter release by ciliary neurotrophic factor requires somatic signaling. Science. 1995;267:695–699. doi: 10.1126/science.7839148. [DOI] [PubMed] [Google Scholar]
- 44.Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang C, Shi Q, Katchman A, Lynch G. Modulation of the time course of fast EPSCs and glutamate channel kinetics by aniracetam. Science. 1991;254:288–290. doi: 10.1126/science.254.5029.288. [DOI] [PubMed] [Google Scholar]
- 47.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–596. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 48.Trussell LO, Otis TS. Physiology of AMPA receptors: biophysical characterizations that subserve the integrative role of synapses. In: Conti F, Hicks TP, editors. Excitatory amino acids and the cerebral cortex. MIT; Cambridge, MA: 1996. pp. 63–72. [Google Scholar]
- 49.Wang T, Xie KW, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wigstrom H, Gustafsson B. Two types of synaptic facilitation recorded in pyramidal cells of in vitro hippocampal slices from guinea pigs. Neurosci Lett. 1981;26:73–78. doi: 10.1016/0304-3940(81)90428-6. [DOI] [PubMed] [Google Scholar]
- 51.Xie K, Wang T, Olafsson P, Mizuno K, Lu B. Activity-dependent expression of NT-3 in muscle cells in culture: implication in the development of neuromuscular junctions. J Neurosci. 1997;17:2947–2958. doi: 10.1523/JNEUROSCI.17-09-02947.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamada KA, Rothman SM. Diazoxide reversibly blocks glutamate desensitization and prolongs excitatory postsynaptic currents in rat hippocampal neurons. J Physiol (Lond) 1992;458:385–407. doi: 10.1113/jphysiol.1992.sp019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]